Sexual Dimorphism of NADPH Oxidase/H2O2 System in Rat Thyroid Cells; Effect of Exogenous 17β-Estradiol


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
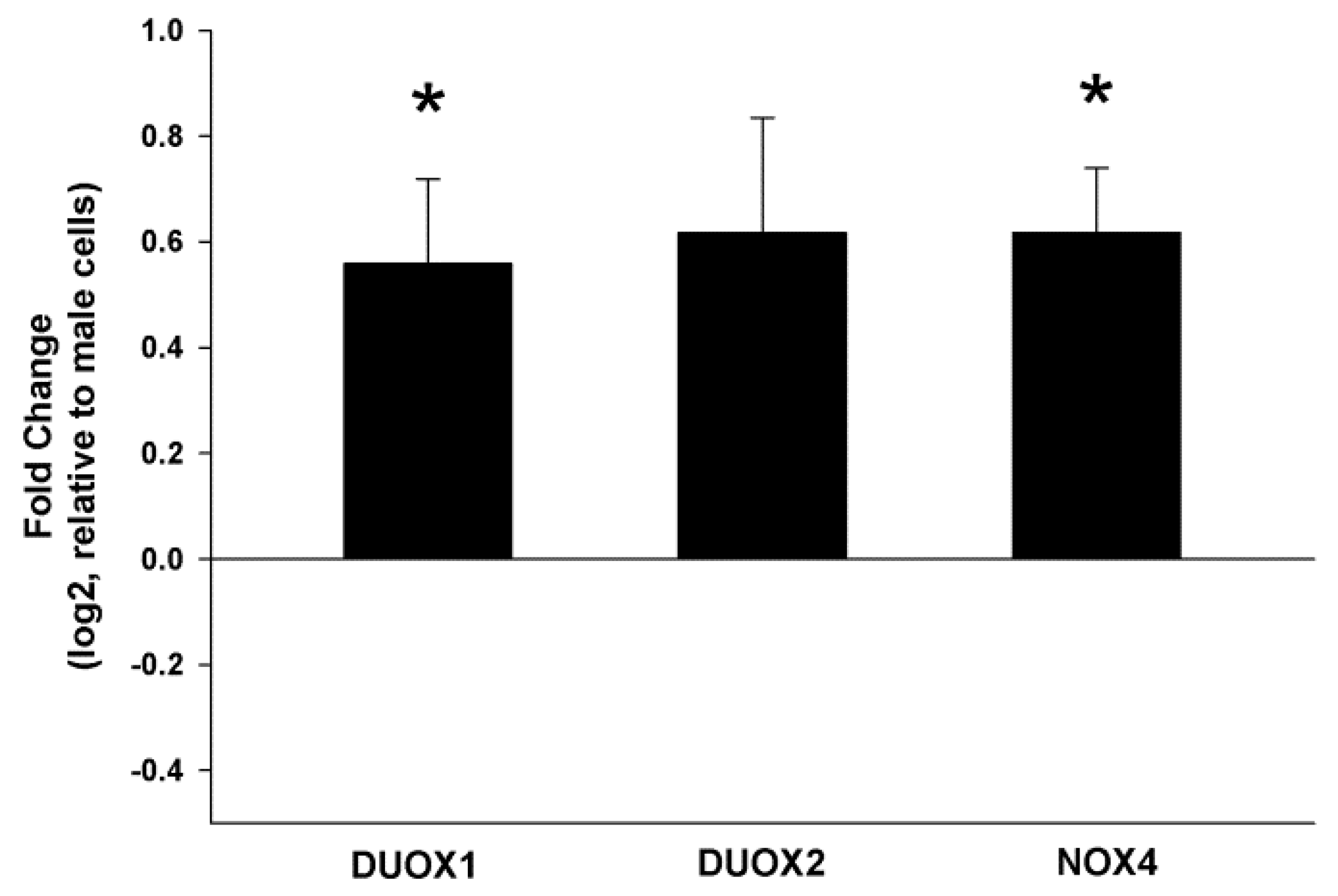
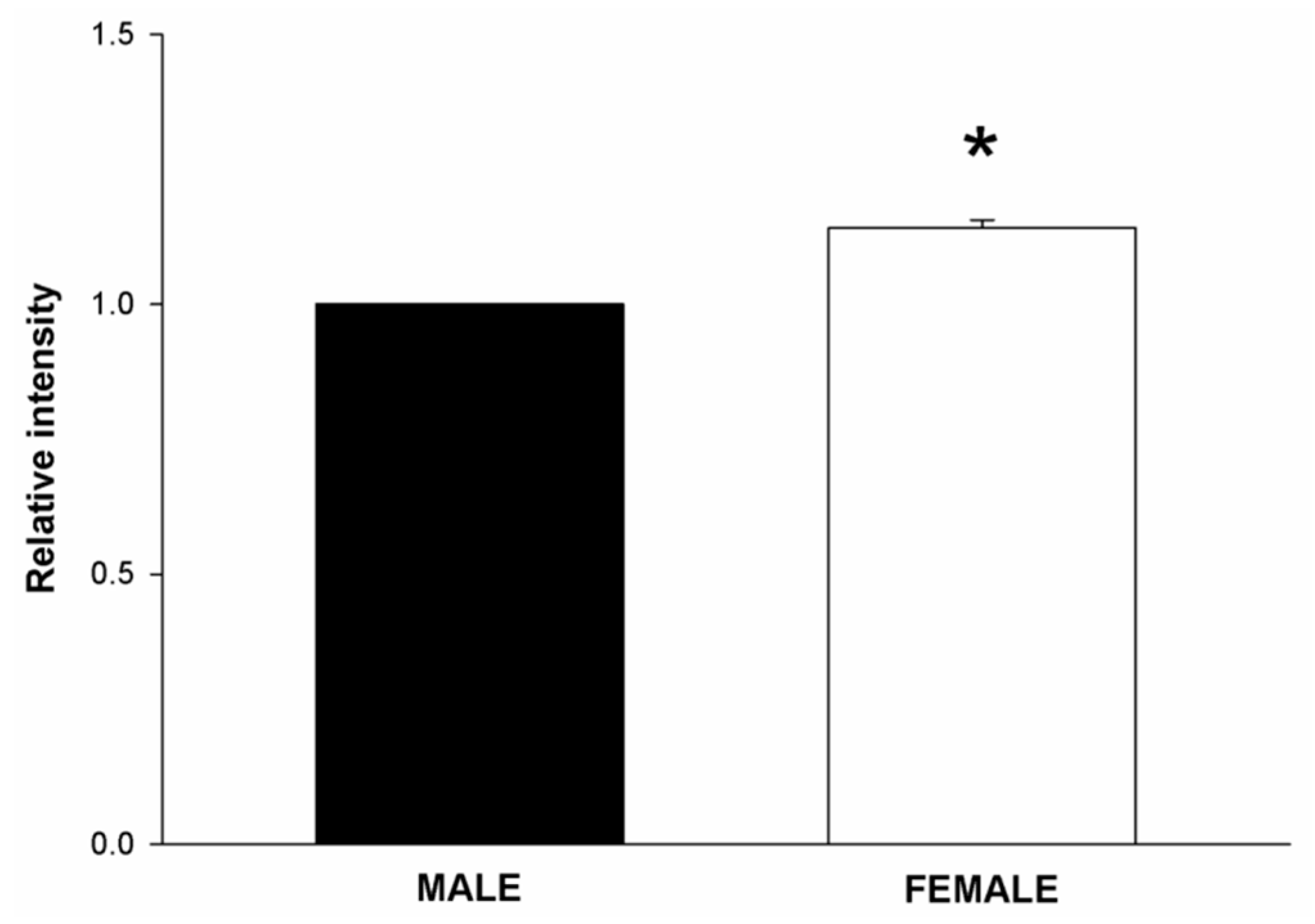
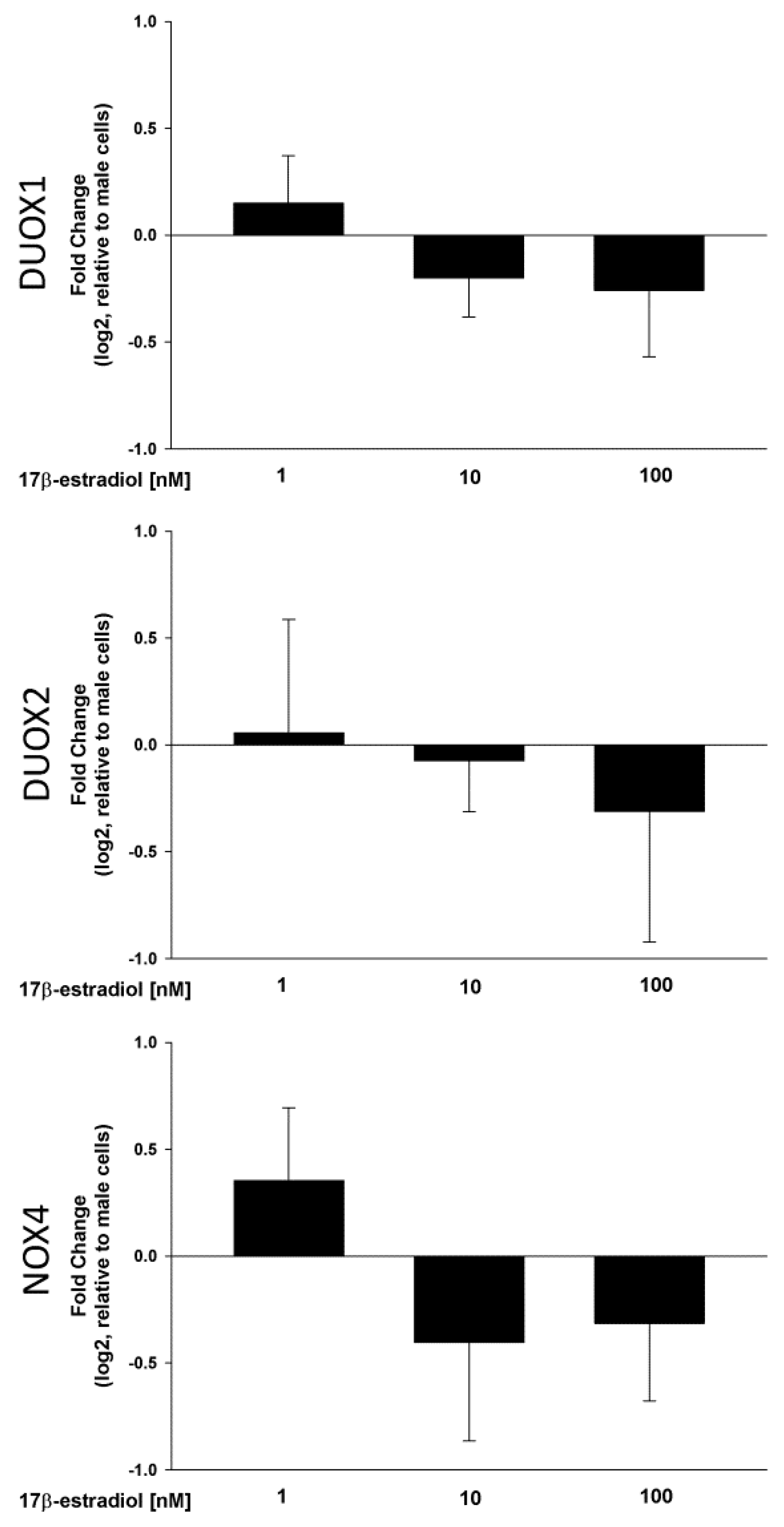
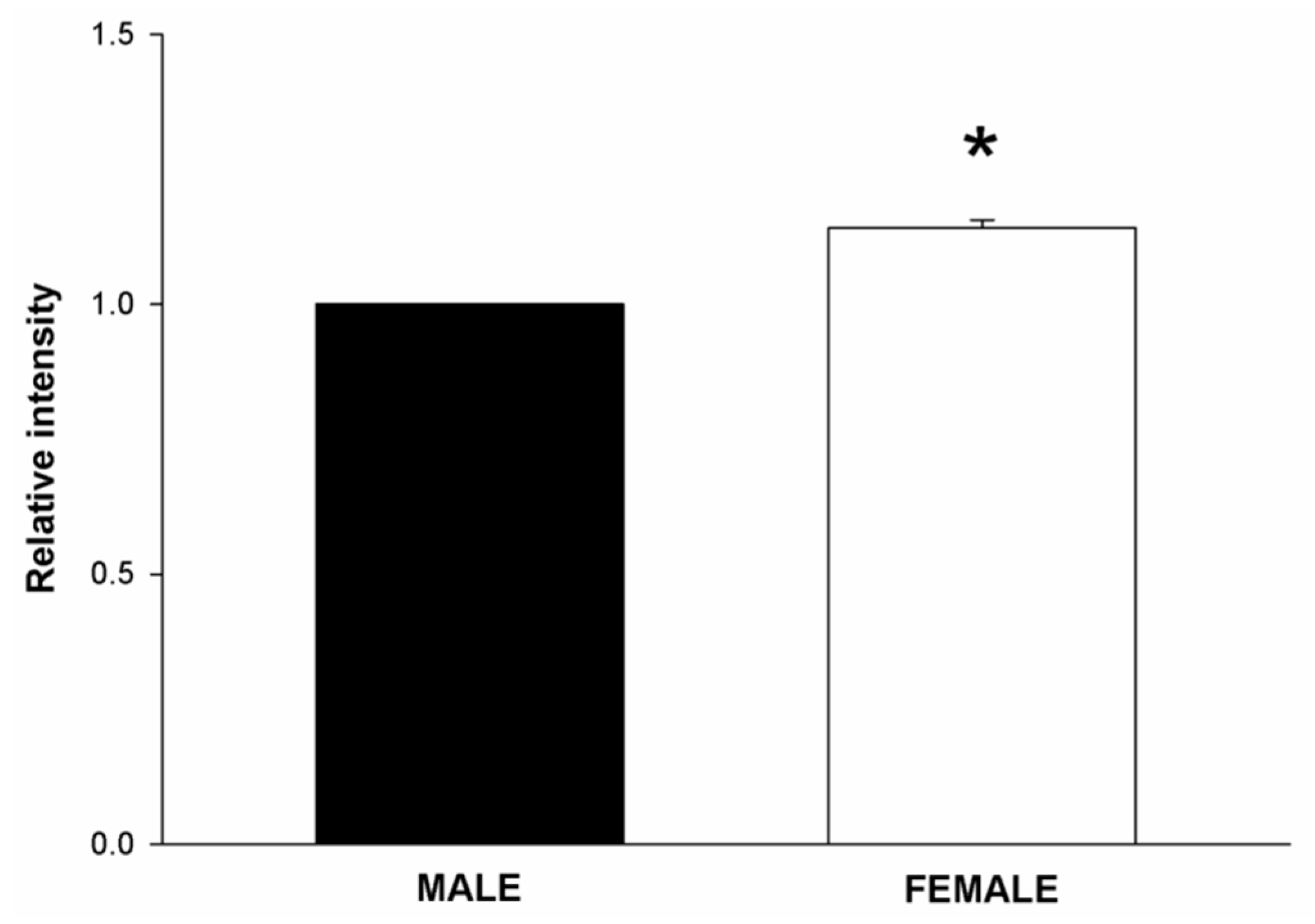
2.1. NOX/DUOX Expression and H2O2 Level in Primary Cells Derived from Male and Female Rat Thyroids
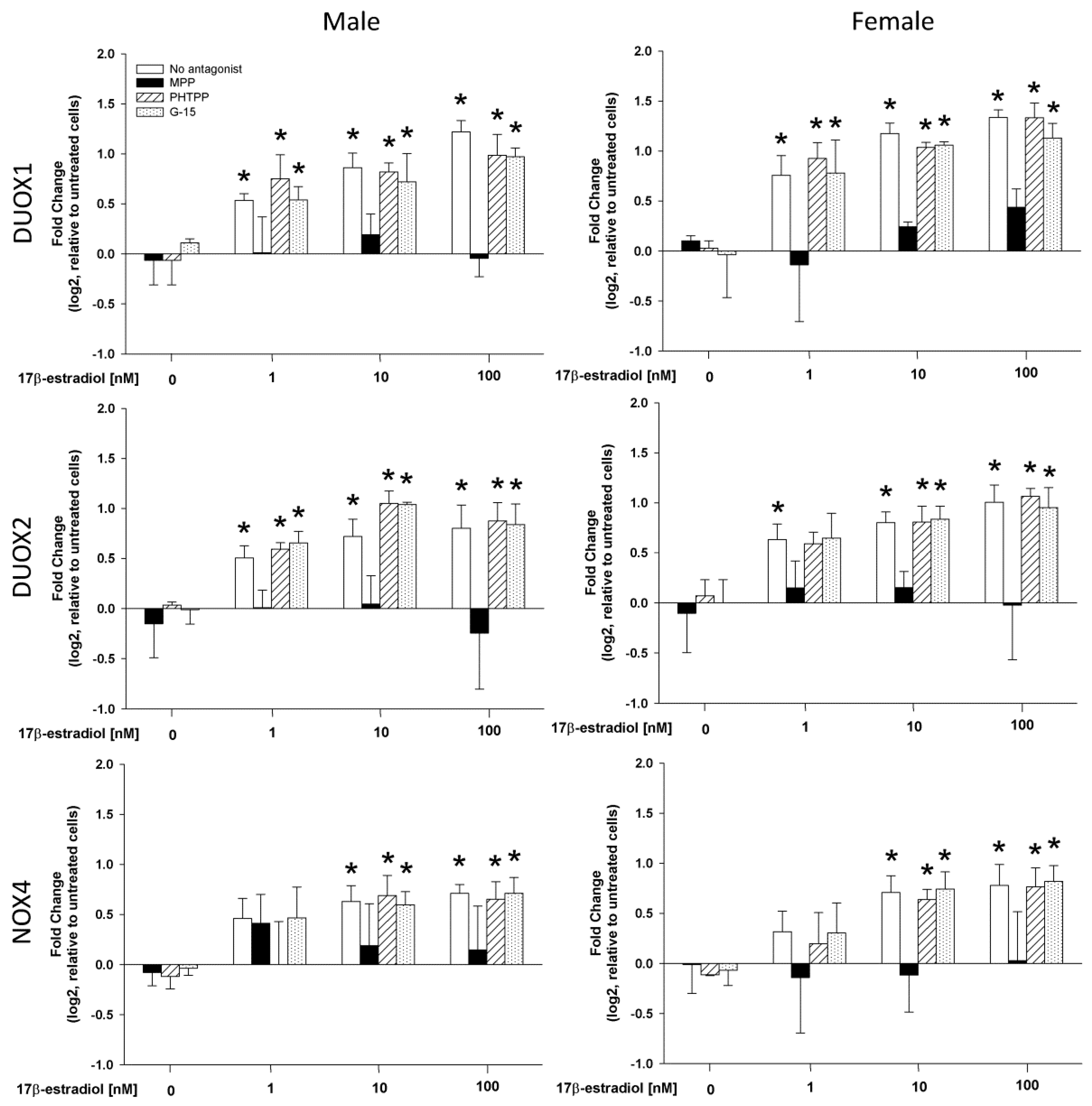
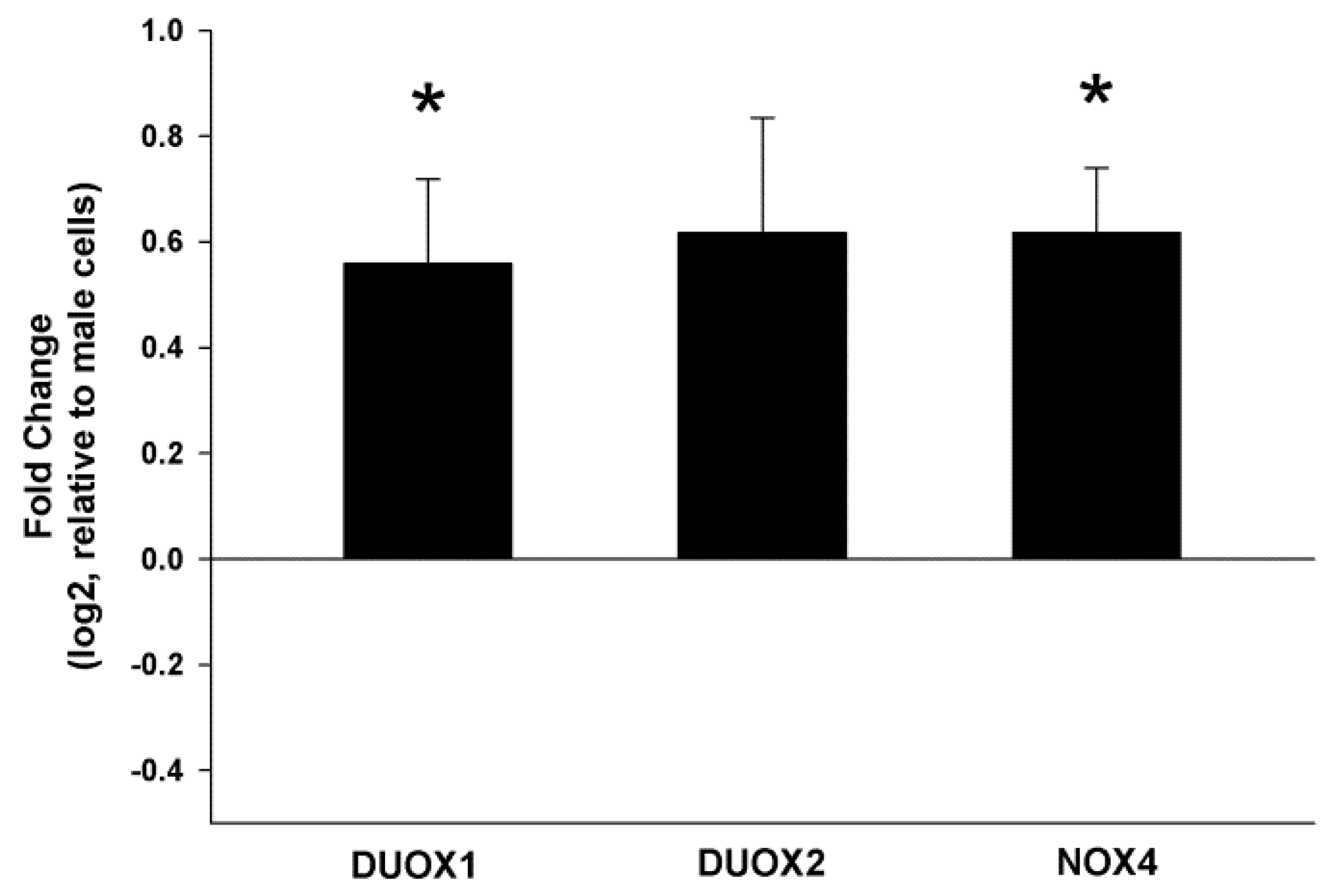
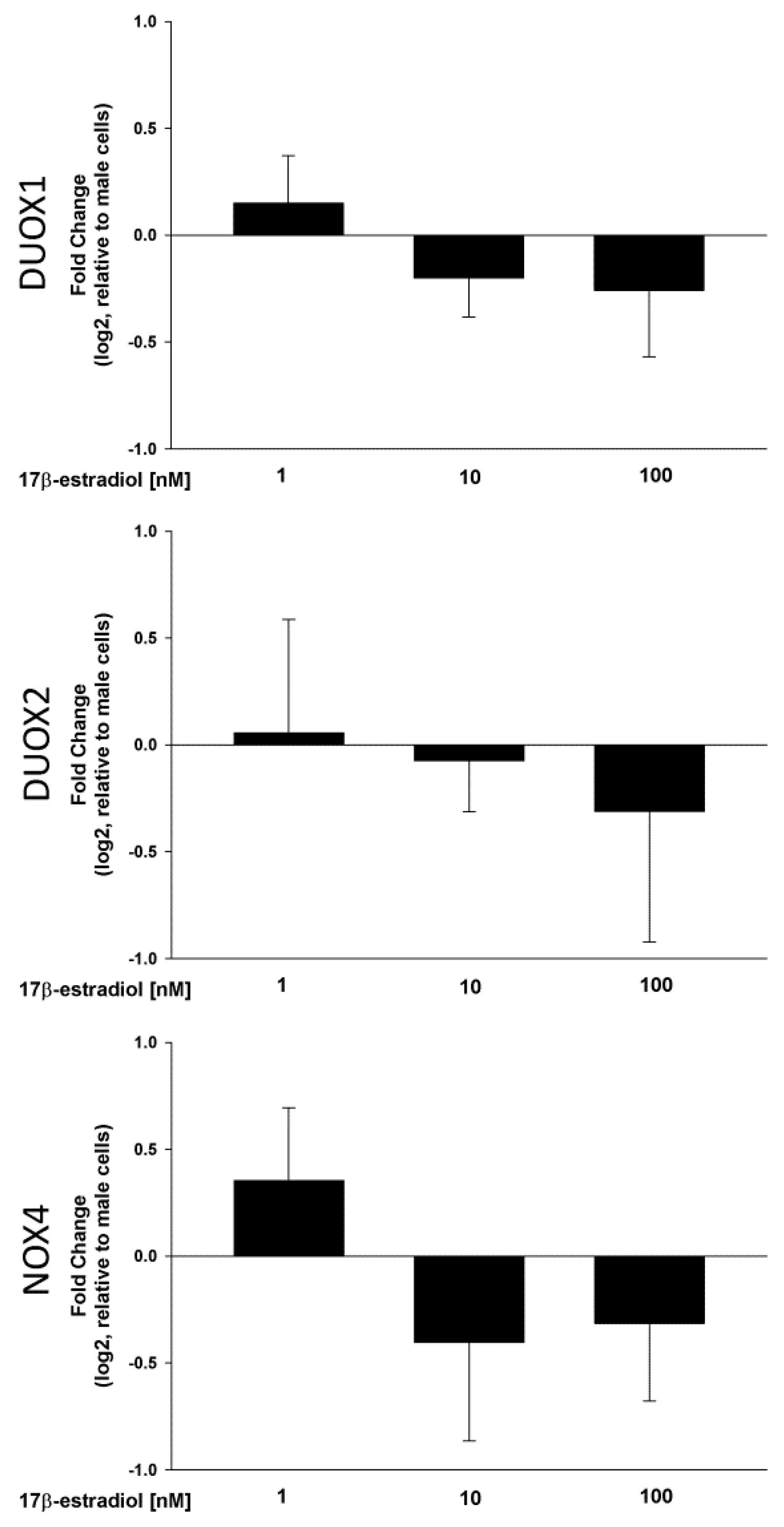
2.2. Stimulation of NOX/DUOX Expression and H2O2 Production by 17β-Estradiol
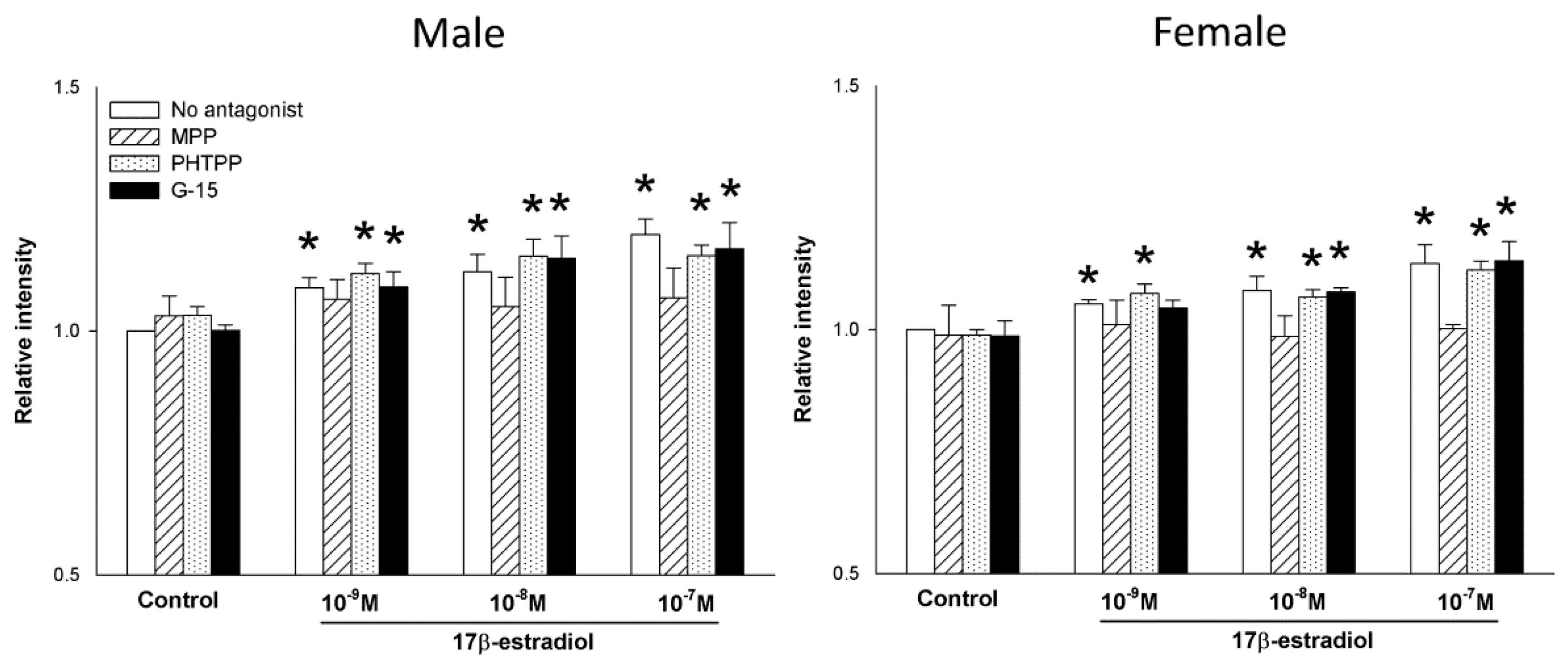
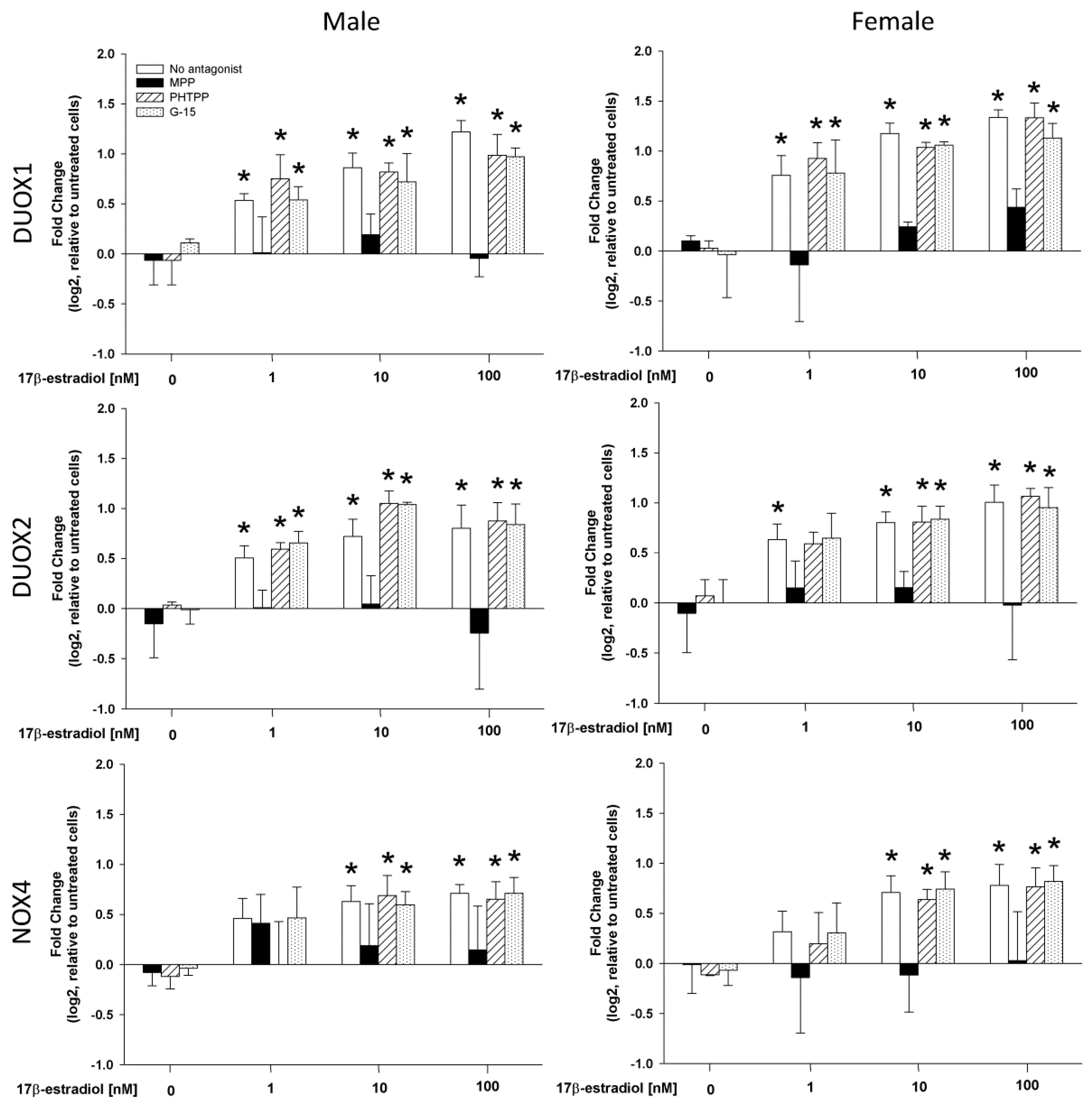
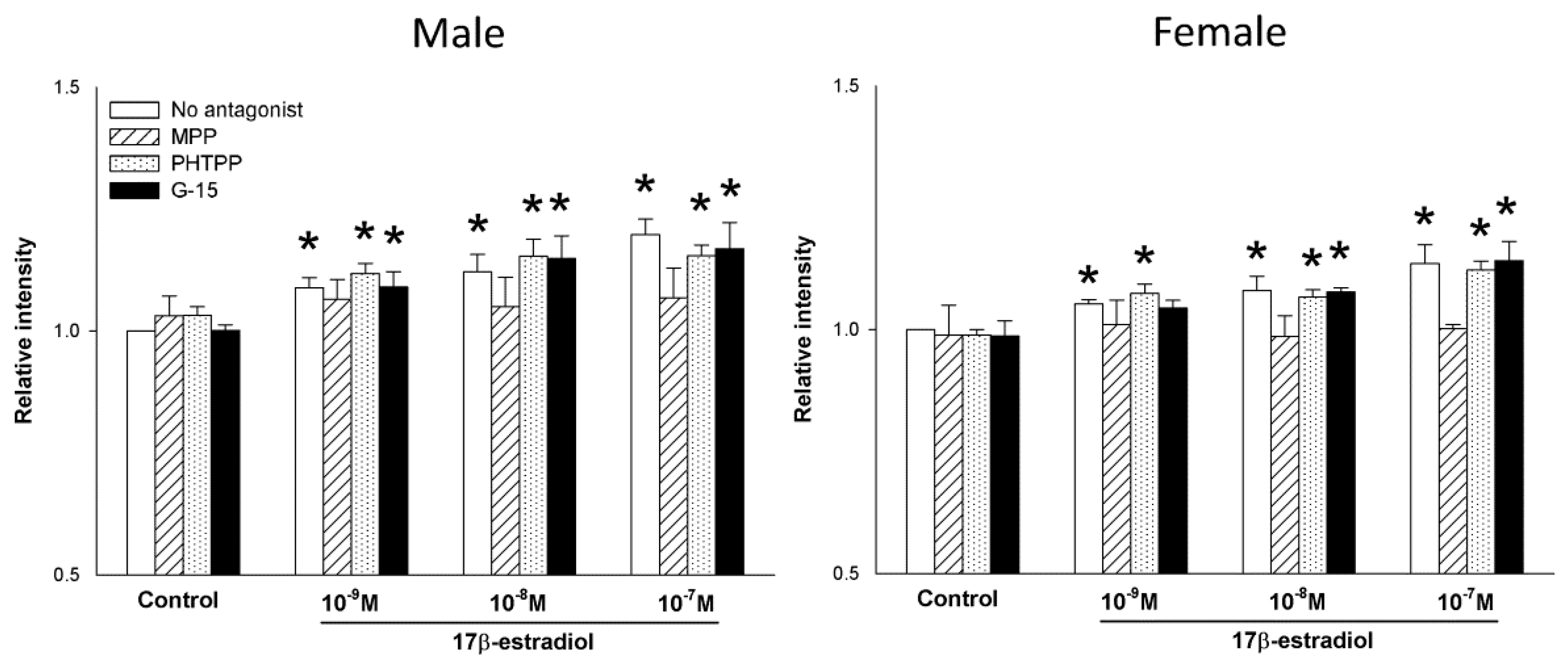
2.3. The Role of Estrogen Receptors in the 17β-Estradiol-Dependent Increase in NOX/DUOX Expression and H2O2 Production
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Treatment
4.4. Cell Viability Assay
4.5. Evaluation of H2O2 Level
4.6. mRNA Analysis by qRT-PCR
4.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- James, B.C.; Mitchell, J.M.; Jeon, H.D.; Vasilottos, N.; Grogan, R.H.; Aschebrook-Kilfoy, B. An update in international trends in incidence rates of thyroid cancer, 1973–2007. Cancer Causes Control 2018, 29, 465–473. [Google Scholar] [CrossRef]
- Hollowell, J.G.; Staehling, N.W.; Flanders, W.D.; Hannon, W.H.; Gunter, E.W.; Spencer, C.A.; Braverman, L.E. Serum TSH, T4, and thyroid antibodies in the United States population (1988–1994): National Health and Nutrition Examination Survey (NHANES III). J. Clin. Endocrinol. Metab. 2002, 87, 489–499. [Google Scholar] [CrossRef]
