Novel Clofarabine-Based Combinations with Polyphenols Epigenetically Reactivate Retinoic Acid Receptor Beta, Inhibit Cell Growth, and Induce Apoptosis of Breast Cancer Cells

 , ,
, ,
Abstract
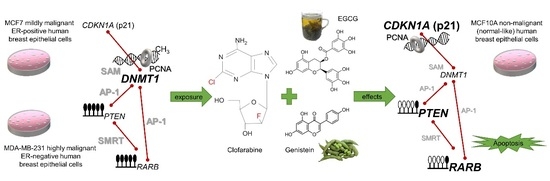
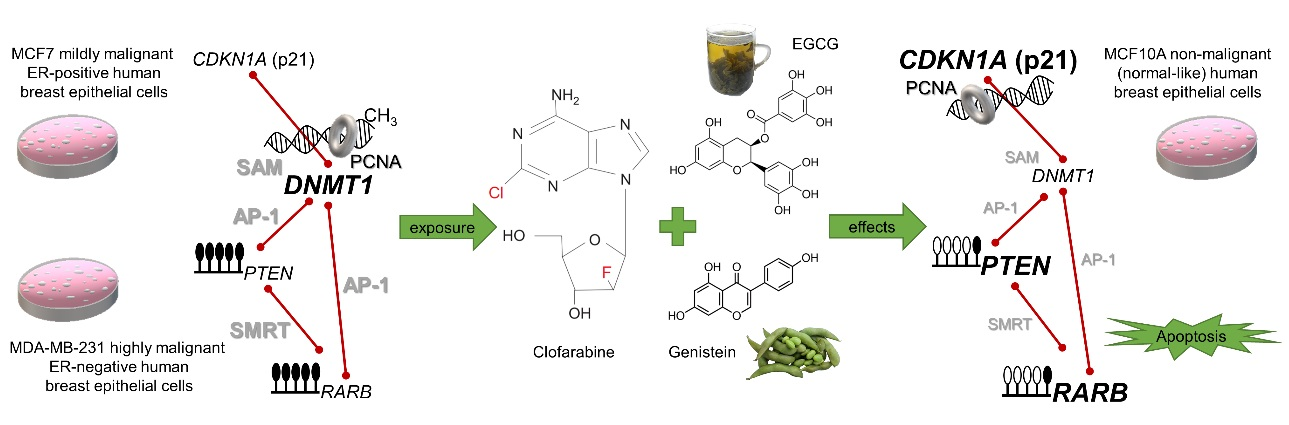
:
1. Introduction
2. Results and Discussion
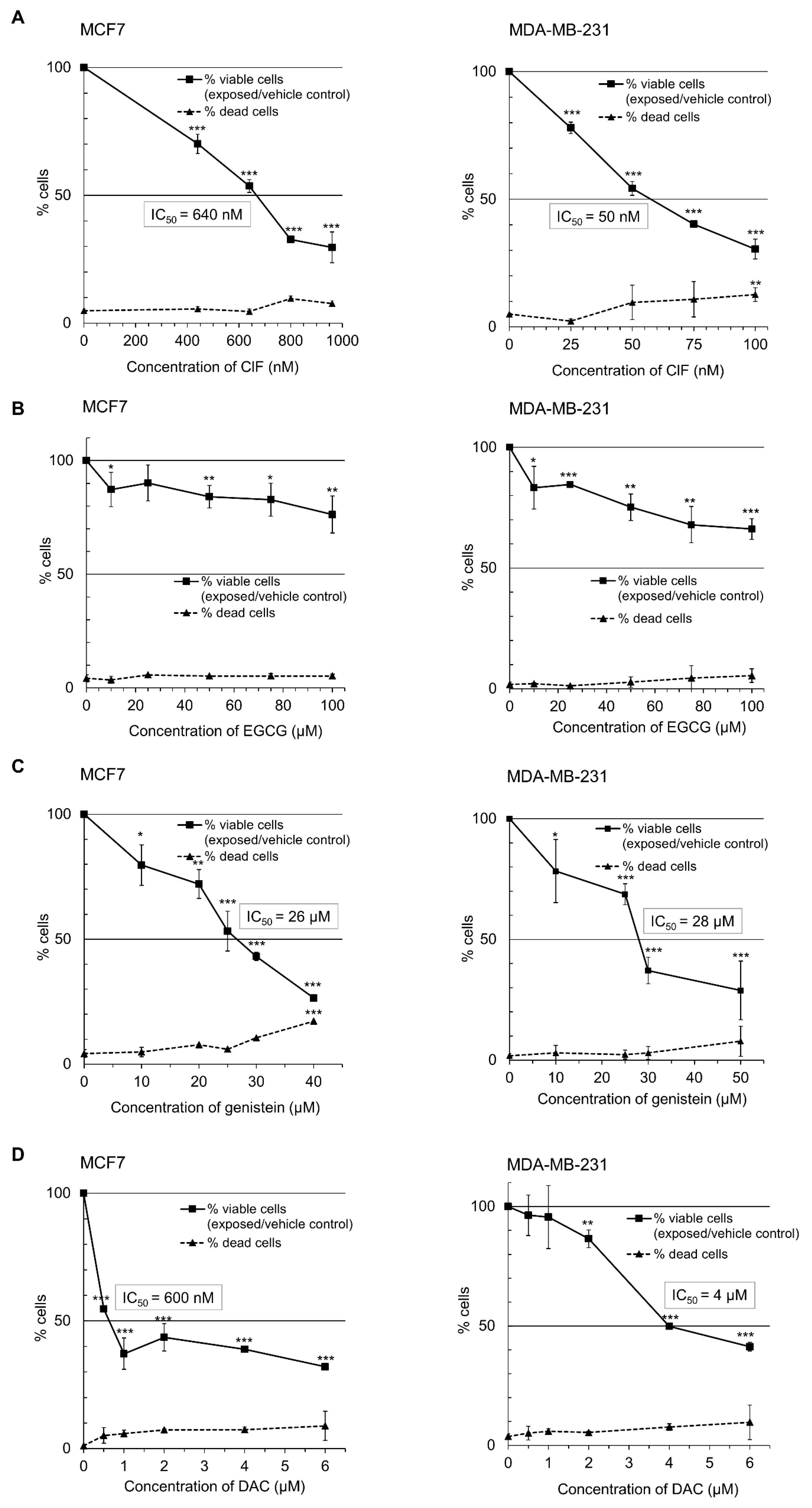
2.1. ClF-Based Combinations with Polyphenols Inhibits Breast Cancer Cell Growth
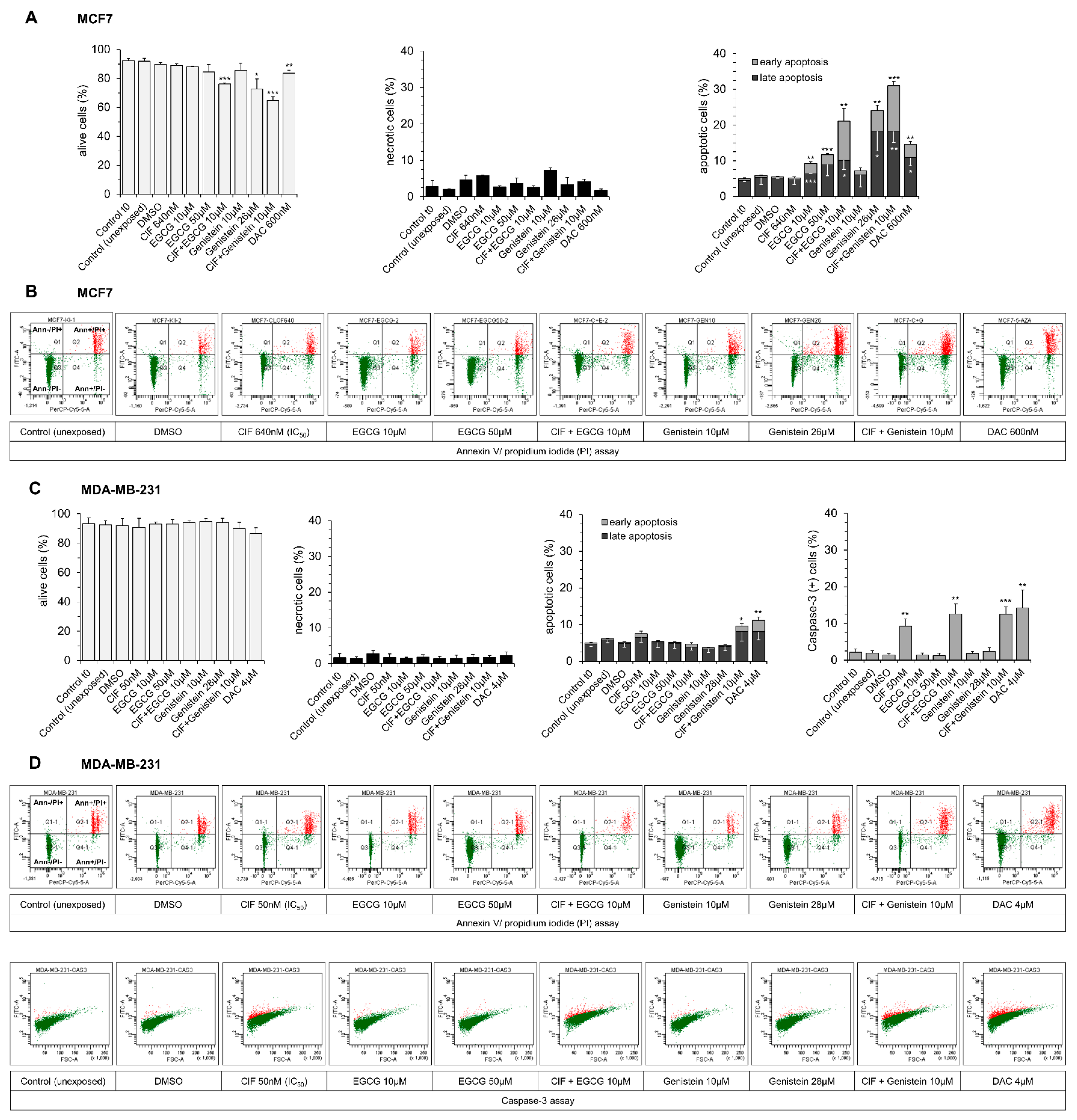
2.2. Both EGCG and Genistein Enhances ClF Pro-Apoptotic Effects Mostly in Mildly Malignant MCF7 Cells
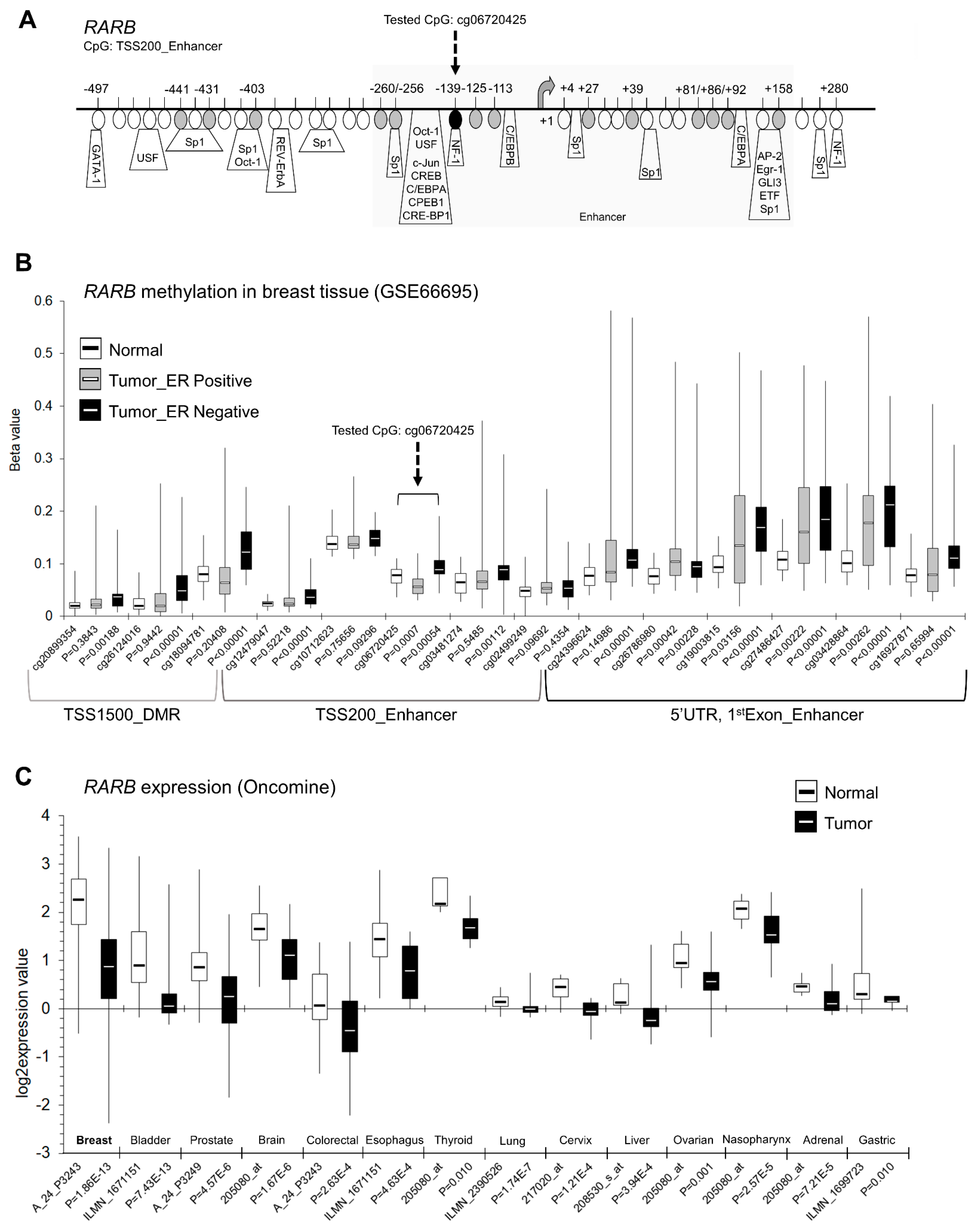
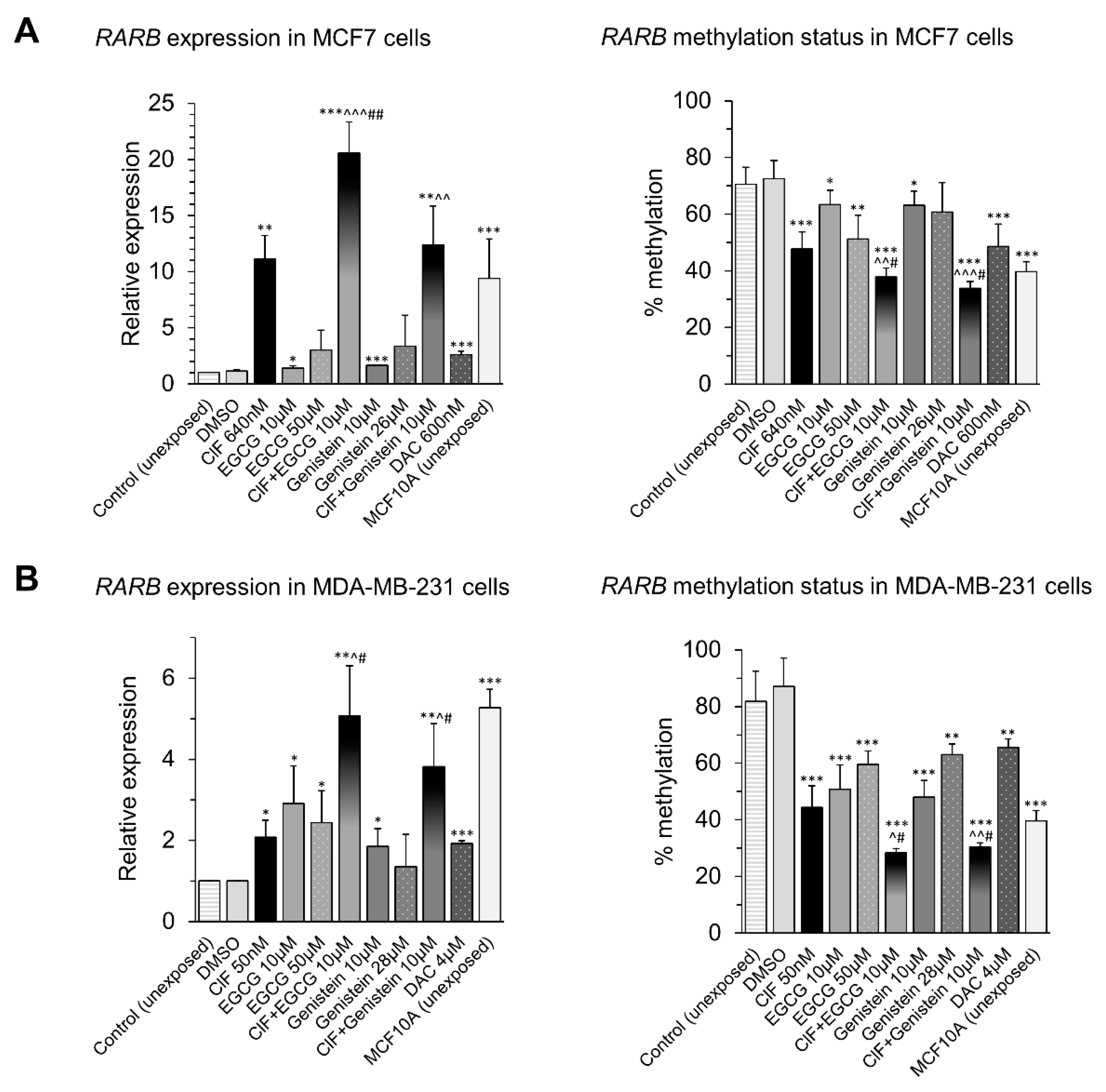
2.3. ClF, EGCG, and Genistein Used Alone and in Combinations Exert Profound Effects on RARB Promoter Methylation and Expression in Breast Cancer Cells
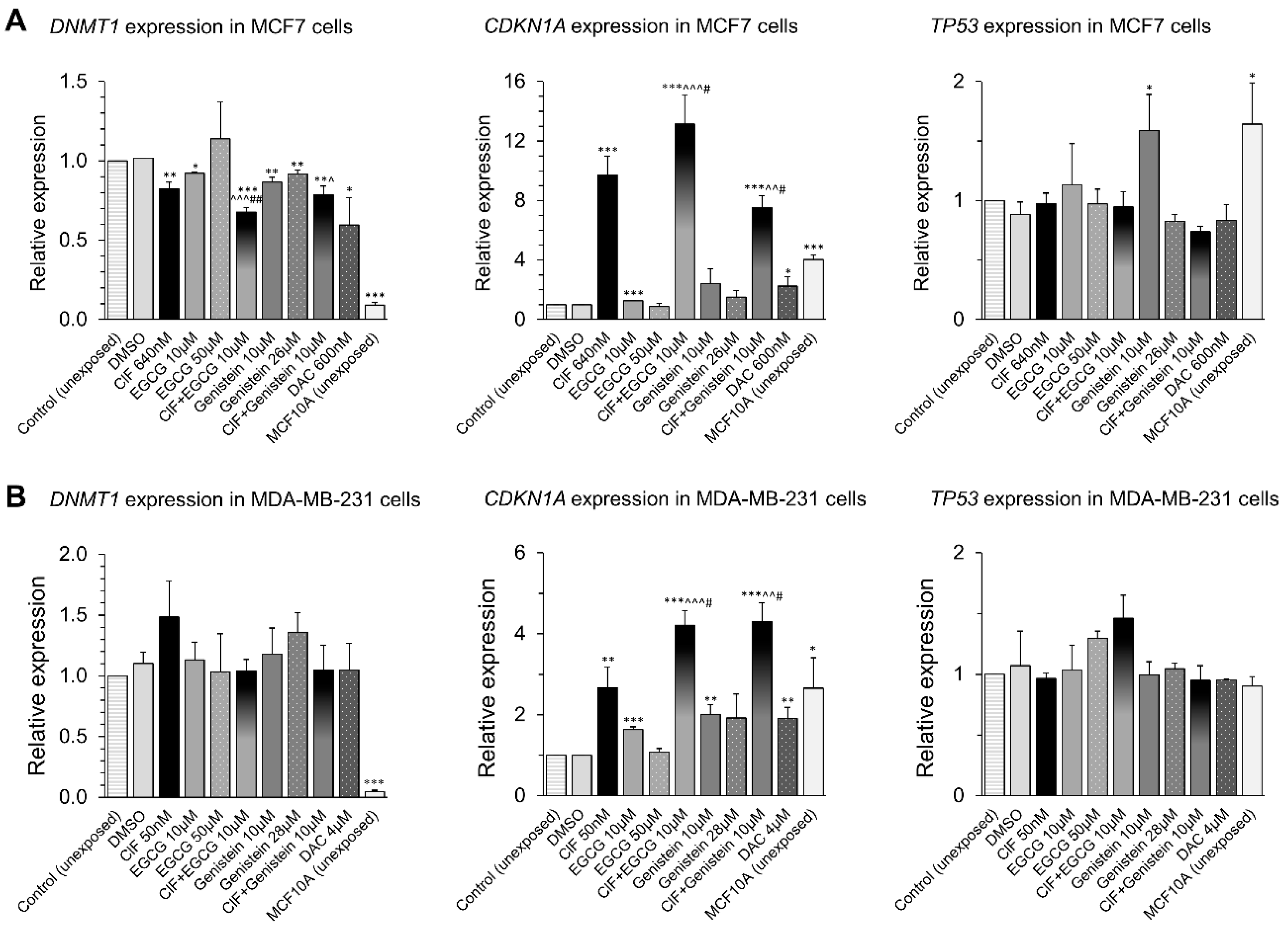
2.4. RARB Transcriptional Reactivation in Response to the ClF Combinations with Polyphenols Is Associated with Robust CDKN1A Upregulation
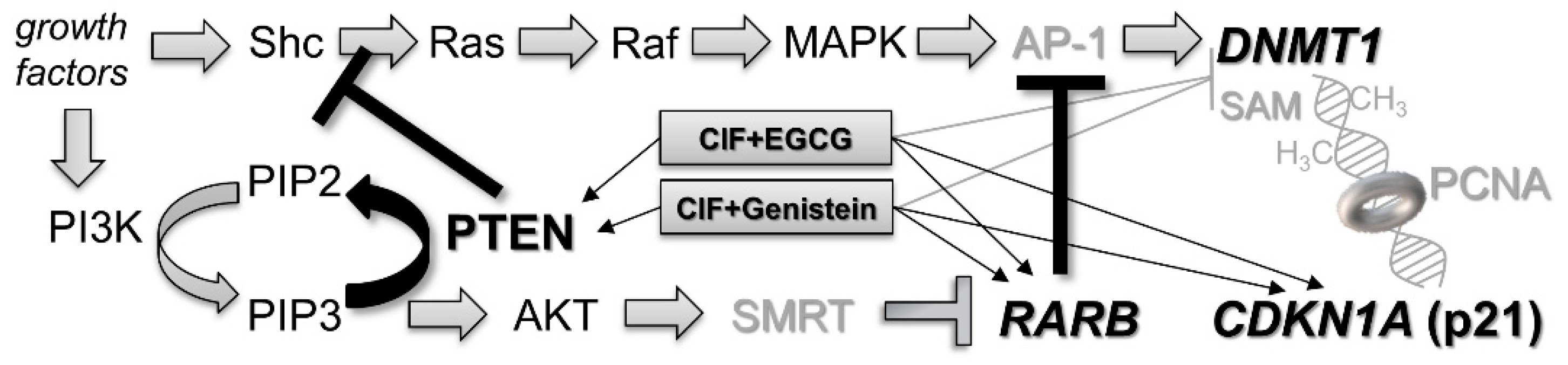
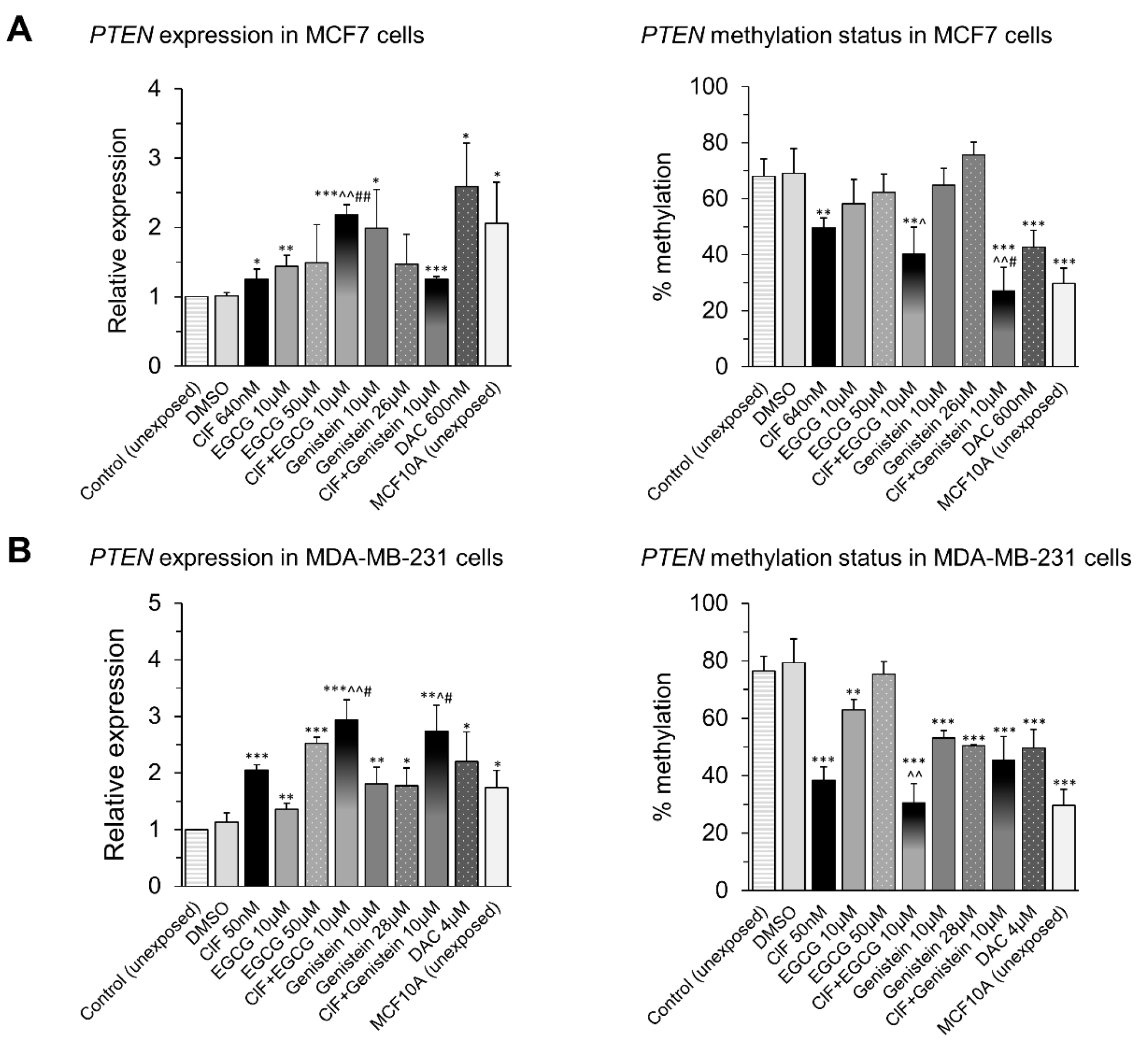
2.5. PTEN Upregulation upon Combinatorial Exposures Is Partly Involved in Robust RARB Transcriptional Reactivation
3. Conclusions
4. Materials and Methods
4.1. Compounds
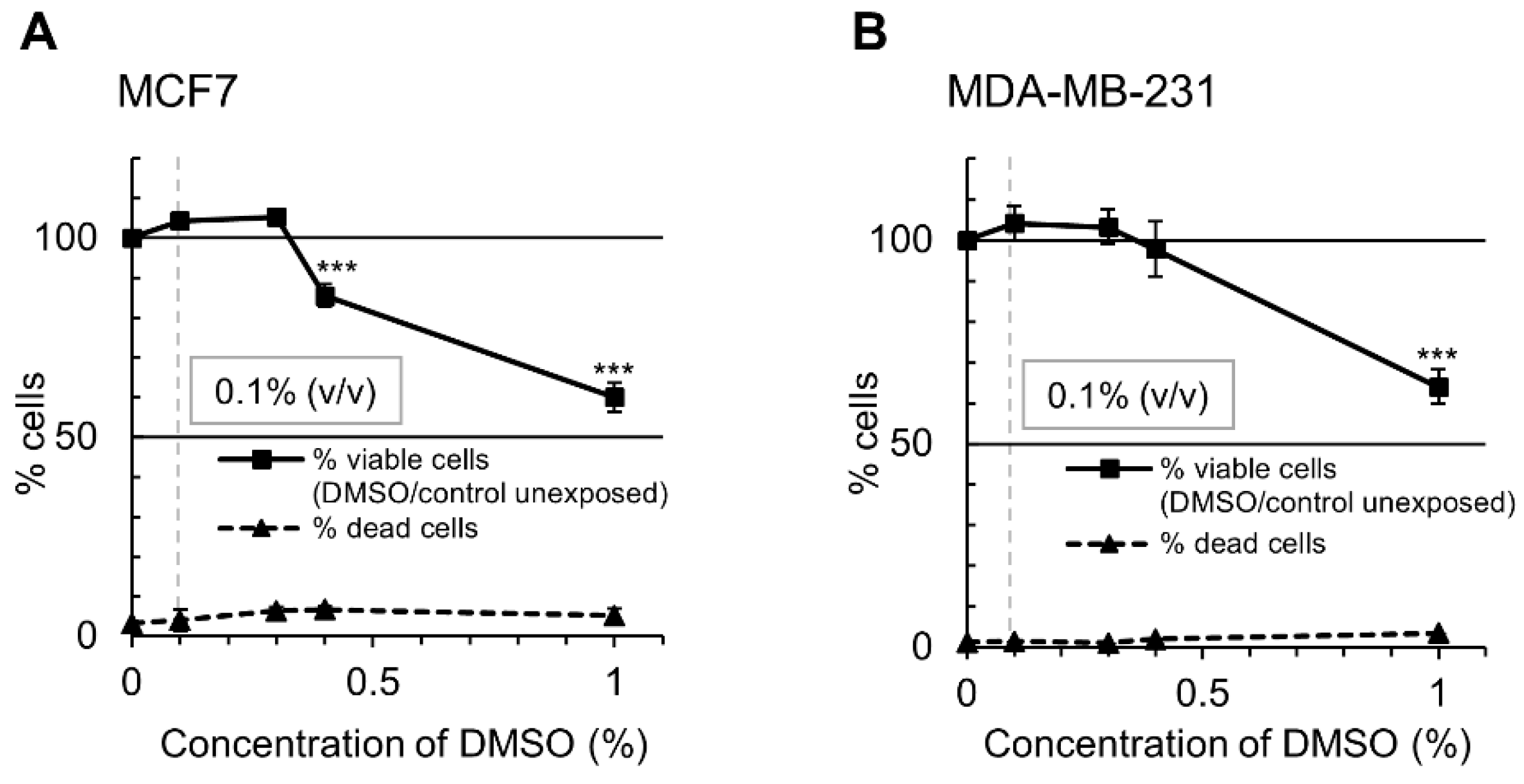
4.2. Cell Culture
4.3. Cell Viability and Apoptosis
4.4. CompuSyn
4.5. DNA and RNA Isolation
4.6. Methylation-Sensitive Restriction Analysis (MSRA)
4.7. Quantitative Real-Time PCR (qPCR)
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ClF | Clofarabine, 2-chloro-2′-fluoro-2′-deoxyarabinosylade |
| DAC | Decitabine, 5-aza-2′-deoxycytidine |
| EGCG | (−)-Epigallocatechin gallate |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018. [Google Scholar] [CrossRef] [PubMed]
- Danaei, G.; Vander Hoorn, S.; Lopez, A.D.; Murray, C.J.; Ezzati, M.; Comparative Risk Assessment collaborating group (Cancers). Causes of cancer in the world: Comparative risk assessment of nine behavioural and environmental risk factors. Lancet 2005, 366, 1784–1793. [Google Scholar] [CrossRef]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Clofarabine, a novel adenosine analogue, reactivates DNA methylation-silenced tumour suppressor genes and inhibits cell growth in breast cancer cells. Eur. J. Pharmacol. 2014, 723, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Cebula-Obrzut, B.; Smolewski, P.; Fabianowska-Majewska, K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. J. Nutrigenet. Nutrigenom. 2015, 8, 91–101. [Google Scholar] [CrossRef]
