From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish

Abstract
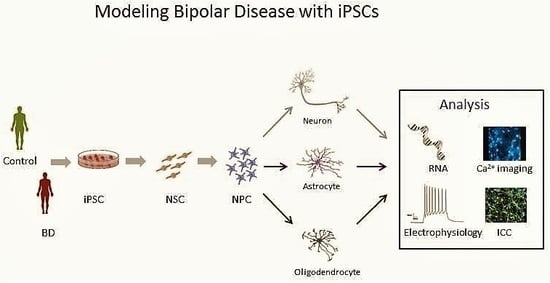
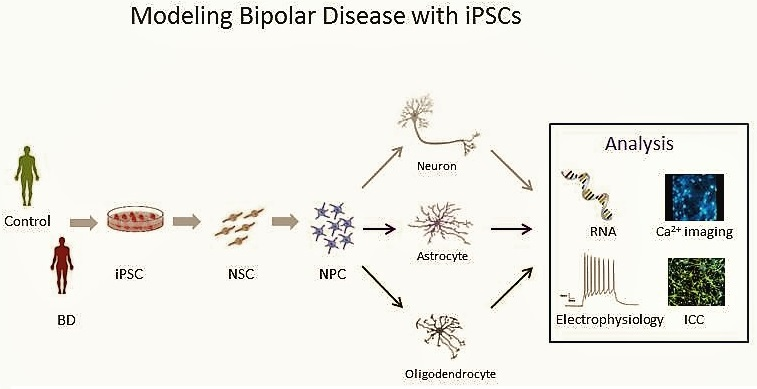
:
1. Introduction
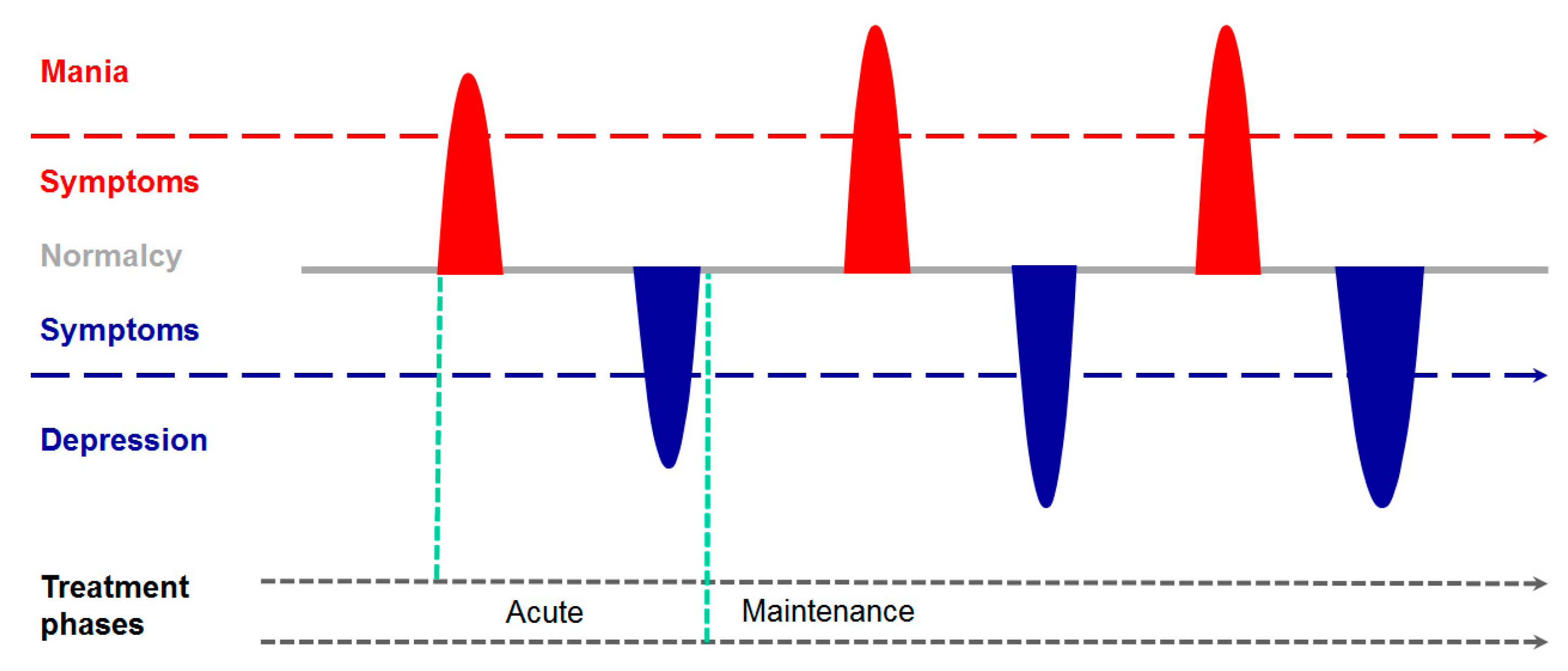
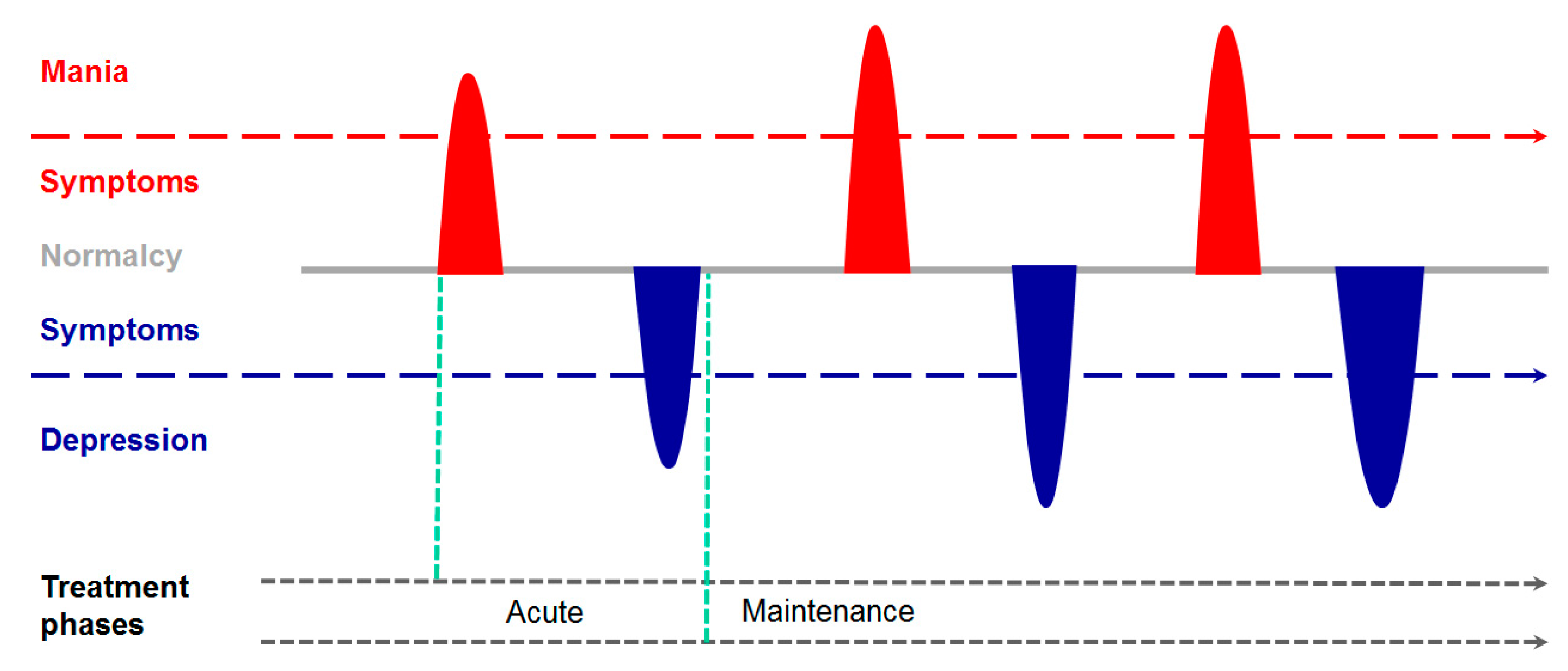
2. From the Psychiatrist’s Couch
3. The Genetic Architecture of BD
4. Neurodevelopmental Anomalies in BD
5. The Promise of iPSCs
6. BD in a Dish
6.1. Neurodevelopmental Alterations in BD-derived iPSCs
6.2. Neuronal Activity in BD-derived iPSCs
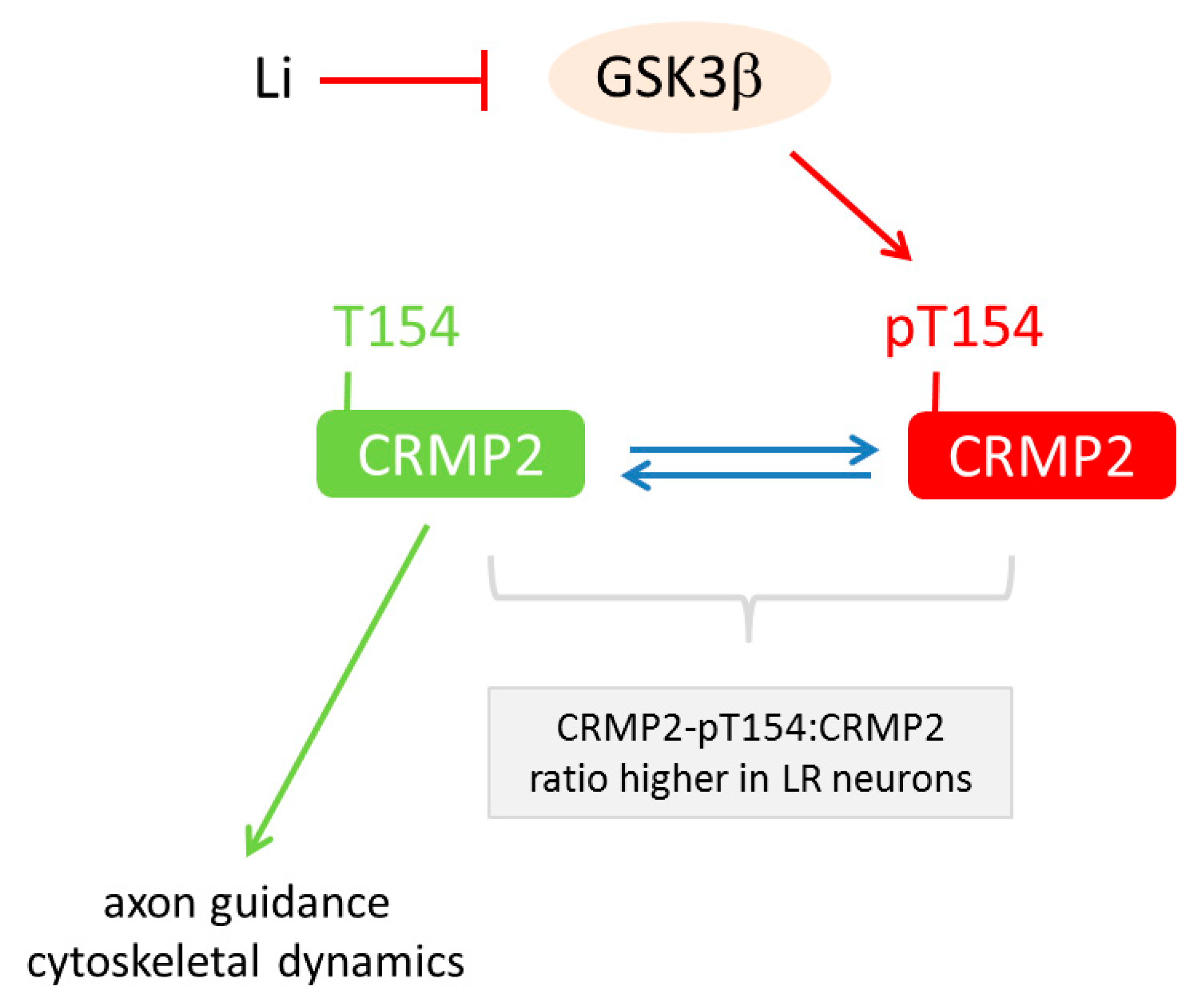
6.3. Lithium-Response Pathways in BD-derived iPSCs
6.4. Select Changes in Gene Expression in BD-derived iPSCs
7. The Road Ahead: Promises, Caveats, and Challenges
7.1. Karyotype Analysis
7.2. Differentiation Capacity
7.3. Study Size
7.4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Strakowski, S.M. (Ed.) The Bipolar Brain: Integrating Neuroimaging with Genetics; Oxford University Press: New York, NY, USA, 2012; ISBN 978-0-19-979760-8. [Google Scholar]
- Institute for Health Metrices and Evaluation. 2017 Global Burden of Disease Study Group 2016. Available online: http://www.healthdata.org/ (accessed on 2 February 2018).