- Kabat, G.C.; Kim, M.Y.; Wactawski-Wende, J.; Lane, D.; Wassertheil-Smoller, S.; Rohan, T.E. Menstrual and reproductive factors, exogenous hormone use, and risk of thyroid carcinoma in postmenopausal women. Cancer Causes Control 2012, 23, 2031–2040. [Google Scholar] [CrossRef]
- Rahbari, R.; Zhang, L.; Kebebew, E. Thyroid cancer gender disparity. Future Oncol. 2010, 6, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Schonfeld, S.J.; Neta, G.; Sturgis, E.M.; Pfeiffer, R.M.; Hutchinson, A.A.; Xu, L.; Wheeler, W.; Guénel, P.; Rajaraman, P.; de Vathaire, F.; et al. Common genetic variants in sex hormone pathway genes and papillary thyroid cancer risk. Thyroid 2012, 22, 151–156. [Google Scholar] [CrossRef]
- Cooke, P.S.; Nanjappa, M.K.; Ko, C.; Prins, G.S.; Hess, R.A. Estrogens in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Stanczyk, F.Z.; Clarke, N.J. Measurement of estradiol--challenges ahead. J. Clin. Endocrinol. Metab. 2014, 99, 56–58. [Google Scholar] [CrossRef]
- Hammes, S.R.; Levin, E.R. Extranuclear steroid receptors: Nature and actions. Endocr. Rev. 2007, 28, 726–741. [Google Scholar] [CrossRef]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 2009, 20, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Maggiolini, M.; Picard, D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol. 2010, 204, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Capelli, V.; Gaiti, M.; Villani, L.; Zerbini, F.; La Manna, L.; Rotondi, M.; Chiovato, L. ER-alpha and ER-beta expression in differentiated thyroid cancer: Relation with tumor phenotype across the TNM staging and peri-tumor inflammation. Endocrine 2015, 49, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, G.; Meng, X.Y.; Liu, Z.H.; Dong, S. Serum levels of sex hormones and expression of their receptors in thyroid tissue in female patients with various types of thyroid neoplasms. Pathol. Res. Pract. 