- Cheishvili, D.; Stefanska, B.; Yi, C.; Li, C.C.; Yu, P.; Arakelian, A.; Tanvir, I.; Khan, H.A.; Rabbani, S.; Szyf, M. A common promoter hypomethylation signature in invasive breast, liver and prostate cancer cell lines reveals novel targets involved in cancer invasiveness. Oncotarget 2015, 6, 33253–33268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubecka, K.; Kaufman-Szymczyk, A.; Fabianowska-Majewska, K. Inhibition of breast cancer cell growth by the combination of clofarabine and sulforaphane involves epigenetically mediated CDKN2A upregulation. Nucleosides Nucleotides Nucleic Acids 2018, 37, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Lubecka, K.; Kurzava, L.; Flower, K.; Buvala, H.; Zhang, H.; Teegarden, D.; Camarillo, I.; Suderman, M.; Kuang, S.; Andrisani, O.; et al. Stilbenoids remodel the DNA methylation patterns in breast cancer cells and inhibit oncogenic NOTCH signaling through epigenetic regulation of MAML2 transcriptional activity. Carcinogenesis 2016, 37, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, B.; Fabianowska-Majewska, K. Alteration of DNA methylation status in K562 and MCF-7 cancer cell lines by nucleoside analogues. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1029–1032. [Google Scholar] [CrossRef]
- Krawczyk, B.; Rudnicka, K.; Fabianowska-Majewska, K. The effects of nucleoside analogues on promoter methylation of selected tumor suppressor genes in MCF-7 and MDA-MB-231 breast cancer cell lines. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1043–1046. [Google Scholar] [CrossRef]
- Stefanska, B.; Rudnicka, K.; Bednarek, A.; Fabianowska-Majewska, K. Hypomethylation and induction of retinoic acid receptor beta 2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur. J. Pharmacol. 2010, 638, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Salamé, P.; Bednarek, A.; Fabianowska-Majewska, K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br. J. Nutr. 2012, 107, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Gandhi, V.; Keating, M.J.; Jeha, S.; Plunkett, W.; Kantarjian, H.M. The role of clofarabine in hematologic and solid malignancies--development of a next-generation nucleoside analog. Cancer 2005, 103, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Majda, K.; Kaufman-Szymczyk, A.; Lubecka-Pietruszewska, K.; Bednarek, A.; Fabianowska-Majewska, K. Influence of clofarabine on transcriptional activity of PTEN, APC, RARB2, ZAP70 genes in K562 cells. Anticancer Res. 2010, 30, 4601–4606. [Google Scholar] [PubMed]
