Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies


1
iBB-Institute for Bioengineering and Biosciences, Biological Sciences Research Group, Av. Rovisco Pais 1, 1049-001 Lisbon, Portugal
2
Department of Bioengineering, Instituto Superior Técnico, University of Lisbon, 1049-001 Lisbon, Portugal
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(12), 3871; https://doi.org/10.3390/ijms19123871
Submission received: 4 October 2018
/
Revised: 27 November 2018
/
Accepted: 3 December 2018
/
Published: 4 December 2018
(This article belongs to the Special Issue Receptor-Targeted Cancer Therapy)
Abstract
:Cancer is a multi-process disease where different mechanisms exist in parallel to ensure cell survival and constant adaptation to the extracellular environment. To adapt rapidly, cancer cells re-arrange their plasma membranes to sustain proliferation, avoid apoptosis and resist anticancer drugs. In this review, we discuss novel approaches based on the modifications and manipulations that new classes of molecules can exert in the plasma membrane lateral organization and order of cancer cells, affecting growth factor signaling, invasiveness, and drug resistance. Furthermore, we present azurin, an anticancer protein from bacterial origin, as a new approach in the development of therapeutic strategies that target the cell membrane to improve the existing standard therapies.
1. Introduction
The role played by the plasma membrane and its biophysical status in cancer is increasingly being recognized in the field of anticancer therapies. Usually, genetic alterations are the trigger for oncogenic transformation, which in many cases leads to changes in the lipid profile and biophysical properties of cells, impacting the response to the various conventional therapies [1]. Most anticancer drugs act on membrane surface receptors or have intracellular targets (e.g., DNA) needing to interact and/or cross cellular membranes to achieve their target sites. Hence, membranes act as additional barriers upon malignant transformation promoting lipid and/or protein–drug interactions and modulating their efficacy. Limited efficacy of the drugs can have as consequence the development of the multidrug resistance (MDR) phenomenon and development of metastasis [2,3]. Multiple factors contribute to MDR such as the heterogeneity of the population which can cause intrinsic resistance in some clonal populations over others, but resistance can also be acquired. The result is often the same, i.e., the reduction in the effective concentration of drug in the cell before it reaches its cellular target. Among others, the mechanisms reported comprise overexpression of drug efflux pumps; alterations in lipid metabolism (ceramide–Cer–pathway); elimination of the drugs by cellular detoxification systems; sequestration of drugs inside lysosomes and endosomes; reduced drug uptake caused by altered surface receptors (G-protein family and/or solute carries); or altered signal transduction pathways regulated by cell surface receptors such as growth factor receptors and/or integrin receptors (for a more extended review see [2]).
Another feature adding to these is the reduced free diffusion of anticancer drugs through the plasma membrane [1]. Therefore, studying the alterations occurring at membranes during cancer progression is a growing field of research which may impact the design of new drugs and therapeutic approaches in the near future. In fact, several anticancer drugs are being evaluated based on their capacity to exert therapeutic effect by modulating the properties of tumor membranes, thus being the basis for membrane-lipid therapies [4,5,6,7].
2. Membrane Biophysics of Cancer Cells
The rapidly dividing nature of cancer cells creates a need for altered biosynthetic pathways and the synthesis of fatty acids is frequently up-regulated in these cells to form new membranes [8,9,10]. However, the biophysical constraints imposed by the nature of the lipids present in the membranes frequently produce alterations in cancer cells, although the nature of the changes differs between different tumors types, stage or sensitivity to drugs [11,12].