2014, 210, 830–835. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Rogan, E.G.; Zahid, M. Critical depurinating DNA adducts: Estrogen adducts in the etiology and prevention of cancer and dopamine adducts in the etiology and prevention of Parkinson’s disease. Int. J. Cancer 2017, 141, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Spencer, W.A.; Vadhanam, M.V.; Jeyabalan, J.; Gupta, R.C. Oxidative DNA damage following microsome/Cu(II)-mediated activation of the estrogens, 17β-estradiol, equilenin, and equilin: Role of reactive oxygen species. Chem. Res. Toxicol. 2012, 25, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Zahid, M.; Goldner, W.; Beseler, C.L.; Rogan, E.G.; Cavalieri, E.L. Unbalanced estrogen metabolism in thyroid cancer. Int. J. Cancer 2013, 133, 2642–2649. [Google Scholar] [CrossRef]
- Zane, M.; Parello, C.; Pennelli, G.; Townsend, D.M.; Merigliano, S.; Boscaro, M.; Toniato, A.; Baggio, G.; Pelizzo, M.R.; Rubello, D.; et al. Estrogen and thyroid cancer is a stem affair: A. preliminary study. Biomed. Pharmacother. 2017, 85, 399–411. [Google Scholar] [CrossRef]
- Zane, M.; Catalano, V.; Scavo, E.; Bonanno, M.; Pelizzo, M.R.; Todaro, M.; Stassi, G. Estrogens and stem cells in thyroid cancer. Front. Endocrinol. 2014, 5, 124. [Google Scholar] [CrossRef]
- Fortunato, R.S.; Braga, W.M.; Ortenzi, V.H.; Rodrigues, D.C.; Andrade, B.M.; Miranda-Alves, L.; Rondinelli, E.; Dupuy, C.; Ferreira, A.C.; Carvalho, D.P. Sexual dimorphism of thyroid reactive oxygen species production due to higher NADPH oxidase 4 expression in female thyroid glands. Thyroid 2013, 23, 111–119. [Google Scholar] [CrossRef]
