Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines

 , , , , and
, , , , and 
Abstract
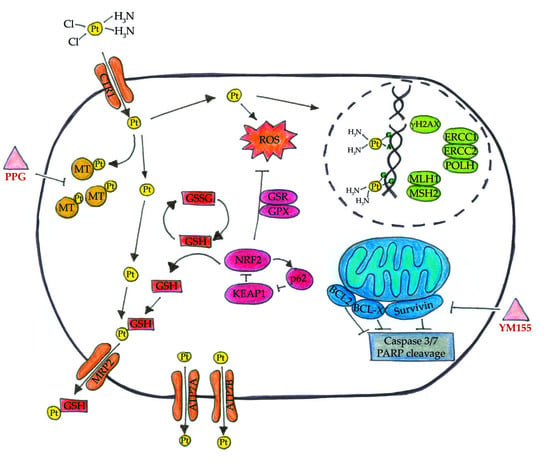
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
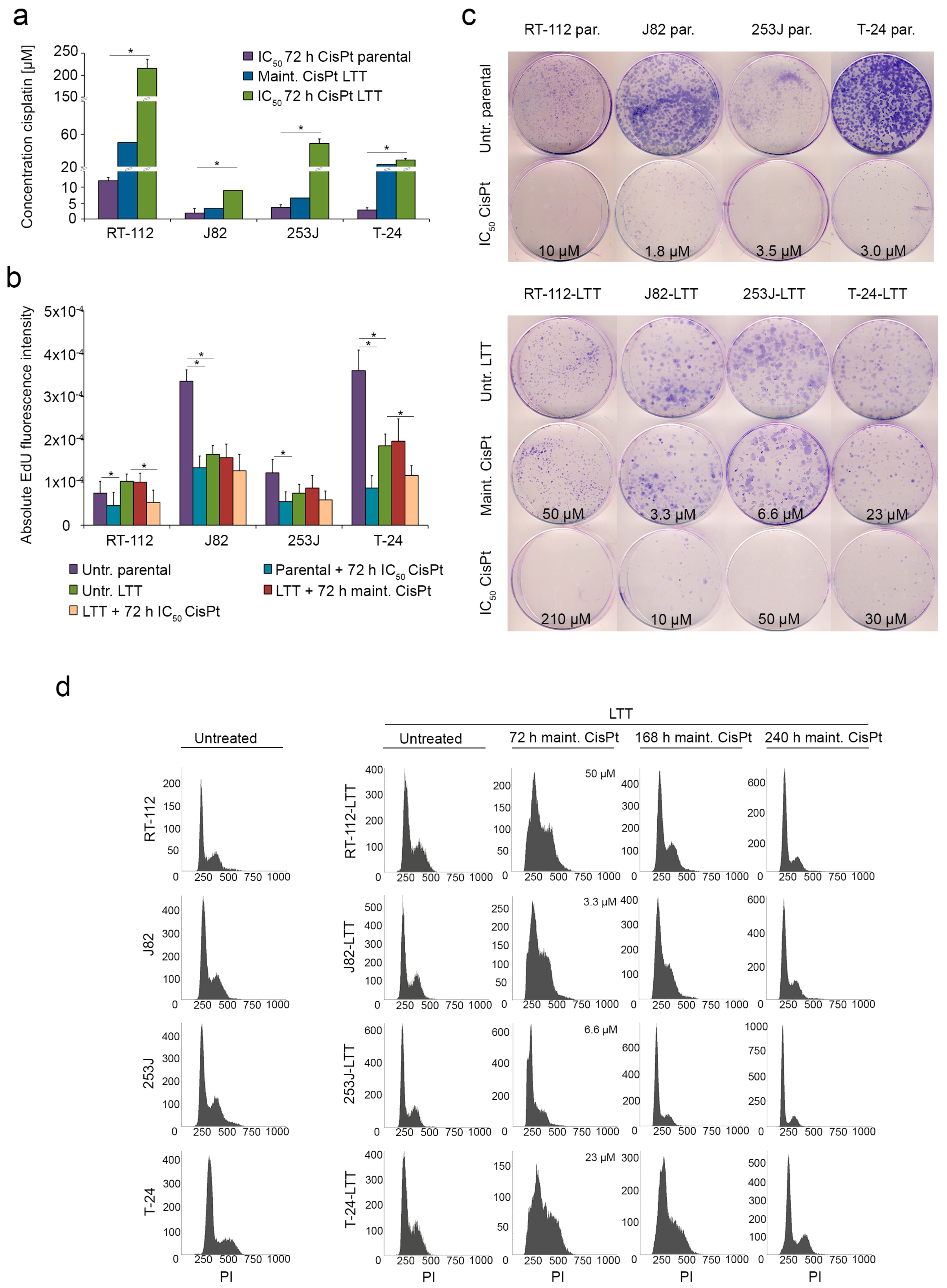
2.1. Long-Term Cisplatin Treated (LTTs) Urothelial Carcinoma Cell Lines Recover from Cisplatin-Induced Stress
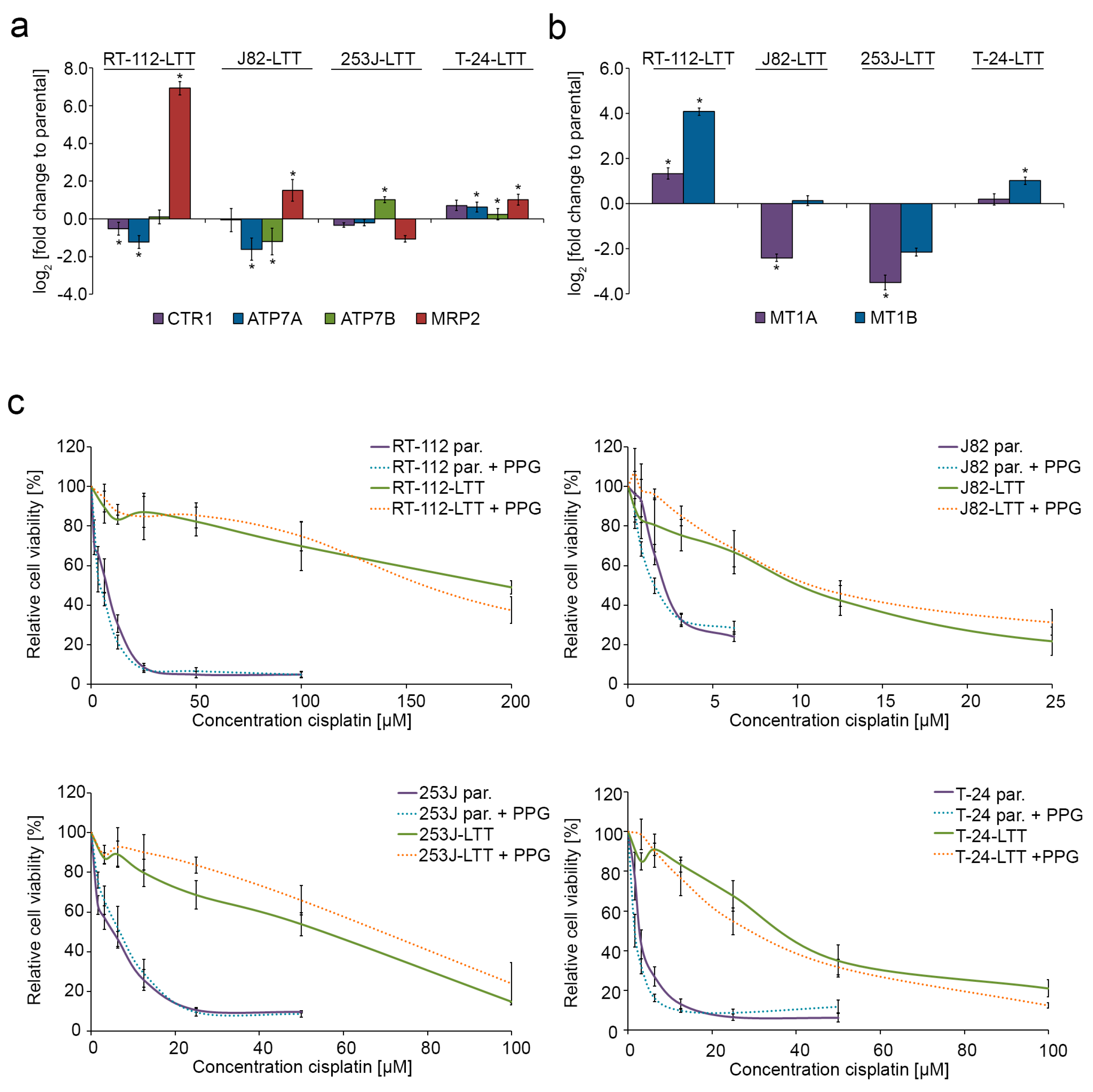
2.2. Cisplatin Exporter and Detoxifying Molecules Are Differentially Expressed in LTT Lines
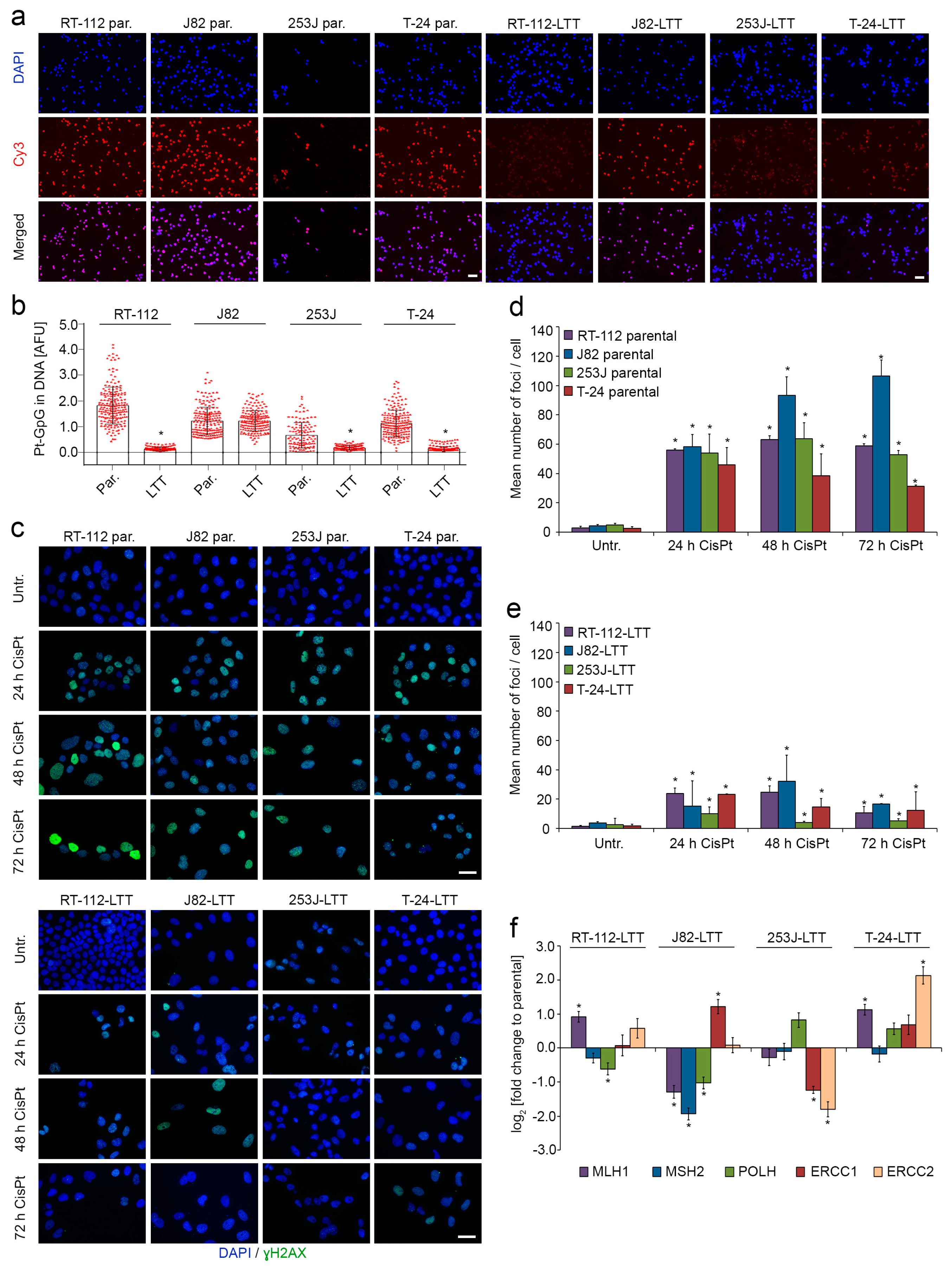
2.3. DNA-Cisplatin Adduct Formation and Extent of DNA Damage Is Reduced in LTTs
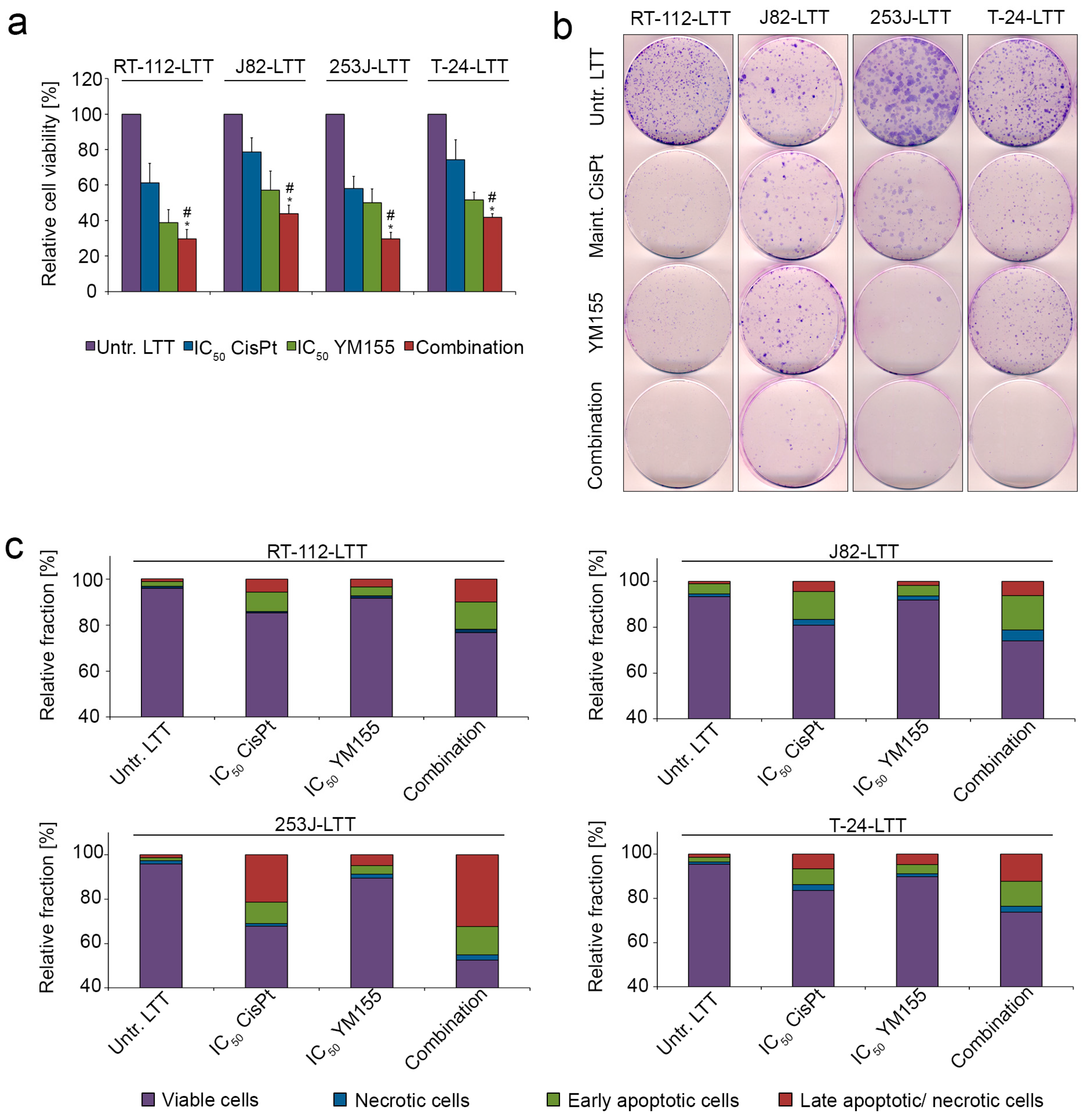
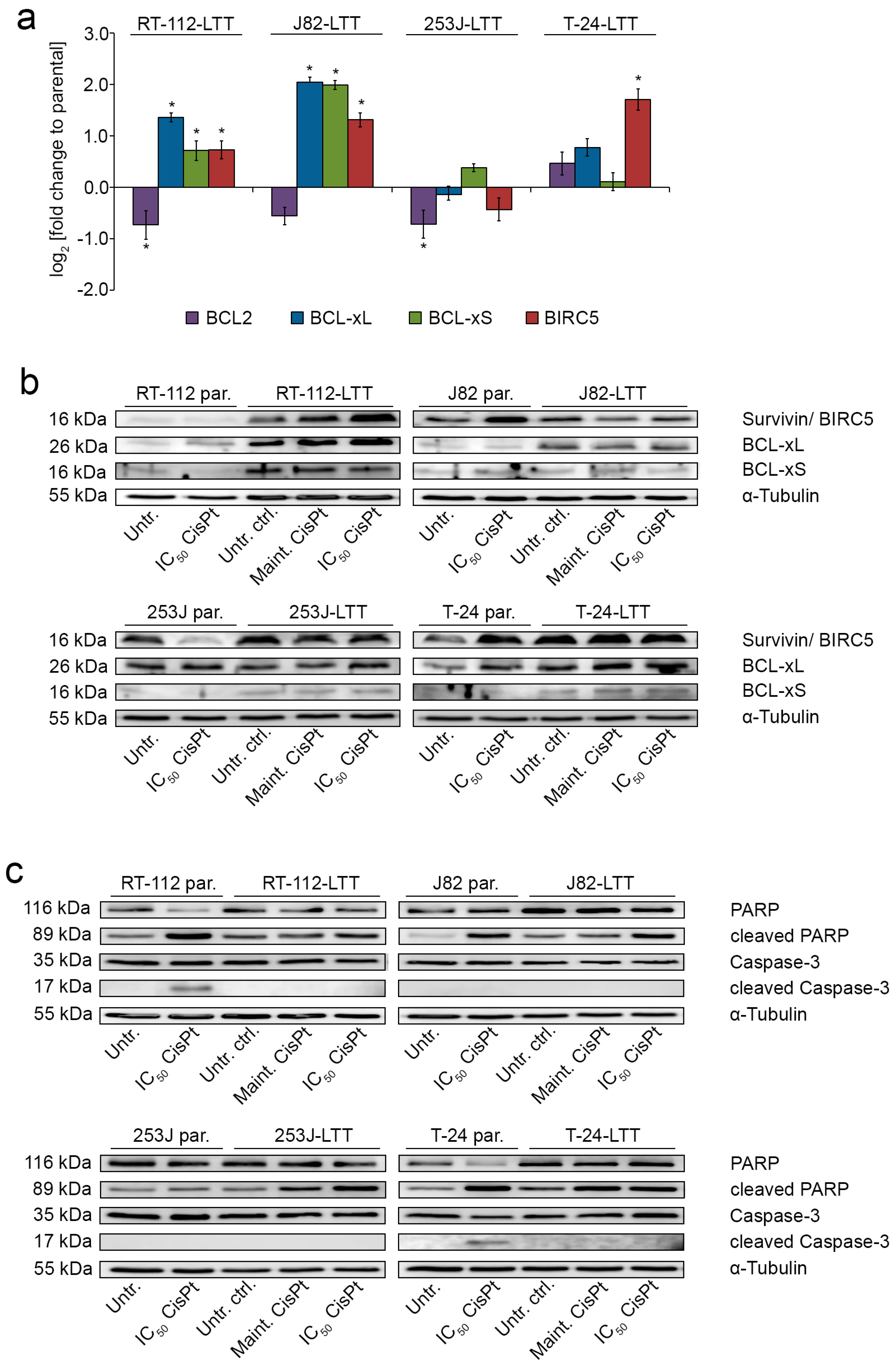
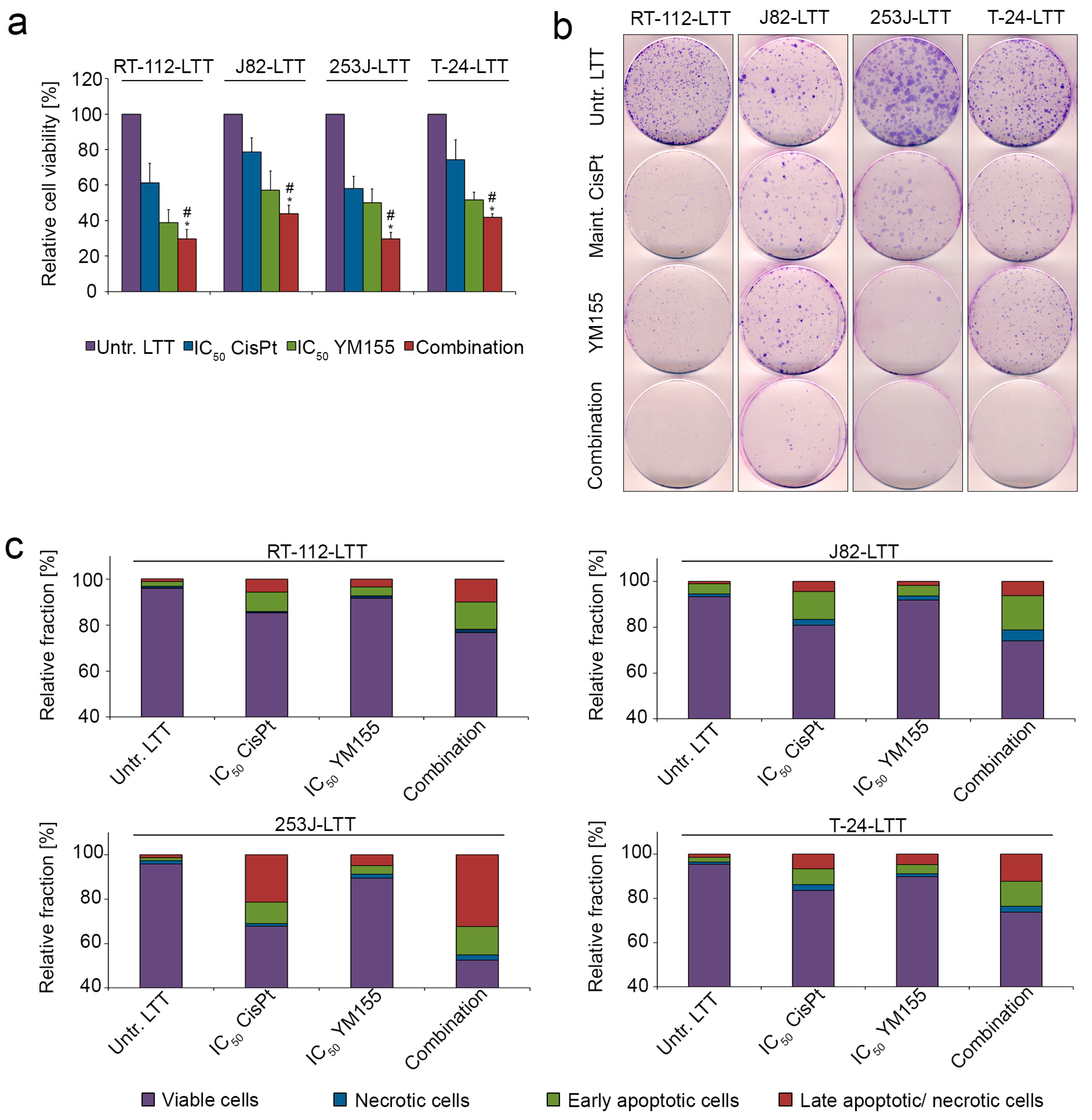
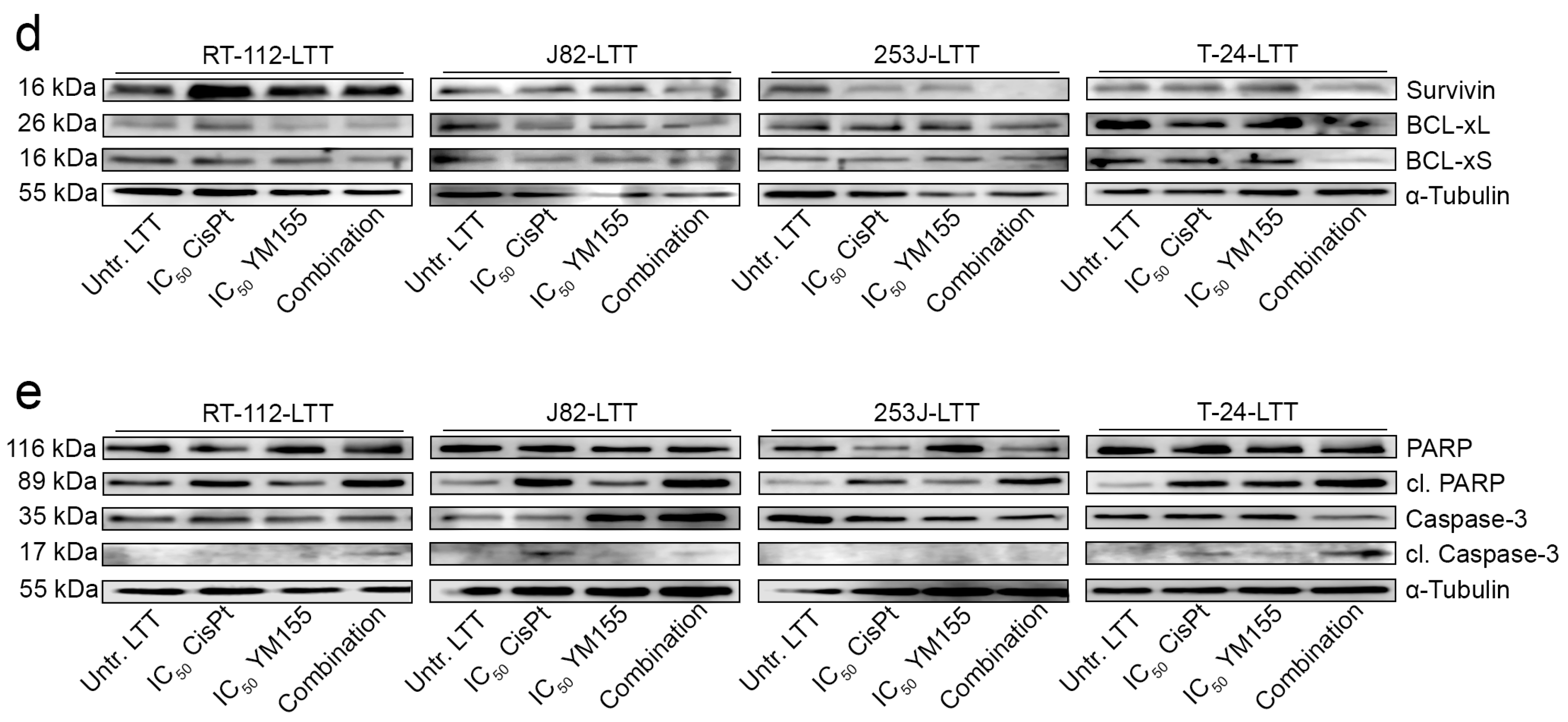
2.4. LTTs Are More Resistant to Cisplatin-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Measurements of Cell Viability, Clonogenicity, and Proliferation
4.3. Molecular Analyses
4.4. Measurement of Caspase 3/7 Activity
4.5. Immunofluorescence
4.6. Quantification of Pt-(GpG) Adducts
4.7. Western Blot Analysis
4.8. Flow Cytometry
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| UCC | Urothelial carcinoma cell line |
| SDHA | Succinate dehydrogenase complex |
| CTR1 | Solute carrier family 31 member 1 |
| ATP7A | ATPase copper transporting alpha |
| ATP7B | ATPase copper transporting beta |
| MT | Metallothionein |
| MRP2 | Multi-drug resistance protein 2 |
| ERCC1 | ERCC excision repair 1, Endonuclease non-catalytic subunit |
| ERCC2 | ERCC excision repair 2, TFIIH core complex helicase subunit |
| MLH1 | MutL homolog 1 |