Lipid molecules comprise a polar head and a hydrophobic tail with a relatively long hydrocarbon chain length. The thermodynamic forces regulating the behavior of these molecules give rise to a spontaneous association in structures such as micelles or bilayered sheets which protects the hydrophobic tails surrounded by the hydrophilic heads [6,13]. The differences in the lipid polar heads groups or the hydrocarbon chain then results in different properties such as overall structure, charge, packing, and thus, the behavior of the membrane. For example, phophatidylcholine (PC) and sphingomyelins (SM) have a cylindrical shape. Due to its large polar head and amphiphilicity, PC usually forms a lipid bilayer in a water-environment. On the contrary, most phosphatidylethanolamine (PE) lipids are considered cone-shaped lipids and do not form lipid bilayers by themselves, a property that is dependent on the length and level of unsaturation of the acyl chains, which is critical for the embedding of membrane proteins. Poliphosphoinositides (PI) however, due to the higher head to tail proportion have an inverted cone-shape form, causing positive membrane curvature. Sphingolipids contain very long fatty acid chains forming solid gel phases fluidized by sterols. Therefore, changes in the lipid composition strongly affect the overall behavior of the membrane [6,14].
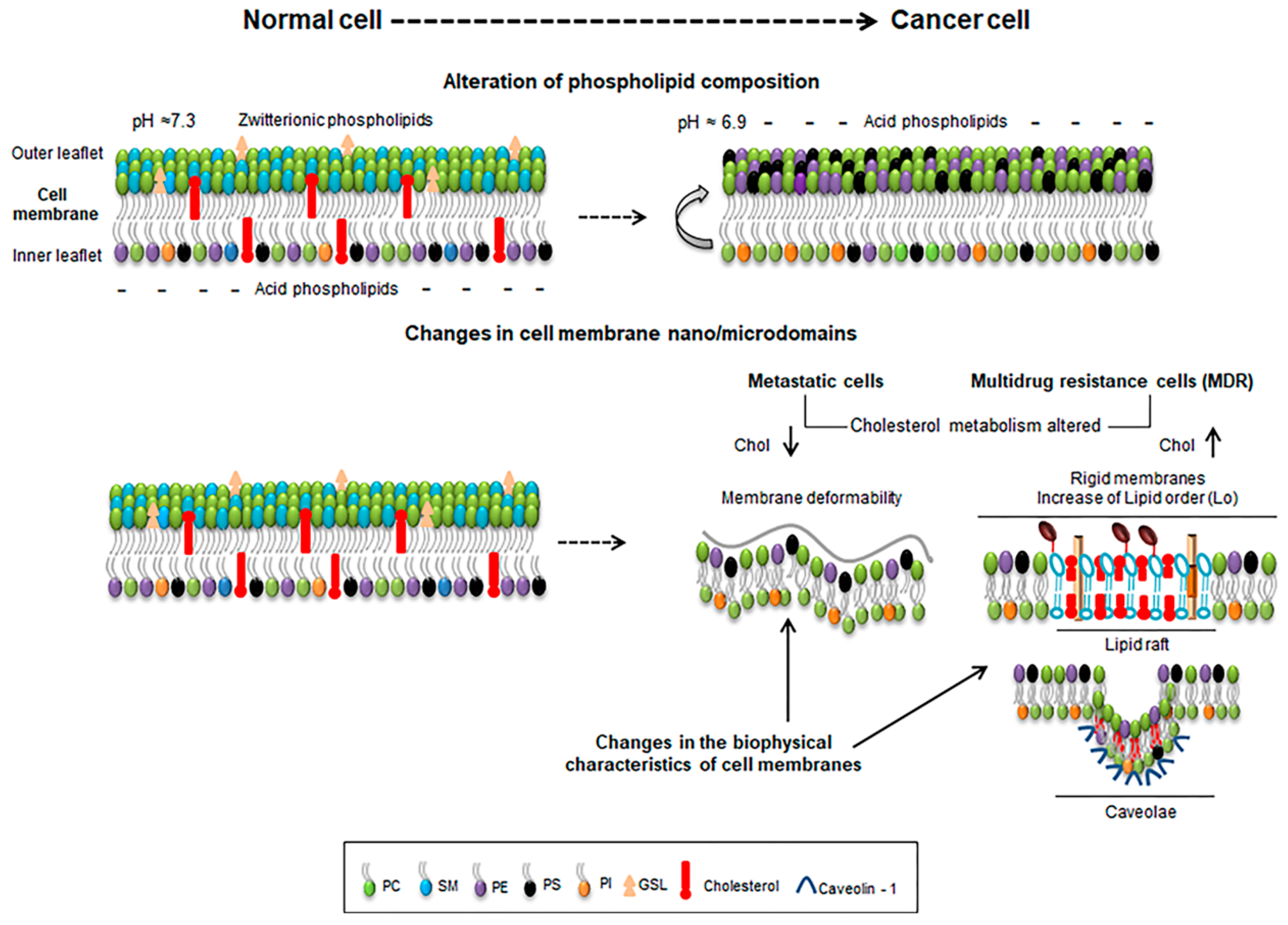
Membranes of normal cells are characterized by a lipid asymmetry between the inner and outer leaflets. Zwittterioninc lipids such as PC and SM are prevalent in the outer leaflet while phospholipids containing amine (phosphatidylethanolamine) (PE) or serine (PS) are more represented in the inner leaflet (Figure 1). This asymmetry is maintained by enzymes such as flipases, scramblases and translocases being an active and energy-dependent process [6,15]. The negative surface charge due to the prevalence of aninonic PS in the inner leaflet influences numerous signaling mechanisms such as the hydrolysis of signaling lipids such as phosphatidylinositol (PI) by phospholipase C into inositol 1,4,5-triphosphate (IP3) and Diacylglycerol (DAG) that act as secondary messengers. Also, the enrichment of PE and PS in the inner leaflet stabilizes interactions with the cytoskeleton by