- Ruggeri, R.M.; Vicchio, T.M.; Cristani, M.; Certo, R.; Caccamo, D.; Alibrandi, A.; Giovinazzo, S.; Saija, A.; Campennì, A.; Trimarchi, F.; et al. Oxidative Stress and Advanced Glycation End Products in Hashimoto’s Thyroiditis. Thyroid 2016, 26, 504–511. [Google Scholar] [CrossRef]
- Bednarek, J.; Wysocki, H.; Sowiński, J. Oxidative stress peripheral parameters in Graves’ disease: The effect of methimazole treatment in patients with and without infiltrative ophthalmopathy. Clin. Biochem. 2005, 38, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Trenti, A.; Tedesco, S.; Boscaro, C.; Trevisi, L.; Bolego, C.; Cignarella, A. Estrogen, Angiogenesis, Immunity and Cell Metabolism: Solving the Puzzle. Int. J. Mol. Sci. 2018, 19, E859. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachucki, J.; Wang, D.; Christophe, D.; Miot, F. Structural and functional characterization of the two human ThOX/Duox genes and their 5′-flanking regions. Mol. Cell. Endocrinol. 2004, 214, 53–62. [Google Scholar] [CrossRef]
- Aycan, Z.; Cangul, H.; Muzza, M.; Bas, V.N.; Fugazzola, L.; Chatterjee, V.K.; Persani, L.; Schoenmakers, N. Digenic DUOX1 and DUOX2 Mutations in Cases With Congenital Hypothyroidism. J. Clin. Endocrinol. Metab. 2017, 102, 3085–3090. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Cordeiro, J.V.; Jacinto, A. The role of transcription-independent damage signals in the initiation of epithelial wound healing. Nat. Rev. Mol. Cell Biol. 2013, 14, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal. 2009, 11, 1289–1299. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Coclet, J.; Foureau, F.; Ketelbant, P.; Galand, P.; Dumont, J.E. Cell population kinetics in dog and human adult thyroid. Clin. Endocrinol. 1989, 31, 655–665. [Google Scholar] [CrossRef]