- Moreira, A.L.R.; Van Meter, A.; Genzlinger, J.; Youngstrom, E.A. Review and Meta-Analysis of Epidemiologic Studies of Adult Bipolar Disorder. J. Clin. Psychiatry 2017, 78, e1259–e1269. [Google Scholar] [CrossRef] [PubMed]
- WHO. 2017 Mental Disorders Fact Sheet 2017. Available online: http://www.who.int/mediacentre/factsheets/fs397/en/ (accessed on 20 November 2017).
- Ferrari, A.J.; Stockings, E.; Khoo, J.-P.; Erskine, H.E.; Degenhardt, L.; Vos, T.; Whiteford, H.A. The prevalence and burden of bipolar disorder: Findings from the Global Burden of Disease Study 2013. Bipolar Disord. 2016, 18, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.; Isometsä, E.T.; Tondo, L.; Moreno, D.H.; Turecki, G.; Reis, C.; Cassidy, F.; Sinyor, M.; Azorin, J.-M.; Kessing, L.V.; et al. International Society for Bipolar Disorders Task Force on Suicide: Meta-analyses and meta-regression of correlates of suicide attempts and suicide deaths in bipolar disorder. Bipolar Disord. 2015, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tidemalm, D.; Haglund, A.; Karanti, A.; Landén, M.; Runeson, B. Attempted suicide in bipolar disorder: Risk factors in a cohort of 6086 patients. PLoS ONE 2014, 9, e94097. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G.S.; Outhred, T.; Das, P.; Morris, G.; Hamilton, A.; Mannie, Z. Modeling suicide in bipolar disorders. Bipolar Disord. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Beaulieu, S.; Alda, M.; O’Donovan, C.; Macqueen, G.; McIntyre, R.S.; Sharma, V.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: Update 2013. Bipolar Disord. 2013, 15, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Judd, L.L.; Akiskal, H.S.; Schettler, P.J.; Endicott, J.; Maser, J.; Solomon, D.A.; Leon, A.C.; Rice, J.A.; Keller, M.B. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch. Gen. Psychiatry 2002, 59, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Kupka, R.W.; Altshuler, L.L.; Nolen, W.A.; Suppes, T.; Luckenbaugh, D.A.; Leverich, G.S.; Frye, M.A.; Keck, P.E.; McElroy, S.L.; Grunze, H.; et al. Three times more days depressed than manic or hypomanic in both bipolar I and bipolar II disorder. Bipolar Disord. 2007, 9, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Tohen, M.; Greil, W.; Calabrese, J.R.; Sachs, G.S.; Yatham, L.N.; Oerlinghausen, B.M.; Koukopoulos, A.; Cassano, G.B.; Grunze, H.; Licht, R.W.; et al. Olanzapine versus lithium in the maintenance treatment of bipolar disorder: A 12-month, randomized, double-blind, controlled clinical trial. Am. J. Psychiatry 2005, 162, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Viguera, A.C.; Tondo, L.; Baldessarini, R.J. Sex differences in response to lithium treatment. Am. J. Psychiatry 2000, 157, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, F.K.; Jamison, K.R. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression, 2nd ed.; Oxford University Press: Oxford, UK, 2007; Chapter 20; ISBN 978-0-19-513579-4. [Google Scholar]
- Nurnberger, J.I. General genetics of bipolar disorders. In The Bipolar Brain: Integrating Neuroimaging and Genetics; Stephen, S., Ed.; Oxford University Press: New York, NY, USA, 2012; Chapter 9. [Google Scholar]
- Bienvenu, O.J.; Davydow, D.S.; Kendler, K.S. Psychiatric “diseases” versus behavioral disorders and degree of genetic influence. Psychol. Med. 2011, 41, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Badner, J.A.; Koller, D.; Foroud, T.; Edenberg, H.; Nurnberger, J.I.; Zandi, P.P.; Willour, V.L.; McMahon, F.J.; Potash, J.B.; Hamshere, M.; et al. Genome-wide linkage analysis of 972 bipolar pedigrees using single-nucleotide polymorphisms. Mol. Psychiatry 2012, 17, 818–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgi, B.; Craig, D.; Kember, R.L.; Liu, W.; Lindquist, I.; Nasser, S.; Brown, C.; Egeland, J.A.; Paul, S.M.; Bućan, M. Genomic View of Bipolar Disorder Revealed by Whole Genome Sequencing in a Genetic Isolate. PLoS Genet. 2014, 10, e1004229. [Google Scholar] [CrossRef] [PubMed]
- Ament, S.A.; Szelinger, S.; Glusman, G.; Ashworth, J.; Hou, L.; Akula, N.; Shekhtman, T.; Badner, J.A.; Brunkow, M.E.; Mauldin, D.E.; et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc. Natl. Acad. Sci. USA 2015, 112, 3576–3581. [Google Scholar] [CrossRef] [PubMed]