- Majda, K.; Lubecka, K.; Kaufman-Szymczyk, A.; Fabianowska-Majewska, K. Clofarabine (2-chloro-2′-fluoro-2′-deoxyarabinosyladenine)-biochemical aspects of anticancer activity. Acta Pol. Pharm. 2011, 68, 459–466. [Google Scholar]
- Mannargudi, M.B.; Deb, S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer therapy? J. Cancer Res. Clin. Oncol. 2017, 143, 1499–1529. [Google Scholar] [CrossRef]
- Linnekamp, J.F.; Butter, R.; Spijker, R.; Medema, J.P.; van Laarhoven, H.W.M. Clinical and biological effects of demethylating agents on solid tumours—A systematic review. Cancer Treat. Rev. 2017, 54, 10–23. [Google Scholar] [CrossRef]
- Warzocha, K.; Fabianowska-Majewska, K.; Błoński, J.; Krakowski, E.; Robak, T. 2-chloro-deoxyadenosine inhibits activity of adenosine deaminase and S-adenosylhomocysteine hydrolase in patients with chronic lymphocytic leukemia. Eur. J. Cancer 1997, 33, 170–173. [Google Scholar] [CrossRef]
- Chiang, P.K. Biological effects of inhibitors of S-adenosylhomocysteine hydrolase. Pharmacol. Ther. 1998, 77, 115–134. [Google Scholar] [CrossRef]
- Fabianowska-Majewska, K.; Ruckemann, K.; Duley, J.A.; Simmonds, H.A. Effect of cladribine, fludarabine, and 5-aza-deoxycytidine on S-adenosylmethionine (SAM) and nucleotides pools in stimulated human lymphocytes. Adv. Exp. Med. Biol. 1998, 431, 531–535. [Google Scholar]
- Wyczechowska, D.; Fabianowska-Majewska, K. The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem. Pharmacol. 2003, 65, 219–225. [Google Scholar] [CrossRef]
- Arapshian, A.; Kuppumbatti, Y.S.; Mira-y-Lopez, R. Methylation of conserved CpG sites neighboring the beta retinoic acid response element may mediate retinoic acid receptor beta gene silencing in MCF-7 breast cancer cells. Oncogene 2000, 19, 4066–4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widschwendter, M.; Berger, M.; Hermann, M.; Müller, H.M.; Amberger, A.; Zeschnigk, M.; Widschwendter, A.; Abendstein, B.; Zeimet, A.G.; Daxenbichler, G.; et al. Methylation and silencing of the retinoic acid receptor-beta2 gene in breast cancer. J. Natl. Cancer Inst. 2000, 92, 826–832. [Google Scholar] [CrossRef] [PubMed]