the amine and serine moieties, forming a fence that limits the free lipid movement within the membrane and contributes to its curvature and mechanical properties [16]. A de-regulation of this asymmetry is often encountered in pathological conditions such as cancer in which cells often expose PS in the outer membrane resulting in a negative surface charge of cancer cells, which also correlates with a more acidic pH of their external media [17,18]. In tumor endothelial cells, PE was also found exposed in the outer leaflet of the cells [19].
The different structure and physical properties of the lipids in the membrane also causes changes in its fluidity. Depending on the temperature, lipids change from gel solid-ordered phase (So) with extended hydrophobic tails, occurring a change at the transition temperature (Tm), where lipids form a liquid-disordered state (Ld), where the hydrocarbon tails are not stretched. At the physiological body temperature, most membranes are at a Ld state due to its lipid composition [20,21], but membrane fluidity is also strongly influenced by cholesterol content and the lipid unsaturation degree [22,23]. Cholesterol (Cho) is a major component of plasma membranes. When its content is between 8–15% membranes remain in the Ld state, although at different possible degrees. Increasing values of cholesterol (20–40%), however, cause a change to a more rigid liquid-ordered (Lo) membrane state [6,24]. Both membrane states co-exist in plasma membranes in which we can found areas with an enrichment in certain lipids, forming domains. This mode of organization of the membranes is essential for its role in several cellular processes such as polarization, trafficking, and endocytosis, and mechanotransduction of external cues to intracellular signaling. The main lipid domains found in plasma membranes are caveolae and lipid rafts [25]. Lipid rafts are nanodomains located on the outer leaflet of the membranes, enriched in sphingolipids (SL) and cholesterol [13,26]. Their assembly and dynamics occur very rapidly through van der Waals forces and hydrogen bonds between the OH- group of cholesterol and the SL [27,28]. These membrane domains are associated with the Lo phase of the membrane providing a platform for certain proteins [6,29] (Figure 1).