- Goes, F.S.; Pirooznia, M.; Parla, J.S.; Kramer, M.; Ghiban, E.; Mavruk, S.; Chen, Y.-C.; Monson, E.T.; Willour, V.L.; Karchin, R.; et al. Exome Sequencing of Familial Bipolar Disorder. JAMA Psychiatry 2016, 73, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.E.; Akula, N.; Cabanero, M.; Cardona, I.; Corona, W.; Klemens, B.; Schulze, T.G.; Cichon, S.; Rietschel, M.; Nöthen, M.M.; et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry 2008, 13, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.T.; Jiang, X.; Akula, N.; Shugart, Y.Y.; Wendland, J.R.; Steele, C.J.M.; Kassem, L.; Park, J.-H.; Chatterjee, N.; Jamain, S.; et al. Genome-wide association study meta-analysis of European and Asian-ancestry samples identifies three novel loci associated with bipolar disorder. Mol. Psychiatry 2013, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.R.; O’Donovan, M.C.; Meng, Y.A.; Jones, I.R.; Ruderfer, D.M.; Jones, L.; Fan, J.; Kirov, G.; Perlis, R.H.; Green, E.K.; et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat. Genet. 2008, 40, 1056–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mühleisen, T.W.; Leber, M.; Schulze, T.G.; Strohmaier, J.; Degenhardt, F.; Treutlein, J.; Mattheisen, M.; Forstner, A.J.; Schumacher, J.; Breuer, R.; et al. Genome-wide association study reveals two new risk loci for bipolar disorder. Nat. Commun. 2014, 5, 3339. [Google Scholar] [CrossRef] [PubMed]
- Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Saito, T.; Kondo, K.; Iwata, N. Genome-wide association studies of bipolar disorder: A systematic review of recent findings and their clinical implications. Psychiatry Clin. Neurosci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- CONVERGE Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 2015, 523, 588–591. [Google Scholar] [CrossRef]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Power, R.A.; Steinberg, S.; Bjornsdottir, G.; Rietveld, C.A.; Abdellaoui, A.; Nivard, M.M.; Johannesson, M.; Galesloot, T.E.; Hottenga, J.J.; Willemsen, G.; et al. Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat. Neurosci. 2015, 18, 953–955. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet Lond. Engl. 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium; Lee, S.H.; Ripke, S.; Neale, B.M.; Faraone, S.V.; Purcell, S.M.; Perlis, R.H.; Mowry, B.J.; Thapar, A.; Goddard, M.E.; et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 2013, 45, 984–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Schizophrenia Consortium; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Jablensky, A.; McNeil, T.F.; Morgan, V.A. Barbara Fish and a Short History of the Neurodevelopmental Hypothesis of Schizophrenia. Schizophr. Bull. 2017, 43, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.W.; Murray, R.M. Obstetric complications, neurodevelopmental deviance, and risk of schizophrenia. J. Psychiatr. Res. 1987, 21, 413–421. [Google Scholar] [CrossRef]
- Murray, R.M.; Lewis, S.W. Is schizophrenia a neurodevelopmental disorder? Br. Med. J. Clin. Res. Ed. 1987, 295, 681–682. [Google Scholar] [CrossRef] [PubMed]
- Agnew-Blais, J.; Danese, A. Childhood maltreatment and unfavourable clinical outcomes in bipolar disorder: A systematic review and meta-analysis. Lancet Psychiatry 2016, 3, 342–349. [Google Scholar] [CrossRef]
- Aldinger, F.; Schulze, T.G. Environmental factors, life events, and trauma in the course of bipolar disorder. Psychiatry Clin. Neurosci. 2017, 71, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Dualibe, A.L.; Osório, F.L. Bipolar Disorder and Early Emotional Trauma: A Critical Literature Review on Indicators of Prevalence Rates and Clinical Outcomes. Harv. Rev. Psychiatry 2017, 25, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Etain, B.; Lajnef, M.; Henry, C.; Aubin, V.; Azorin, J.M.; Bellivier, F.; Bougerol, T.; Courtet, P.; Gard, S.; Kahn, J.P.; et al. Childhood trauma, dimensions of psychopathology and the clinical expression of bipolar disorders: A pathway analysis. J. Psychiatr. Res. 2017, 95, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Strakowski, S.M. Bipolar Disorder; Oxford University Press: Oxford, UK, 2014; ISBN 978-0-19-999568-4. [Google Scholar]