- García, J.M.; Silva, J.; Peña, C.; Garcia, V.; Rodríguez, R.; Cruz, M.A.; Cantos, B.; Provencio, M.; España, P.; Bonilla, F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer 2004, 41, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kumagai, T.; Vora, J.; Bose, N.; Sehgal, I.; Koeffler, P.H.; Bose, S. PTEN promoter is methylated in a proportion of invasive breast cancers. Int. J. Cancer 2004, 112, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Connolly, R.; Stearns, V. Epigenetics as a Therapeutic Target in Breast Cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 191–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, S.; Germain, P.; Alvarez, R.; Rodríguez-Barrios, F.; Gronemeyer, H.; de Lera, A.R. Structure, function and modulation of retinoic acid receptor beta, a tumor suppressor. Int. J. Biochem. Cell Biol. 2007, 39, 1406–1415. [Google Scholar] [CrossRef]
- Lin, F.; Xiao, D.; Kolluri, S.K.; Zhang, X. Unique anti-activator protein-1 activity of retinoic acid receptor beta. Cancer Res. 2000, 60, 3271–3280. [Google Scholar]
- Yang, L.; Kim, H.T.; Munoz-Medellin, D.; Reddy, P.; Brown, P.H. Induction of retinoid resistance in breast cancer cells by overexpression of cJun. Cancer Res. 1997, 57, 4652–4661. [Google Scholar]
- Cantley, L.C.; Neel, B.G. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 4240–4245. [Google Scholar] [CrossRef]
- Gu, J.; Tamura, M.; Yamada, K.M. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J. Cell Biol. 1998, 143, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Bigey, P.; Ramchandani, S.; Theberge, J.; Araujo, F.D.; Szyf, M. Transcriptional regulation of the human DNA Methyltransferase (DNMT1) gene. Gene 2000, 242, 407–418. [Google Scholar] [CrossRef]
- Qin, W.; Leonhardt, H.; Pichler, G. Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2011, 2, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefebvre, B.; Brand, C.; Flajollet, S.; Lefebvre, P. Down-regulation of the tumour suppressor gene retinoic acid receptor beta2 through the phosphoinisitide 3- knase/Akt signaling pathway. Mol. Endocrinol. 2006, 20, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buckhaults, P.; Cui, X.; Tollefsbol, T.O. Combinatorial epigenetic mechanisms and efficacy of early breast cancer inhibition by nutritive botanicals. Epigenomics 2016, 8, 1019–1037. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tollefsbol, T.O. Combinational Proanthocyanidins and Resveratrol Synergistically Inhibit Human Breast Cancer Cells and Impact Epigenetic⁻Mediating Machinery. Int. J. Mol. Sci. 2018, 19, 2204. [Google Scholar] [CrossRef] [PubMed]