Cer-enriched lipid rafts are micro or macrodomains in size. The small polar head of Cer promotes a self-association stabilized by intermolecular hydrogen bonds, forming stable and highly ordered domains [30]. The presence of even small amounts of Cer is sufficient to displace cholesterol from lipid raft Lo nanodomains which improves the overall order of the membrane [6]. Combined with a higher lifetime than Lo nanodomains and lower lateral mobility, Cer lipid rafts establish longer associations to embedded proteins. To clarify the role of these two lipid raft domains, some authors have proposed the denomination of the two different lipid rafts, being Type 1 the cholesterol and shingolipid-enriched domains and Type 2 the Cer-enriched domains [6,31,32]. Both domains are considered to be hot spots for signaling since important proteins are associated with them through different post-transcriptional modifications [13,25,33,34,35]. GPI anchored proteins [13,36], growth factor receptors such as EGFR (epidermal growth factor receptor) [37,38] and IGF-1 (insulin-like receptor) [12] are proteins known to associate to Type 1 lipid rafts, while proteins involved in apoptosis, such as Fas (CD95), TNF-R1 or TRAIL receptor, are associated with Type 2 lipid rafts. The caveolae domains are enriched in cholesterol, sphingolipids and lipid-anchored proteins in which the polymerization of the coat protein caveolin-1 is needed to form surface invaginations with 50–100 nm of diameter [39,40].
Cholesterol metabolism is frequently altered in cancer cells resulting in either lower or higher values that may vary according to the type of cancer, stage and sensitivity [1,6,12]. Indeed, in metastatic cells lower cholesterol levels correlate well with a more deformable membrane increasing their ability to enter in blood vessels [41,42]. However, in multidrug resistant cells (MDR), higher values of cholesterol can be found, turning the membrane more rigid and, thus, less permeable for drugs [1] (Figure 1). Besides cholesterol, also PC, PE and PI are more abundant in MDR cells than normal cells. The increase in cholesterol promotes the presence of Lo lipid raft domains contributing to malignant transformations, hyper growth and invasiveness [6,12]. Also, in general a decrease in drug influx correlates to the membrane biophysical properties. Doxorubicin-resistant P338 cell line had a decreased PC/SM ratio and an increase in the membrane order [1,43]; Vinblastine-resistant leukemia T cells showed an increase of up to 60% in protein/lipid content compared to drug-sensitive cells [44]. Peetla et al. reported that lipids isolated from doxorubicin-resistant MCF-7 breast cancer cells (MCF-7/ADR) had higher concentrations of SM, PI, and cholesterol than sensitive cells and that membrane lipids are more rigid than sensitive cells, showing also that doxorubicin had strong hydrophobic interactions with resistant cell membrane lipids when compared to lower and ionic interactions that established with membrane lipids from sensitive cells [45].
Apart from the growth factors already referred, Lo lipid rafts also harbor other proteins such as integrins, CD44 and CD24 receptors that are involved in tumor progression [36,46]. In particular, in MDR cells transporters such as P-glycoprotein (P-gp) that have a role in the efflux of chemotherapeutic drugs from the cytoplasm to the extracellular space are located within lipid rafts [47,48,49]. Several studies relate not only the presence of P-gp in lipid rafts but also the direct influence of this membrane environment in its activity (for a more extended review see [1]). Using model membranes, Alves et al. [50] studied the interaction of doxorubicin with lipids to evaluate the impact of this drug in the organization of the plasma membrane. Doxorubicin was found to locate preferentially in ordered domains of the membrane, which could then contribute to its enhanced efflux by P-glycoprotein.
The metabolism of Cer is also an effective drug resistance mechanism used by cells [51,52]. In cells, Cer is an intermediate metabolite of more complex SL or is the result of Sphingomyelinase (SMase) activity (which produces Cer from SM). The activity of SMase produces then a very tight control on Cer levels and controls different processes such as differentiation, proliferation, and apoptosis. Low levels of Cer are found in MDR cells either by lowering the activity of SMase or by increasing the SM levels. Therefore, low levels of Type 2 lipid rafts are formed, preventing apoptosis [46].