- Faedda, G.L.; Marangoni, C.; Serra, G.; Salvatore, P.; Sani, G.; Vázquez, G.H.; Tondo, L.; Girardi, P.; Baldessarini, R.J.; Koukopoulos, A. Precursors of bipolar disorders: A systematic literature review of prospective studies. J. Clin. Psychiatry 2015, 76, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.L.; Swartz, H.A. A critical appraisal of neuroimaging studies of bipolar disorder: Toward a new conceptualization of underlying neural circuitry and a road map for future research. Am. J. Psychiatry 2014, 171, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Passos, I.C.; Mwangi, B.; Vieta, E.; Berk, M.; Kapczinski, F. Areas of controversy in neuroprogression in bipolar disorder. Acta Psychiatr. Scand. 2016, 134, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Selemon, L.D.; Rajkowska, G. Cellular pathology in the dorsolateral prefrontal cortex distinguishes schizophrenia from bipolar disorder. Curr. Mol. Med. 2003, 3, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Benes, F.M.; Vincent, S.L.; Todtenkopf, M. The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol. Psychiatry 2001, 50, 395–406. [Google Scholar] [CrossRef]
- Sanches, M.; Keshavan, M.S.; Brambilla, P.; Soares, J.C. Neurodevelopmental basis of bipolar disorder: A critical appraisal. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1617–1627. [Google Scholar] [CrossRef] [PubMed]
- Folsom, T.D.; Fatemi, S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology 2013, 68, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Estrada, J.; Benítez-King, G.; Berlanga, C.; Meza, I. Altered subcellular distribution of the 75-kDa DISC1 isoform, cAMP accumulation, and decreased neuronal migration in schizophrenia and bipolar disorder: Implications for neurodevelopment. CNS Neurosci. Ther. 2015, 21, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Sindhu, C.; Meissner, A. Molecular features of cellular reprogramming and development. Nat. Rev. Mol. Cell Biol. 2016, 17, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Turksen, K.; Nagy, A. (Eds.) Induced Pluripotent Stem (iPS) Cells: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2016; ISBN 978-1-4939-3055-5. [Google Scholar]
- Verma, P.J.; Sumer, H. (Eds.) Cell Reprogramming: Methods and Protocols; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; ISBN 978-1-4939-2847-7. [Google Scholar]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Paşca, S.P.; Panagiotakos, G.; Dolmetsch, R.E. Generating human neurons in vitro and using them to understand neuropsychiatric disease. Annu. Rev. Neurosci. 2014, 37, 479–501. [Google Scholar] [CrossRef] [PubMed]
- Ardhanareeswaran, K.; Mariani, J.; Coppola, G.; Abyzov, A.; Vaccarino, F.M. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat. Rev. Neurol. 2017, 13, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Gascón, S.; Masserdotti, G.; Russo, G.L.; Götz, M. Direct Neuronal Reprogramming: Achievements, Hurdles, and New Roads to Success. Cell Stem Cell 2017, 21, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; DeLong, C.J.; Bame, M.; Rajapakse, I.; Herron, T.J.; McInnis, M.G.; O’Shea, K.S. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl. Psychiatry 2014, 4, e375. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Shamah, S.M.; Sun, A.X.; Waldman, I.D.; Haggarty, S.J.; Perlis, R.H. Label-free, live optical imaging of reprogrammed bipolar disorder patient-derived cells reveals a functional correlate of lithium responsiveness. Transl. Psychiatry 2014, 4, e428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madison, J.M.; Zhou, F.; Nigam, A.; Hussain, A.; Barker, D.D.; Nehme, R.; van der Ven, K.; Hsu, J.; Wolf, P.; Fleishman, M.; et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol. Psychiatry 2015, 20, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Wang, Q.-W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
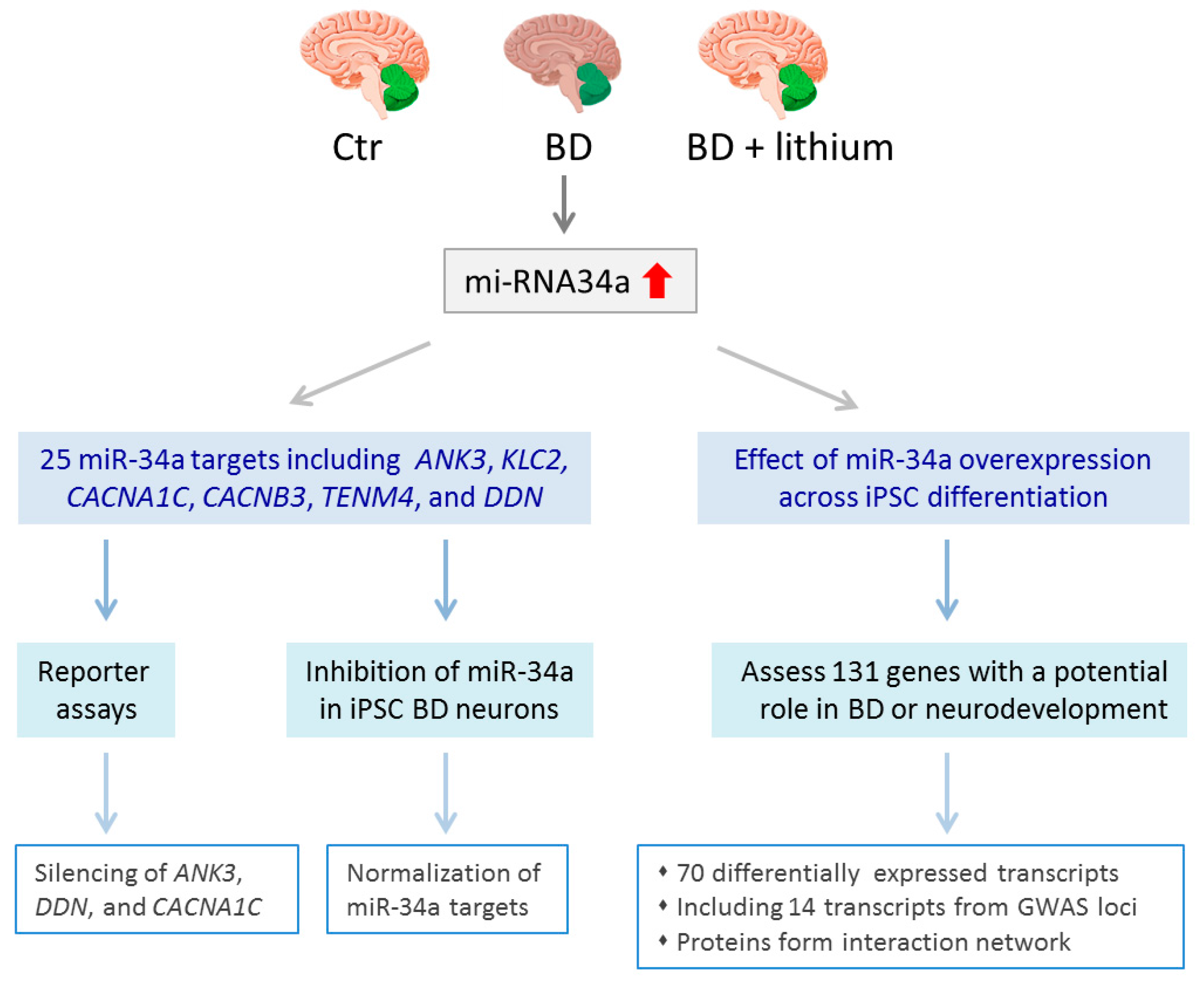
- Bavamian, S.; Mellios, N.; Lalonde, J.; Fass, D.M.; Wang, J.; Sheridan, S.D.; Madison, J.M.; Zhou, F.; Rueckert, E.H.; Barker, D.; et al. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol. Psychiatry 2015, 20, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Liu, J.; Sells Galvin, R.J.; Dage, J.L.; Egeland, J.A.; Smith, R.C.; Merchant, K.M.; Paul, S.M. Transcriptomic Analysis of Induced Pluripotent Stem Cells Derived from Patients with Bipolar Disorder from an Old Order Amish Pedigree. PLoS ONE 2015, 10, e0142693. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.; Santos, R.; Marchetto, M.C.; Mendes, A.P.D.; Rouleau, G.A.; Biesmans, S.; Wang, Q.-W.; Yao, J.; Charnay, P.; Bang, A.G.; et al. Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients’ responsiveness to lithium. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