- Ateeq, B.; Unterberger, A.; Szyf, M.; Rabbani, S.A. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 2008, 10, 266–278. [Google Scholar] [CrossRef]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef]
- Lacroix, M.; Toillon, R.A.; Leclercq, G. p53 and breast cancer, an update. Endocr. Relat. Cancer 2006, 13, 293–325. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 170325. [Google Scholar] [CrossRef]
- Peterson, E.J.; Bögler, O.; Taylor, S.M. p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding. Cancer Res. 2003, 63, 6579–6582. [Google Scholar] [PubMed]
- Chuang, L.S.; Ian, H.I.; Koh, T.W.; Ng, H.H.; Xu, G.; Li, B.F. Human DNA-(cytosine-5)-methyl-transferase-PCNA complex as a target for p21WAF1. Science 1997, 277, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Suetake, I.; Tajima, S.; Morioka, H.; Ohta, S.; Obuse, C.; Tsurimoto, T. PCNA clamp facilitates action of DNA cytosine methyltransferase 1 on hemimethylated DNA. Genes Cells 2002, 7, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARβ, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res. 2005, 11, 7033–7041. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Chen, D.; Yang, C.S. Dietary polyphenols may affect DNA methylation. J. Nutr. 2007, 137, 223S–228S. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Zhu, B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis 2006, 27, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Tanaka, T.; Kawashima, H.; Nakatani, T. Involvement of the estrogen receptor beta in genistein-induced expression of p21(waf1/cip1) in PC-3 prostate cancer cells. Anticancer Res. 2008, 28, 709–714. [Google Scholar]
- Pethe, V.; Shekhar, P.V. Estrogen inducibility of c-Ha-ras transcription in breast cancer cells. Identification of functional estrogen-responsive transcriptional regulatory elements in exon 1/intron 1 of the c-Ha-ras gene. J. Biol. Chem. 1999, 274, 30969–30978. [Google Scholar] [CrossRef]
- Chung, J.H.; Ostrowski, M.C.; Romigh, T.; Minaguchi, T.; Waite, K.A.; Eng, C. The ERK1/2 pathway modulates nuclear PTEN-mediated cell cycle arrest by cyclin D1 transcriptional regulation. Hum. Mol. Genet. 2006, 15, 2553–2559. [Google Scholar] [CrossRef]