- Maier, J.; van Steeg, H.; van Oostrom, C.; Karger, S.; Paschke, R.; Krohn, K. Deoxyribonucleic acid damage and spontaneous mutagenesis in the thyroid gland of rats and mice. Endocrinology 2006, 147, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Karbownik-Lewinska, M.; Stepniak, J.; Milczarek, M.; Lewinski, A. Protective effect of KI in mtDNA in porcine thyroid: Comparison with KIO3 and nDNA. Eur. J. Nutr. 2015, 54, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Gutscher, M.; Sobotta, M.C.; Wabnitz, G.H.; Ballikaya, S.; Meyer, A.J.; Samstag, Y.; Dick, T.P. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009, 284, 31532–31540. [Google Scholar] [CrossRef]
- Kumar, A.; Klinge, C.M.; Goldstein, R.E. Estradiol-induced proliferation of papillary and follicular thyroid cancer cells is mediated by estrogen receptors α and β. Int. J. Oncol. 2010, 36, 1067–1080. [Google Scholar]
- Chen, G.G.; Vlantis, A.C.; Zeng, Q.; van Hasselt, C.A. Regulation of cell growth by estrogen signaling and potential targets in thyroid cancer. Curr. Cancer Drug Targets 2008, 8, 367–377. [Google Scholar] [CrossRef]
- Sipos, J.A.; Mazzaferri, E.L. Thyroid cancer epidemiology and prognostic variables. Clin. Oncol. 2010, 22, 395–404. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Xia, F.; Wang, N.; Chen, C.; Nie, X.; Li, Q.; Han, B.; Zhai, H.; Jiang, B.; et al. A Higher Ratio of Estradiol to Testosterone Is Associated with Autoimmune Thyroid Disease in Males. Thyroid 2017, 27, 960–966. [Google Scholar] [CrossRef]
- Luo, J.; Hendryx, M.; Manson, J.E.; Liang, X.; Margolis, K.L. Hysterectomy, Oophorectomy, and Risk of Thyroid Cancer. J. Clin. Endocrinol. Metab. 2016, 101, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Burek, C.L.; Rose, N.R. Autoimmune thyroiditis and ROS. Autoimmun. Rev. 2008, 7, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Ameziane-El-Hassani, R.; Schlumberger, M.; Dupuy, C. NADPH oxidases: New actors in thyroid cancer? Nat. Rev. Endocrinol. 