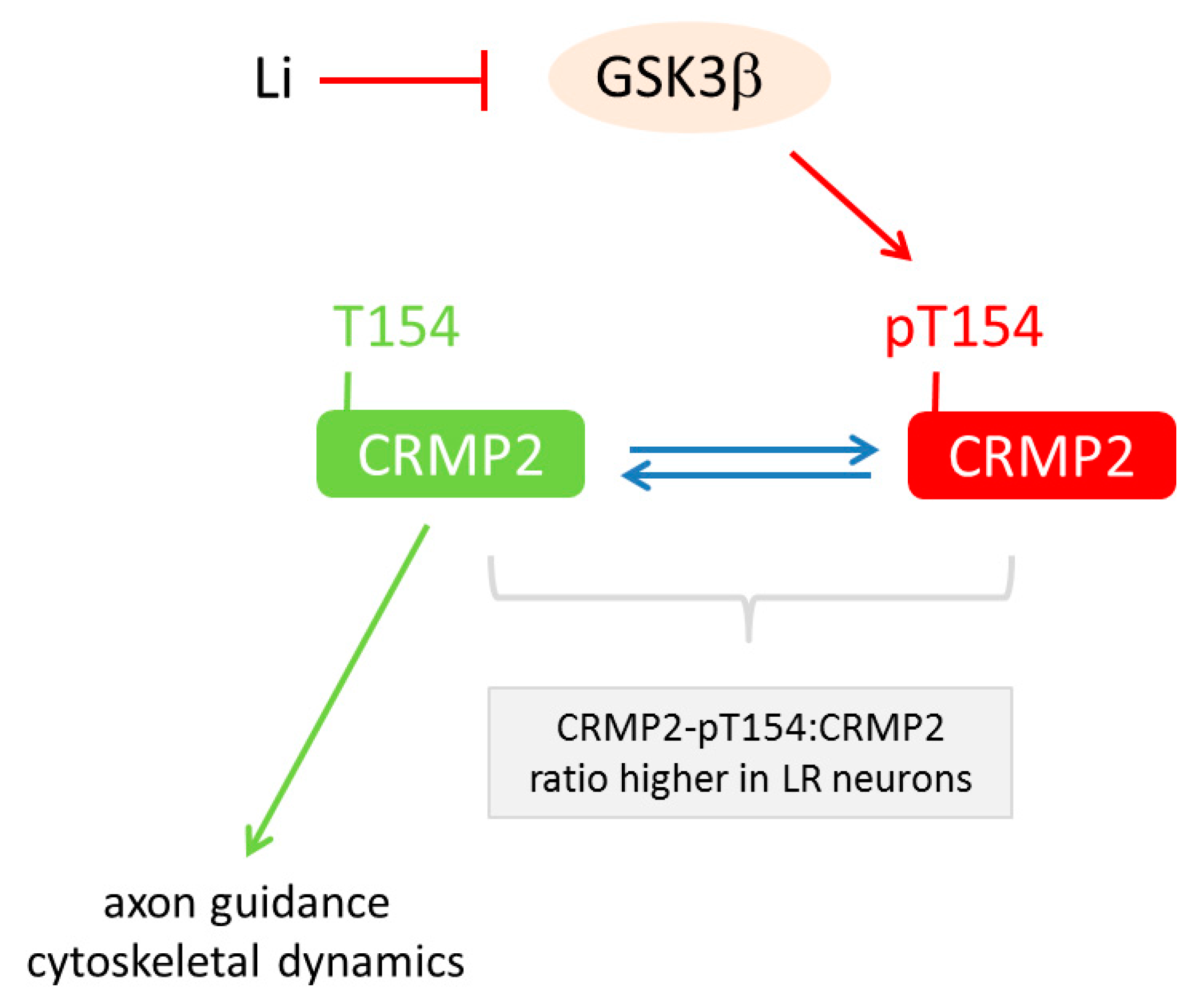
- Tobe, B.T.D.; Crain, A.M.; Winquist, A.M.; Calabrese, B.; Makihara, H.; Zhao, W.-N.; Lalonde, J.; Nakamura, H.; Konopaske, G.; Sidor, M.; et al. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E4462–E4471. [Google Scholar] [CrossRef] [PubMed]
- Vizlin-Hodzic, D.; Zhai, Q.; Illes, S.; Södersten, K.; Truvé, K.; Parris, T.Z.; Sobhan, P.K.; Salmela, S.; Kosalai, S.T.; Kanduri, C.; et al. Early onset of inflammation during ontogeny of bipolar disorder: The NLRP2 inflammasome gene distinctly differentiates between patients and healthy controls in the transition between iPS cell and neural stem cell stages. Transl. Psychiatry 2017, 7, e1010. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.-J.; Schuldt, B.M.; Williams, R.; Mason, D.; Altun, G.; Papapetrou, E.P.; Danner, S.; Goldmann, J.E.; Herbst, A.; Schmidt, N.O.; et al. A bioinformatic assay for pluripotency in human cells. Nat. Methods 2011, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Kiskinis, E.; Verstappen, G.; Gu, H.; Boulting, G.; Smith, Z.D.; Ziller, M.; Croft, G.F.; Amoroso, M.W.; Oakley, D.H.; et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011, 144, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. The developing cortex. Handb. Clin. Neurol. 2013, 111, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Lee, P.R.; Cohen, J.E. Temporal integration of intracellular Ca2+ signaling networks in regulating gene expression by action potentials. Cell Calcium 2005, 37, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lei, Q.; Oosterveen, T.; Ericson, J.; Matise, M.P. Tcf/Lef repressors differentially regulate Shh-Gli target gene activation thresholds to generate progenitor patterning in the developing CNS. Development 2011, 138, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Brylka, H.; Schwegler, H.; Venkataramanappa, S.; Andratschke, J.; Wiegreffe, C.; Liu, P.; Fuchs, E.; Jenkins, N.A.; Copeland, N.G.; et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J. 2012, 31, 2922–2936. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, B.J.; Arlotta, P.; Menezes, J.R.L.; Macklis, J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007, 8, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y. Neuro-archaeology: Pre-symptomatic architecture and signature of neurological disorders. Trends Neurosci. 2008, 31, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, C.; Walter, H.; Kirsch, P.; Erk, S.; Schnell, K.; Arnold, C.; Haddad, L.; Mier, D.; Opitz von Boberfeld, C.; Raab, K.; et al. Neural mechanisms of a genome-wide supported psychosis variant. Science 2009, 324, 605. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.J.; Jeffries, A.R.; Dobson, R.J.B.; Price, J.; Bray, N.J. Knockdown of the psychosis susceptibility gene ZNF804A alters expression of genes involved in cell adhesion. Hum. Mol. Genet. 2012, 21, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Rajman, M.; Schratt, G. MicroRNAs in neural development: From master regulators to fine-tuners. Development 2017, 144, 2310–2322. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.X.; Crabtree, G.R.; Yoo, A.S. MicroRNAs: Regulators of neuronal fate. Curr. Opin. Cell Biol. 2013, 25, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kwon, E.J.; Tsai, L.-H. MicroRNAs in learning, memory, and neurological diseases. Learn. Mem. 2012, 19, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Issler, O.; Chen, A. Determining the role of microRNAs in psychiatric disorders. Nat. Rev. Neurosci. 2015, 16, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Newman, R.; Chandler, S.; Stehlik, C.; Reed, J.C. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J. Biol. Chem. 2004, 279, 51897–51907. [Google Scholar] [CrossRef] [PubMed]