- Xie, Q.; Bai, Q.; Zou, L.Y.; Zhang, Q.Y.; Zhou, Y.; Chang, H.; Yi, L.; Zhu, J.D.; Mi, M.T. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014, 53, 422–431. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Geutjes, E.J.; Tian, S.; Roepman, P.; Bernards, R. Deoxycytidine kinase is overexpressed in poor outcome breast cancer and determines responsiveness to nucleoside analogs. Breast Cancer Res. Treat. 2012, 131, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Abramczyk, H.; Surmacki, J.; Kopeć, M.; Olejnik, A.K.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The role of lipid droplets and adipocytes in cancer. Raman imaging of cell cultures: MCF10A, MCF7, and MDA-MB-231 compared to adipocytes in cancerous human breast tissue. Analyst 2015, 140, 2224–2235. [Google Scholar] [CrossRef] [PubMed]
- Lubecka-Pietruszewska, K.; Kaufman-Szymczyk, A.; Stefanska, B.; Fabianowska-Majewska, K. Folic acid enforces DNA methylation-mediated transcriptional silencing of PTEN, APC and RARbeta2 tumour suppressor genes in breast cancer. Biochem. Biophys. Res. Commun. 2013, 430, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Han, Z.; Li, X.; Xie, H.H.; Zhu, S.S. Mechanism of EGCG promoting apoptosis of MCF-7 cell line in human breast cancer. Oncol. Lett. 2017, 14, 3623–3627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, O.Y.; Noh, E.M.; Jang, H.Y.; Lee, Y.R.; Lee, B.K.; Jung, S.H.; Kim, J.S.; Youn, H.J. Epigallocatechin gallate inhibits the growth of MDA-MB-231 breast cancer cells via inactivation of the β-catenin signaling pathway. Oncol. Lett. 2017, 14, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]
- Xu, J.; Loo, G. Different effects of genistein on molecular markers related to apoptosis in two phenotypically dissimilar breast cancer cell lines. J. Cell Biochem. 2001, 82, 78–88. [Google Scholar] [CrossRef]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-Epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, CS. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Sundaram, M.K.; Ansari, M.Z.; Al Mutery, A.; Ashraf, M.; Nasab, R.; Rai, S.; Rais, N.; Hussain, A. Genistein Induces Alterations of Epigenetic Modulatory Signatures in Human Cervical Cancer Cells. Anticancer Agents Med. Chem. 2018, 18, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Saramaki, A.; Banwell, C.M.; Campbell, M.J.; Carlberg, C. Regulation of the human p21(waf1/cip1) gene promoter via multiple binding sites for p53 and the vitamin D3 receptor. Nucleic Acids Res. 2006, 34, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Iwase, H.; Omoto, Y.; Iwata, H.; Toyama, T.; Hara, Y.; Ando, Y.; Ito, Y.; Fujii, Y.; Kobayashi, S. DNA methylation analysis at distal and proximal promoter regions of the oestrogen receptor gene in breast cancers. Br. J. Cancer 1999, 80, 1982–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, F.; Gundersen, G.; Lopez, R.; Prydz, H. CpG islands as gene markers in the human genome. Genomics 1992, 13, 1095–1107. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Untergrasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Saldaña-Meyer, R.; Recillas-Targa, F. Transcriptional and epigenetic regulation of the p53 tumor suppressor gene. Epigenetics 2011, 6, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Srivastava, G.; Gao, Z.; Zhang, X.; Murray, P.; Ambinder, R.; Tao, Q. Promoter hypermethylation of the cyclin-dependent kinase inhibitor (CDKI) gene p21WAF1/CIP1/SDI1 is rare in various lymphomas and carcinomas. Blood 2004, 103, 743–746. [Google Scholar] [CrossRef]
- Zheng, S.; Rollet, M.; Yang, K.; Pan, Y.X. A gestational low-protein diet represses p21(WAF1/Cip1) expression in the mammary gland of offspring rats through promoter histone modifications. Br. J. Nutr 2012, 108, 998–1007. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, 1–10. [Google Scholar] [CrossRef]
- Potemski, P.; Pluciennik, E.; Bednarek, A.K.; Kusinska, R.; Kubiak, R.; Kordek, R. Evaluation of oestrogen receptor expression in breast cancer by quantification of mRNA. Histopathology 2007, 51, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Żelazowski, M.J.; Płuciennik, E.; Pasz-Walczak, G.; Potemski, P.; Kordek, R.; Bednarek, A.K. WWOX expression in colorectal cancer--a real-time quantitative RT-PCR study. Tumour Biol. 2011, 32, 551–560. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | Cell Viability | |||
| MCF7 | MDA-MB-231 | |||
| ClF (IC50) | 53.6 ± 2.5 *** | 54.2 ± 2.7 *** | ||
| EGCG 10 µM | 87.2 ± 7.6 * | 83.3 ± 8.9 * | ||
| Genistein 10 µM | 79.7 ± 8.1 * | 78.3 ± 13.0 * | ||
| ClF + EGCG | 37.1 ± 3.1 ***^^^## | 45.2 ± 2.7 ***^^# | ||
| ClF + Genistein | 39.9 ± 1.1 ***^^### | 44.9 ± 3.6 ***^# | ||
| B | MCF7 | MDA-MB-231 | ||
| ClF (IC50) | + | + | + | + |
| EGCG 10 µM | + | − | + | − |
| Genistein 10 µM | − | + | − | + |
| Average CI | 0.8021 ± 0.0458 *** | 1.1439 ± 0.0913 | 0.7825 ± 0.0593 ** | 1.0808 ± 0.1065 |