2016, 12, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Antico-Arciuch, V.G.; Dima, M.; Liao, X.H.; Refetoff, S.; Di Cristofano, A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene 2010, 29, 5678–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyemi, U.; Caillou, B.; Talbot, M.; Ameziane-El-Hassani, R.; Lacroix, L.; Lagent-Chevallier, O.; Al Ghuzlan, A.; Roos, D.; Bidart, J.M.; Virion, A.; et al. Intracellular expression of reactive oxygen species-generating NADPH oxidase NOX4 in normal and cancer thyroid tissues. Endocr. Relat. Cancer 2010, 17, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameziane-El-Hassani, R.; Talbot, M.; de Souza Dos Santos, M.C.; Al Ghuzlan, A.; Hartl, D.; Bidart, J.M.; De Deken, X.; Miot, F.; Diallo, I.; de Vathaire, F.; et al. NADPH oxidase DUOX1 promotes long-term persistence of oxidative stress after an exposure to irradiation. Proc. Natl. Acad. Sci. USA 2015, 112, 5051–5056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, L.; Nocera, M.; Mian, C.; Caillou, B.; Virion, A.; Dupuy, C.; Filetti, S.; Bidart, J.M.; Schlumberger, M. Expression of nicotinamide adenine dinucleotide phosphate oxidase flavoprotein DUOX genes and proteins in human papillary and follicular thyroid carcinomas. Thyroid 2001, 11, 1017–1023. [Google Scholar] [CrossRef]
- Eskalli, Z.; Achouri, Y.; Hahn, S.; Many, M.C.; Craps, J.; Refetoff, S.; Liao, X.H.; Dumont, J.E.; Van Sande, J.; Corvilain, B.; et al. Overexpression of Interleukin-4 in the Thyroid of Transgenic Mice Upregulates the Expression of Duox1 and the Anion Transporter Pendrin. Thyroid 2016, 26, 1499–1512. [Google Scholar] [CrossRef]