- Altamura, A.C.; Buoli, M.; Pozzoli, S. Role of immunological factors in the pathophysiology and diagnosis of bipolar disorder: Comparison with schizophrenia. Psychiatry Clin. Neurosci. 2014, 68, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Trépanier, M.O.; Hopperton, K.E.; Mizrahi, R.; Mechawar, N.; Bazinet, R.P. Postmortem evidence of cerebral inflammation in schizophrenia: A systematic review. Mol. Psychiatry 2016, 21, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Patino, G.A.; Isom, L.L. Electrophysiology and beyond: Multiple roles of Na+ channel β subunits in development and disease. Neurosci. Lett. 2010, 486, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.M.; Connelly, J.P.; Hansen, N.F.; Donovan, F.X.; Winkler, T.; Davis, B.W.; Alkadi, H.; Chandrasekharappa, S.C.; Dunbar, C.E.; Mullikin, J.C.; et al. iPSCs and fibroblast subclones from the same fibroblast population contain comparable levels of sequence variations. Proc. Natl. Acad. Sci. USA 2017, 114, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Chou, B.-K.; Gu, H.; Gao, Y.; Dowey, S.N.; Wang, Y.; Shi, J.; Li, Y.; Ye, Z.; Cheng, T.; Cheng, L. A facile method to establish human induced pluripotent stem cells from adult blood cells under feeder-free and xeno-free culture conditions: A clinically compliant approach. Stem Cells Transl. Med. 2015, 4, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Burrows, C.K.; Banovich, N.E.; Pavlovic, B.J.; Patterson, K.; Gallego Romero, I.; Pritchard, J.K.; Gilad, Y. Genetic Variation, Not Cell Type of Origin, Underlies the Majority of Identifiable Regulatory Differences in iPSCs. PLoS Genet. 2016, 12, e1005793. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, F.; Kumasaka, N.; de Brito, M.C.; Bradley, A.; Vallier, L.; Gaffney, D. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 2014, 10, e1004432. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Gu, H.; Müller, F.; Donaghey, J.; Tsai, L.T.-Y.; Kohlbacher, O.; De Jager, P.L.; Rosen, E.D.; Bennett, D.A.; Bernstein, B.E.; et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013, 500, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banovich, N.E.; Li, Y.I.; Raj, A.; Ward, M.C.; Greenside, P.; Calderon, D.; Tung, P.Y.; Burnett, J.E.; Myrthil, M.; Thomas, S.M.; et al. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Res. 2018, 28, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Carcamo-Orive, I.; Hoffman, G.E.; Cundiff, P.; Beckmann, N.D.; D’Souza, S.L.; Knowles, J.W.; Patel, A.; Papatsenko, D.; Abbasi, F.; Reaven, G.M.; et al. Analysis of Transcriptional Variability in a Large Human iPSC Library Reveals Genetic and Non-genetic Determinants of Heterogeneity. Cell Stem Cell 2017, 20, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.E.; Hartley, B.J.; Flaherty, E.; Ladran, I.; Gochman, P.; Ruderfer, D.M.; Stahl, E.A.; Rapoport, J.; Sklar, P.; Brennand, K.J. Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun. 2017, 8, 2225. [Google Scholar] [CrossRef] [PubMed]
- Tsankov, A.M.; Akopian, V.; Pop, R.; Chetty, S.; Gifford, C.A.; Daheron, L.; Tsankova, N.M.; Meissner, A. A qPCR ScoreCard quantifies the differentiation potential of human pluripotent stem cells. Nat. Biotechnol. 2015, 33, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340. [Google Scholar] [CrossRef] [PubMed]
- Osafune, K.; Caron, L.; Borowiak, M.; Martinez, R.J.; Fitz-Gerald, C.S.; Sato, Y.; Cowan, C.A.; Chien, K.R.; Melton, D.A. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 2008, 26, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, J.; Pang, Z.P.; Ge, W.; Kim, K.J.; Blanchi, B.; Chen, C.; Südhof, T.C.; Sun, Y.E. Integrative genomic and functional analyses reveal neuronal subtype differentiation bias in human embryonic stem cell lines. Proc. Natl. Acad. Sci. USA 2007, 104, 13821–13826. [Google Scholar] [CrossRef] [PubMed]
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. Switch-Like Roles for Polycomb Proteins from Neurodevelopment to Neurodegeneration. Epigenomes 2017, 1, 21. [Google Scholar] [CrossRef]
- Soldner, F.; Jaenisch, R. Medicine. iPSC disease modeling. Science 2012, 338, 1155–1156. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Foskolou, S.; Kilpinen, H.; Rodrigues, J.; Alasoo, K.; Knights, A.J.; Patel, M.; Goncalves, A.; Ferreira, R.; Benn, C.L.; et al. Molecular and functional variation in iPSC-derived sensory neurons. Nat. Genet. 2018, 50, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Paull, D.; Sevilla, A.; Zhou, H.; Hahn, A.K.; Kim, H.; Napolitano, C.; Tsankov, A.; Shang, L.; Krumholz, K.; Jagadeesan, P.; et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat. Methods 2015, 12, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Gandal, M.J.; Leppa, V.; Won, H.; Parikshak, N.N.; Geschwind, D.H. The road to precision psychiatry: Translating genetics into disease mechanisms. Nat. Neurosci. 2016, 19, 1397–1407. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Source | Factors | Methods | N° | Auth | Karyo | Pluripotency |