| MSH2 | MutS homolog 2 |
| POLH | DNA polymerase eta |
| BCL-2 | B-Cell CLL/Lymphoma 2 |
| BCL-XL | BCL2 like 1 |
| BIRC5 | Baculoviral IAP repeat containing 5 |
| γH2AX | Phosphorylated H2A histone family member X |
| Pt-adduct | Platinum-adduct |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder cancer incidence and mortality: A global overview and recent trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Crul, M.; Schellens, J.H.; Beijnen, J.H.; Maliepaard, M. Cisplatin resistance and DNA repair. Cancer Treat. Rev. 1997, 23, 341–366. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Köberle, M.; Piee-Staffa, A. The molecular basis of cisplatin resistance in bladder cancer cells. In Bladder Cancer—From Basic Science to Robotic Surgery; Canda, A.E., Ed.; InTech: London, UK, 2012; pp. 265–290. ISBN 978-953-307-839-7. [Google Scholar]
- Drayton, R.M.; Catto, J.W.F. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev. Anticancer Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Matyunina, L.V.; Wagner, G.T.; McDonald, J.F. Acquired resistance of pancreatic cancer cells to cisplatin is multifactorial with cell context-dependent involvement of resistance genes. Cancer Gene Ther. 2016, 23, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity protein 1 (Sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression. Mol. Pharmacol. 