As seen, the pathological conditions found in cancer can alter membrane-lipid composition, its biophysical properties and nano/microdomains dynamics with consequences at the intracellular downstream signaling events. Therefore, drugs interaction and regulation of membrane properties can be used to modulate membranes and potentiate new therapeutic strategies.
3. Therapeutic Strategies and Anticancer Drugs to Modulate Membranes
Conventional anticancer drugs are mostly designed to interact with proteins or nucleic acids. Currently, drugs that target lipids or affect membrane-lipid composition are becoming interesting alternatives to target the aforementioned changes observed in cancer cells. The modulation of the membrane biophysics or the targeting of the specific lipid alterations changing its composition and structure may alter the domain dynamics and/or the affinity of one or more proteins involved in the transduction of specific signaling [6,53].
3.1. Modulation of Invasion and Proliferation Associated Pathways by Lowering Cholesterol Content
The removal of cholesterol from the plasma membranes reduces the prevalence of Lo lipid raft domains causing the dissociation of the proteins there located. The use of chemical agents such as filipin which binds cholesterol preventing its association to SL, or chemical remove using methyl-beta-cyclodextrin (MβCD), results in hampering the propagation of their signaling events. Pathways associated with EGFR or estrogen receptors often overexpressed are severely decreased upon cholesterol depletion [6,12,38].
The main biologically active component of green tea, epigallocatechin gallate (EGCG), inhibits the EGFR activation and its downstream signaling pathways in several human cancer cell lines [12,54,55]. In HT-29 colon cancer cell lines deprived of epidermal growth factor (EGF), it was seen that EGFR is mainly located at in Ld domains but upon activation with EGF or TGFα, EGFR translocates to Lo domains, to produce its downstream signaling. However, EGCG disabled the ordered domains thus preventing the proliferative signaling [56]. The same type of preventive effects was seen in head and neck squamous cell carcinomas (HNSCC) [57]. Additionally, in HNSCC EGCG demonstrated a synergistic effect with erlotinib, an inhibitor of EGFR [58]. Other examples of synergistic effect of EGCG are found in trastuzumab-resistant BT474 breast cancer cell line [59] and tamoxifen-resistant breast carcinoma cells. In both cases, EGCG caused dose-dependent apoptosis facilitating the action of each drug.
Malignant cells depend strongly on the ability to synthesize new fatty acids (FA) to provide the required elevated levels of building blocks for the newly synthesized membranes [60]. For that reason, the levels of the FASN (Fatty acid synthase), an enzyme required for this increased lipogenesis is often overexpressed in malignant cells. FASN catalyzes the condensation of one acetyl-coenzyme A (acetyl-coA) with seven molecules of malonyl-coA originating long-chain FA, in particular palmitate. This 16-carbon saturated FA can be further elongated or desaturated forming other FA by other enzymes such as stearoyl-CoA desaturases (SCDs) [10]. Increased levels of SCDs have also been associated with tumorigenesis and poor prognosis in several cancers. Moreover, it has been shown FASN and caveolin-1 can interact both physically and functionally in prostate cancer cells, suggesting an involvement with membrane ordered domains [61]. However, despite its importance in cancer progression several attempts to pharmacologically target FASN have failed until now, but a new FASN antagonist (TVB-2640) is now under evaluation in Phase I and II clinical trials [62].
Another lipogenic pathway is the mevalonate (MVA) pathway, which begins with acetyl-CoA and produces isoprenoids such as cholesterol [60]. Statins lower cholesterol levels by blocking the conversion of 3-hydroxy-methylglutaryl (HMG) coA to mevalonic acid through the inhibition of the HMG coA reductase enzyme to which statins are structurally similar [63]. Cancer cells often present an upregulation of the mevalonate pathway [63,64], therefore acting on the early stages of this pathway will decrease the lipid raft content and membrane rigidity [6]. Statins not only block the synthesis of cholesterol but also of other metabolites such as the isoprenoids geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate that activate farnesylated proteins of the Ras family and geranylgeranylated proteins such as RhoA and Rac1 [12,63]. Lovastatin was able to induce apoptosis in ovarian cancer through a mevalonate-dependent mechanism while also displaying synergistic effects with doxorubicin [65]. Another statin, atorvastatin, in combination with carboplatin (a platinum-based anticancer drug) enhanced the survival of lung cancer cells A549-bearing nude mice compared to carboplatin only through the inhibition of AKT [66].