| Gene | Amplicon Length (bp) | Sequence of Primers (5′ → 3′) F—Forward; R—Reverse | Annealing Temperature (°C) | UCSC RefSeq Gene Accession NM (Human GRCh37/hg19 Assembly) |
|---|---|---|---|---|
| RARB | 295 | F: CTCGCTGCCTGCCTCTCTGG | 58.4 | NM_016152 |
| R: GCGTTCTCGGCATCCCAGTC | chr:3 | |||
| PTEN | 214 | F: CAGCCGTTCGGAGGATTATTC | 61.1 | NM_000314 |
| R: GGGCTTCTTCTGCAGGATGG | chr:10 |
| Gene | Amplicon Length (bp) | Sequence of Primers (5′ → 3′) F—Forward; R—Reverse | Annealing Temperature (°C) |
|---|---|---|---|
| RARB | 92 | F: TTCAAGCAAGCCTCACATGTTTCCA | 58.4 |
| R: AGGTAATTACACGCTCTGCACCTTTAG | |||
| PTEN | 330 | F: CGAACTGGTGTAATGATATGT | 50.0 |
| R: CATGAACTTGTCTTCCCGT | |||
| DNMT1 | 100 | F: ACCGCCCCTGGCCAAAGCCATTG | 60.0 |
| R: AGCAGCTTCCTCCTCCTTTATTTTAGCTGAG | |||
| CDKN1A | 103 | F: GCTCAGGGGAGCAGGCTGAAG | 60.0 |
| R: CGGCGTTTGGAGTGGTAGAAATCTGT | |||
| TP53 | 120 | F: TAACAGTTCCTGCATGGGCGGC | 64.0 |
| R: GGACAGGCACAAACACGCACC | |||
| H3F3A | 76 | F: AGGACTTTAAAACAGATCTGCGCTTCCAGAG | 65.0 |
| R: ACCAGATAGGCCTCACTTGCCTCCTGC | |||
| RPLP0 | 69 | F: ACGGATTACACCTTCCCACTTGCTGAAAAGGTC | 65.0 |
| R: AGCCACAAAGGCAGATGGATCAGCCAAG | |||
| RPS17 | 87 | F: AAGCGCGTGTGCGAGGAGATCG | 64.0 |
| R: TCGCTTCATCAGATGCGTGACATAACCTG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lubecka, K.; Kaufman-Szymczyk, A.; Cebula-Obrzut, B.; Smolewski, P.; Szemraj, J.; Fabianowska-Majewska, K. Novel Clofarabine-Based Combinations with Polyphenols Epigenetically Reactivate Retinoic Acid Receptor Beta, Inhibit Cell Growth, and Induce Apoptosis of Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3970. https://doi.org/10.3390/ijms19123970
Lubecka K, Kaufman-Szymczyk A, Cebula-Obrzut B, Smolewski P, Szemraj J, Fabianowska-Majewska K. Novel Clofarabine-Based Combinations with Polyphenols Epigenetically Reactivate Retinoic Acid Receptor Beta, Inhibit Cell Growth, and Induce Apoptosis of Breast Cancer Cells. International Journal of Molecular Sciences. 2018; 19(12):3970. https://doi.org/10.3390/ijms19123970
Chicago/Turabian StyleLubecka, Katarzyna, Agnieszka Kaufman-Szymczyk, Barbara Cebula-Obrzut, Piotr Smolewski, Janusz Szemraj, and Krystyna Fabianowska-Majewska. 2018. "Novel Clofarabine-Based Combinations with Polyphenols Epigenetically Reactivate Retinoic Acid Receptor Beta, Inhibit Cell Growth, and Induce Apoptosis of Breast Cancer Cells" International Journal of Molecular Sciences 19, no. 12: 3970. https://doi.org/10.3390/ijms19123970