2012, 81, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Bay, B.H.; Jin, R.; Huang, J.; Tan, P.H. Metallothionein as a prognostic biomarker in breast cancer. Exp. Biol. Med. (Maywood) 2006, 231, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, Y.; Kohno, H.; Ueda, S.; Kimoto, T.; Dhar, D.K.; Kubota, H.; Tachibana, M.; Koji, T.; Nagasue, N. Expression of metallothionein in colorectal cancers and synchronous liver metastases. Oncology 2001, 61, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Siegsmund, M.J.; Marx, C.; Seemann, O.; Schummer, B.; Steidler, A.; Toktomambetova, L.; Kohrmann, K.U.; Rassweiler, J.; Alken, P. Cisplatin-resistant bladder carcinoma cells: Enhanced expression of metallothioneins. Urol. Res. 1999, 27, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Banerjee, D.; Khurana, R.J.; Pan, X.; Pflueger, R.; Tannock, I.F.; Moore, M.J. The prognostic role of p53, metallothionein, P-glycoprotein, and MIB-1 in muscle-invasive urothelial transitional cell carcinoma. Clin. Cancer Res. 1998, 4, 559–565. [Google Scholar] [PubMed]
- Skowron, M.A.; Niegisch, G.; Albrecht, P.; van Koeveringe, G.; Romano, A.; Albers, P.; Schulz, W.A.; Hoffmann, M.J. Various mechanisms involve the nuclear factor (erythroid-derived 2)-like (NRF2) to achieve cytoprotection in long-term cisplatin-treated urothelial carcinoma cell lines. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ali-Osman, F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J. Biol. Chem. 1993, 268, 20116–20125. [Google Scholar] [PubMed]
- Materna, V.; Liedert, B.; Thomale, J.; Lage, H. Protection of platinum-DNA adduct formation and reversal of cisplatin resistance by anti-MRP2 hammerhead ribozymes in human cancer cells. Int. J. Cancer 2005, 115, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.A.; Peixoto, A.; Neves, M.; Gaiteiro, C.; Reis, C.A.; Assaraf, Y.G.; Santos, L.L. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug Resist. Updat. 2016, 24, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Nozaki, S.; Kitahara, H.; Ohara, T.; Kato, K.; Kawashiri, S.; Yamamoto, E. Copper efflux transporter (ATP7B) contributes to the acquisition of cisplatin-resistance in human oral squamous cell lines. Oncol. Rep. 2007, 18, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Konkimalla, V.B.; Kaina, B.; Efferth, T. Role of transporter genes in cisplatin resistance. In Vivo 2008, 22, 279–283. [Google Scholar] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Mouw, K.W.; Kim, P.; Iyer, G.; Wagle, N.; Al-Ahmadie, H.; Zhu, C.; Ostrovnaya, I.; Kryukov, G.V.; O’Connor, K.W.; et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014, 4, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Han, C.; Zhao, R.; Cui, T.; Dai, Y.; Mao, C.; Zhao, W.; Zhang, X.; Yu, J.; Wang, Q.E. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4411–4416. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.M.; Sung, J.Y.; Park, S.H.; Kwon, G.Y.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Lee, H.M.; Jo, J.; Choi, H.Y.; et al. ERCC1 as a biomarker for bladder cancer patients likely to benefit from adjuvant chemotherapy. BMC Cancer 2012, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Henkels, K.M.; Turchi, J.J. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999, 59, 3077–3083. [Google Scholar] [PubMed]