In addition to alterations in the synthesis, the β-oxidation of FA also provides energy to the cells, as ATP, NADPH for the reduction of oxidative stress and acetyl-coA for protein acetylation and both mechanisms play roles in cancer cell maintenance since recently cancer has been shown dependent on both FA synthesis and oxidation [60].
Emodin (3-methyl-1,6,8-trihydroxyanthraquinone) is an active component found in the roots and rhizomes of Rheum palmatum L. [12]. This compound was found to have similar effects on cholesterol levels as MβCD, lowering its levels in isolated detergent-resistant membranes (DRM) fractions, as well as of the main SL [67]. Lipid raft-associated events such as translocation of integrin subunit β1 and the formation of focal adhesions were prevented by both emodin and MβCD, an effect that was lost after cholesterol replenishment [67]. In MDA-MB-231 breast cancer cells, emodin also inhibited the co-localization of integrin β1 with lipid rafts [67]; in breast and liver carcinomas it was found to inhibit cell migration by suppressing PI3K-Cdc42/Rac1 signaling [68]; and in lung and prostate carcinoma emodin prevented the invasion and migration by down-regulating the CXCR4 chemokine receptor [69].
3.2. Stabilization of Pro-Apoptotic Membrane Domains
As mentioned before, Type 2 lipid rafts are mainly pro-apoptic Cer-enriched membrane domains. Therefore, in this approach, the strategy is to enhance the Cer rafts, displacing cholesterol from Type 1 lipid rafts therefore inducing apoptosis in cancer cells [6]. Synthetic alkylphospholipids (ALPs) are a class of new anticancer drugs chemically divided in two main classes: alkylysophospholipids (ALP) and alkylphosphocolines (APC). These agents target cell membranes by interfering with lipid homeostasis due to their similarity with endogenous phospholipids. ALPs act by modulating membrane permeability and fluidity altering lipid-linking signaling, phospholipid metabolism, proliferation and, ultimately, leading to apoptosis [5,11]. Edelfosine was the first agent of this class reported to induce apoptosis in cancer cells. In multiple myeloma cells, edelfosine demonstrated great ability to induce modifications in membrane-lipid raft structure clustering Fas/CD95 death receptors and lipid rafts [26,70,71,72]; however, its clinical application has been limited due to gastrointestinal toxicity, hemolytic potential, and metabolic instability [5]. Edelfosine analogue miltefosine lacking the glycerol motif was the first ALP used in clinic for the local treatment of cutaneous metastasis of breast cancer, but as the lead compound edelfosine its hemolytic activity prevented its extension to other tumor types [5]. A modification in miltefosine replacing the choline moiety by a heterocyclic pipepridine group resulted in perifosine which demonstrated increased half-life and stability preventing its rapid degradation [73,74]. It also induces apoptosis in carcinoma cell lines [75], recruiting Fas/CD95 to lipid rafts [76]. Despite phase II clinical trials using perifosine as single-agent still demonstrated high levels of gastrointestinal toxicity, it has had clinical success as an AKT inhibitor in combination with other anticancer agents [5]. Indeed, one of the most studied pathway targeted by ALPs is the PI3K/Akt/mTOR pathway, particularly Aky which is frequently overexpressed in many cancer types (for a detailed review see [5]). The ethyl phosphate 10-(Octyloxy) decyl-2-(trimethylammonium) (ODPC) is another ALP that interacts with the cell membrane targeting high cholesterol domains inducing apoptosis in leukemia cells but not having cytotoxic effects in normal hematopoietic cells [77]. Furthermore, in lipid model systems containing phase coexistence of Lo-Ld domains, the presence of ODPC disrupted the domains [78].