- Peshavariya, H.; Jiang, F.; Taylor, C.J.; Selemidis, S.; Chang, C.W.; Dusting, G.J. Translation-linked mRNA destabilization accompanying serum-induced Nox4 expression in human endothelial cells. Antioxid. Redox Signal. 2009, 11, 2399–2408. [Google Scholar] [CrossRef]
- Stępniak, J.; Lewiński, A.; Karbownik-Lewińska, M. Membrane lipids and nuclear DNA are differently susceptive to Fenton reaction substrates in porcine thyroid. Toxicol. In Vitro 2013, 27, 71–78. [Google Scholar] [CrossRef]
- Karbownik-Lewińska, M.; Stępniak, J.; Lewiński, A. High level of oxidized nucleosides in thyroid mitochondrial DNA; damaging effects of Fenton reaction substrates. Thyroid Res. 2012, 5, 24. [Google Scholar] [CrossRef]
- Karbownik, M.; Lewinski, A. The role of oxidative stress in physiological and pathological processes in the thyroid gland; possible involvement in pineal-thyroid interactions. Neuro Endocrinol. Lett. 2003, 24, 293–303. [Google Scholar] [PubMed]
- Stepniak, J.; Karbownik-Lewinska, M. 17β-estradiol prevents experimentally-induced oxidative damage to membrane lipids and nuclear DNA in porcine ovary. Syst. Biol. Reprod. Med. 2016, 62, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Park, B.O.; Meyer, T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010, 29, 4473–4484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manole, D.; Schildknecht, B.; Gosnell, B.; Adams, E.; Derwahl, M. Estrogen promotes growth of human thyroid tumor cells by different molecular mechanisms. J. Clin. Endocrinol. Metab. 2001, 86, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, Z.M.; González-Flecha, B. TOR kinase and Ran are downstream from PI3K/Akt in H2O2-induced mitosis. J. Cell. Biochem. 2004, 91, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Nozhat, Z.; Hedayati, M. PI3K/AKT Pathway and Its Mediators in Thyroid Carcinomas. Mol. Diagn. Ther. 2016, 20, 13–26. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef]