- Van Oosterwijk, J.G.; Herpers, B.; Meijer, D.; Briaire-de Bruijn, I.H.; Cleton-Jansen, A.M.; Gelderblom, H.; van de Water, B.; Bovee, J.V. Restoration of chemosensitivity for doxorubicin and cisplatin in chondrosarcoma in vitro: BCL-2 family members cause chemoresistance. Ann. Oncol. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Michaud, W.A.; Nichols, A.C.; Mroz, E.A.; Faquin, W.C.; Clark, J.R.; Begum, S.; Westra, W.H.; Wada, H.; Busse, P.M.; Ellisen, L.W.; et al. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Ashfaq, R.; Karakiewicz, P.I.; Saeedi, O.; Sagalowsky, A.I.; Lotan, Y. Survivin expression is associated with bladder cancer presence, stage, progression, and mortality. Cancer 2007, 109, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Margulis, V.; Lotan, Y.; Shariat, S.F. Survivin: a promising biomarker for detection and prognosis of bladder cancer. World J. Urol. 2008, 26, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Niegisch, G.; Fritz, G.; Arent, T.; van Roermund, J.G.; Romano, A.; Albers, P.; Schulz, W.A.; Hoffmann, M.J. Phenotype plasticity rather than repopulation from CD90/CK14+ cancer stem cells leads to cisplatin resistance of urothelial carcinoma cell lines. J. Exp. Clin. Cancer Res. 2015, 34, 144. [Google Scholar] [CrossRef] [PubMed]
- Kilari, D.; Iczkowski, K.A.; Pandya, C.; Robin, A.J.; Messing, E.M.; Guancial, E.; Kim, E.S. Copper transporter-CTR1 expression and pathological outcomes in platinum-treated muscle-invasive bladder cancer patients. Anticancer Res. 2016, 36, 495–501. [Google Scholar] [PubMed]