|---|---|---|---|---|---|---|---|
| [63] | FB | OKSM | RV | ≥5 | nd | nd | ICC, EB and TriL |
| [64] | FB | miR/NAM | LV | na | na | na | ICC |
| [65] | FB | OKSM | RV | ≥3 | SNP | G-Band | ICC, PluriTest, EB and ScoC, Tera |
| [66] | FB | OKSM | SV | 2 | nd | G-Band | ICC, TriL |
| [67] | FB | miR/NAM | LV | na | na | na | ICC |
| [68] | FB | OKSM | SV | 3 | nd | G-Band | ICC, EB and TriL |
| [69] | LCL | OKSM, LIN28 | Epi | ≥3 | STRP | nd | ICC |
| [70] | FB, LB | OKSM | Epi, LV, RV | 1–3 | SNP | nd | ICC, PluriTest, EB, Tera |
| [71] | AP | OKSM | Epi | 1 | nd | nd | ICC, EB and TriL |
| Ref. | Study Design | BD vs. Controls | Model | Major Cell Type(s) |
|---|---|---|---|---|
| [63] | BD I (LR)—control | 3 vs. 3 | iPSC | Forebrain, mixed glutamatergic—GABAergic neurons |
| [64] | BD I—control | 12 vs. 6 | iNLC | NSC, NPC, neuronal-like cells |
| [65] | Familial BD | 2 vs. 2 | iPSC | FACS-sorted NPCs, neurons |
| [66] | BD I—control | 6 vs. 4 | iPSC | Hippocampal dentate gyrus-like granule neurons |
| [67] | BD I—control | 1 vs. 1 5 vs. 3 | iPSC iNLC | NPC, NPC-derived neurons transdifferentiated neurons |
| [68] | Familial BD | 4 vs. 4 | iPSC | PAX6-positive NPC, early and late cortical neurons |
| [69] | BD I (LR-NR)—control | 3 vs. 3 vs. 4 | iPSC | Hippocampal dentate gyrus-like granule neurons |
| [70] | BD I (LR-NR)—control—MDD—PD | 7 vs. 3 vs. 6 vs. 2 vs. 1 | iPSC | Forebrain, mixed glutamatergic—GABAergic neurons |
| [71] | BD I—control | 6 vs. 4 | iPSC | Cortical stem and progenitor cells |
| Ref. | Major Methods | Major Findings in BD-derived iPSCs |
|---|---|---|
| [63] | Microarray, Ca2+ transients | Ventralization, increased expression of membrane bound receptors and ion channels, Li reduces wave altered length amplitude and Ca2+ transients |
| [64] | Morphology, Res-Imag | Cellular adhesion associates with clinical response to Li |
| [65] | NanoString, RNA-seq, WCPC | Impaired early NPCs proliferation that is normalized by GSK3β inhibitor, altered WNT/GSK3β signaling and ion channel expression in NPCs |
| [66] | RNA-seq, WCPC | Altered neuronal excitability, altered mitochondrial function and size, Li reduces hyperexcitability in LR donors and partly normalizes mitochondrial function |
| [67] | qRT-PCR, NanoString, reporter assays | Upregulation of miR-34a in NPC and neurons, reducing miR-34a expression enhances dendritic elaboration and maturation of NPCs |
| [68] | Microarray | Deregulation of receptor-mediated signaling. RNA metabolism, and protein trafficking in late neurons, upregulation of GAD1 |
| [69] | WCPC | Neurons differ according to LR and NR, larger fast after-hyperpolarization |
| [70] | Proteomics | Li-response pathway in BD acts through GSK3β-dependent CRMP2 phosphorylation to alter dendrite and dendritic spine formation, Ca2+ fluxes and neuronal activity |
| [71] | RNA-seq, WCPC | Upregulation of immune-regulatory NLRP2, GABA- and dopamine signaling |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, A.; Sportelli, V.; Ziller, M.; Spengler, D. From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish. Int. J. Mol. Sci. 2018, 19, 770. https://doi.org/10.3390/ijms19030770
Hoffmann A, Sportelli V, Ziller M, Spengler D. From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish. International Journal of Molecular Sciences. 2018; 19(3):770. https://doi.org/10.3390/ijms19030770
Chicago/Turabian StyleHoffmann, Anke, Vincenza Sportelli, Michael Ziller, and Dietmar Spengler. 2018. "From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish" International Journal of Molecular Sciences 19, no. 3: 770. https://doi.org/10.3390/ijms19030770
APA StyleHoffmann, A., Sportelli, V., Ziller, M., & Spengler, D. (2018). From the Psychiatrist’s Couch to Induced Pluripotent Stem Cells: Bipolar Disease in a Dish. International Journal of Molecular Sciences, 19(3), 770. https://doi.org/10.3390/ijms19030770