- Shanle, E.K.; Xu, W. Selectively targeting estrogen receptors for cancer treatment. Adv. Drug Deliv. Rev. 2010, 62, 1265–1276. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Liu, J.; Gu, L.; Ma, X.; Huang, B.; Pan, X. Research progress on the reproductive and non-reproductive endocrine tumors by estrogen-related receptors. J. Steroid Biochem. Mol. Biol. 2016, 158, 22–30. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRα coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal. 2018, 11, eaam5855. [Google Scholar] [CrossRef]
- Zeng, Q.; Chen, G.G.; Vlantis, A.C.; Van Hasselt, C.A. Oestrogen mediates the growth of human thyroid carcinoma cells via an oestrogen receptor-ERK pathway. Cell Prolif. 2007, 40, 921–935. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepniak, J.; Lewinski, A.; Karbownik-Lewinska, M. Sexual Dimorphism of NADPH Oxidase/H2O2 System in Rat Thyroid Cells; Effect of Exogenous 17β-Estradiol. Int. J. Mol. Sci. 2018, 19, 4063. https://doi.org/10.3390/ijms19124063
Stepniak J, Lewinski A, Karbownik-Lewinska M. Sexual Dimorphism of NADPH Oxidase/H2O2 System in Rat Thyroid Cells; Effect of Exogenous 17β-Estradiol. International Journal of Molecular Sciences. 2018; 19(12):4063. https://doi.org/10.3390/ijms19124063
Chicago/Turabian StyleStepniak, Jan, Andrzej Lewinski, and Malgorzata Karbownik-Lewinska. 2018. "Sexual Dimorphism of NADPH Oxidase/H2O2 System in Rat Thyroid Cells; Effect of Exogenous 17β-Estradiol" International Journal of Molecular Sciences 19, no. 12: 4063. https://doi.org/10.3390/ijms19124063