- Lage, H.; Christmann, M.; Kern, M.A.; Dietel, M.; Pick, M.; Kaina, B.; Schadendorf, D. Expression of DNA repair proteins hMSH2, hMSH6, hMLH1, O6-methylguanine-DNA methyltransferase and N-methylpurine-DNA glycosylase in melanoma cells with acquired drug resistance. Int. J. Cancer 1999, 80, 744–750. [Google Scholar] [CrossRef]
- Kunze, D.; Erdmann, K.; Froehner, M.; Wirth, M.P.; Fuessel, S. Enhanced inhibition of bladder cancer cell growth by simultaneous knockdown of antiapoptotic Bcl-xL and survivin in combination with chemotherapy. Int. J. Mol. Sci. 2013, 14, 12297–12312. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.T.; Cheng, C.C.; Lin, T.C.; Chiu, T.H.; Lai, P.C. Therapeutic potential of sepantronium bromide YM155 in gemcitabine-resistant human urothelial carcinoma cells. Oncol. Rep. 2014, 31, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Mir, R.; Stanzani, E.; Martinez-Soler, F.; Villanueva, A.; Vidal, A.; Condom, E.; Ponce, J.; Gil, J.; Tortosa, A.; Gimenez-Bonafe, P. YM155 sensitizes ovarian cancer cells to cisplatin inducing apoptosis and tumor regression. Gynecol. Oncol. 2014, 132, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Dizdar, L.; Oesterwind, K.A.; Riemer, J.C.; Werner, T.A.; Mersch, S.; Mohlendick, B.; Schutte, S.C.; Verde, P.E.; Raba, K.; Topp, S.A.; et al. Preclinical assesement of survivin and XIAP as prognostic biomarkers and therapeutic targets in gastroenteropancreatic neuroendocrine neoplasia. Oncotarget 2017, 8, 8369–8382. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Uehara, S.; Nakahata, K.; Okuyama, H. Survivin selective inhibitor YM155 promotes cisplatin-induced apoptosis in embryonal rhabdomyosarcoma. Int. J. Oncol. 2016, 48, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Shen, D.; Kong, C.; Zhang, Z.; Zeng, Y.; Lin, X.; Liu, X. NF-kappaB suppresses apoptosis and promotes bladder cancer cell proliferation by upregulating survivin expression in vitro and in vivo. Sci. Rep. 2017, 7, 40723. [Google Scholar] [CrossRef] [PubMed]
- Wangpaichitr, M.; Wu, C.; Li, Y.Y.; Nguyen, D.J.M.; Kandemir, H.; Shah, S.; Chen, S.; Feun, L.G.; Prince, J.S.; Kuo, M.T.; et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget 2017, 8, 49275–49292. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef] [PubMed]
- Schlutermann, D.; Skowron, M.A.; Berleth, N.; Bohler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J.; et al. Targeting urothelial carcinoma cells by combining cisplatin with a specific inhibitor of the autophagy-inducing class III PtdIns3K complex. Urol. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hardie, M.E.; Kava, H.W.; Murray, V. Cisplatin analogues with an increased interaction with DNA: Prospects for therapy. Curr. Pharm. Des. 2016, 22, 6645–6664. [Google Scholar] [CrossRef] [PubMed]
- Cetraz, M.; Sen, V.; Schoch, S.; Streule, K.; Golubev, V.; Hartwig, A.; Koberle, B. Platinum(IV)-nitroxyl complexes as possible candidates to circumvent cisplatin resistance in RT112 bladder cancer cells. Arch. Toxicol. 2017, 91, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Pages, B.J.; Ang, D.L.; Wright, E.P.; Aldrich-Wright, J.R. Metal complex interactions with DNA. Dalton. Trans. 2015, 44, 3505–3526. [Google Scholar] [CrossRef] [PubMed]
- Pages, B.J.; Sakoff, J.; Gilbert, J.; Rodger, A.; Chmel, N.P.; Jones, N.C.; Kelly, S.M.; Ang, D.L.; Aldrich-Wright, J.R. Multifaceted studies of the DNA interactions and in vitro cytotoxicity of anticancer polyaromatic platinum(II) complexes. Chemistry 2016, 22, 8943–8954. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Rudner, J.; Jendrossek, V. The Focinator—A new open-source tool for high-throughput foci evaluation of DNA damage. Radiat. Oncol. 2015, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Liedert, B.; Pluim, D.; Schellens, J.; Thomale, J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006, 34, e47. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skowron, M.A.; Melnikova, M.; Van Roermund, J.G.H.; Romano, A.; Albers, P.; Thomale, J.; Schulz, W.A.; Niegisch, G.; Hoffmann, M.J. Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2018, 19, 590. https://doi.org/10.3390/ijms19020590
Skowron MA, Melnikova M, Van Roermund JGH, Romano A, Albers P, Thomale J, Schulz WA, Niegisch G, Hoffmann MJ. Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines. International Journal of Molecular Sciences. 2018; 19(2):590. https://doi.org/10.3390/ijms19020590
Chicago/Turabian StyleSkowron, Margaretha A., Margarita Melnikova, Joep G. H. Van Roermund, Andrea Romano, Peter Albers, Jürgen Thomale, Wolfgang A. Schulz, Günter Niegisch, and Michèle J. Hoffmann. 2018. "Multifaceted Mechanisms of Cisplatin Resistance in Long-Term Treated Urothelial Carcinoma Cell Lines" International Journal of Molecular Sciences 19, no. 2: 590. https://doi.org/10.3390/ijms19020590