The natural polyphenol resveratrol (3,5,4’-trihydroxystilbene) is also capable of inducing apoptosis in a colon cancer cell line by clustering the Fas/CD95 receptors to membrane rafts without ligand activation [79]. It accumulates in lipid rafts before its endocytosis [80] and even for the HT-29 resveratrol-resistant cell line, its pre-treatment enhances greatly apoptosis induced by the natural ligands TNFα, TRAIL or anti-Fas/CD95 antibodies [79].
The equilibrium between Cer and SM is then fundamental for the cells to regulate proliferation and apoptosis. The levels of two enzymes that, among others, regulate the levels of these two lipids, Glucosylceramide synthase (GCS) that converts Cer into SM and SMase that converts SM to Cer phosphate, are frequently de-regulated in cancer cells. Drug resistant cells often overexpress GCS [6,81] or down-regulate SMase [1,52,82,83] to favor proliferation over apoptosis. Several strategies have been used to increase the Cer levels, for instance the activation of SMase, which has demonstrated to be achieved by TNFα, etoposide or cytarabine (cytosine arabinoside—Ara-C) [83]; the epigenetic drug decitabine that demethylates SMase gene [1,6]; siRNA transfection; or analogues of glucosylceramide such as PDMP [52,84].
Table 1 shows some of the aforementioned agents and the different strategies used to target either the membrane directly or different steps in lipid metabolism pathways that ultimately result in changes in the membrane-lipid composition and biophysical status.
3.3. Lipid Replacement
The more rigid and less permeable membranes such it occurs in MDR cells creates an extra difficulty to the diffusion of chemotherapeutics. Since this barrier is essentially caused by increased cholesterol content and non-saturated/saturated lipid ratio, another strategy employed is the replacement of the lipids present in the membrane. Extensive experimental and clinical evidence suggests that the consumption of mono (oleic acid) and polyunsaturated fatty acids (PUFA), including docosahexaenoic acid (DHA), eicosapetaenoic acid (EPA) or linoleic acid contribute to prevent colon tumorigenesis. These fatty acids incorporate in the membrane phospholipids changing their composition, and, thus, its properties such as fluidity and phase behavior. Minerval, the synthetic fatty acid 2-hydroxyoleic acid (2OHOA) is a first-in-class in phase I/II clinical trials being tested for the treatment of glioma [85] (for an extended review see [4]). Also, propofol-DHA is being developed to treat breast cancer cells [7,84].
Apart of these changes in membrane properties, another bottleneck often encountered is the entrapment of chemotherapeutic drugs or protein-based therapeutics within endosomes upon uptake by cells. This may ultimately lead to drugs being released from cells after endosomes recycle back to the membrane or their degradation in lysosomes. Another class of molecules that were found to enhance the endosomal escape are saponins. Glycosylated triterpenoid saponins comprise a pentacyclic C30 perpene backbone structure with one or more covalently bound sugar chains [86]. Saponins have been used to enhance the cytotoxicity of several protein ligands in different cancer cell models (for a review see [87]).
4. The Anticancer Effects of the Bacterial Protein Azurin: A Cell Membrane Targeted Therapy
In the previous section, several molecules that can modulate the plasma membrane-lipid and/or protein contents and biophysical profile in cancer cells were presented. In this section, we present findings regarding a similar behavior of a bacterial protein acting at the membrane of cancer cells. Azurin is a small (14 kDa) membrane-associated protein from the Pseudomonas aeruginosa bacterium from the cupredoxin super family. In recent years, azurin has been intensively studied as an anticancer protein, down-regulating signaling pathways downstream of membrane receptors and functional processes such as adhesion and invasiveness [88,89,90,91,92,93,94].
4.1. Cancer Cells’ Membrane Modulation by Azurin
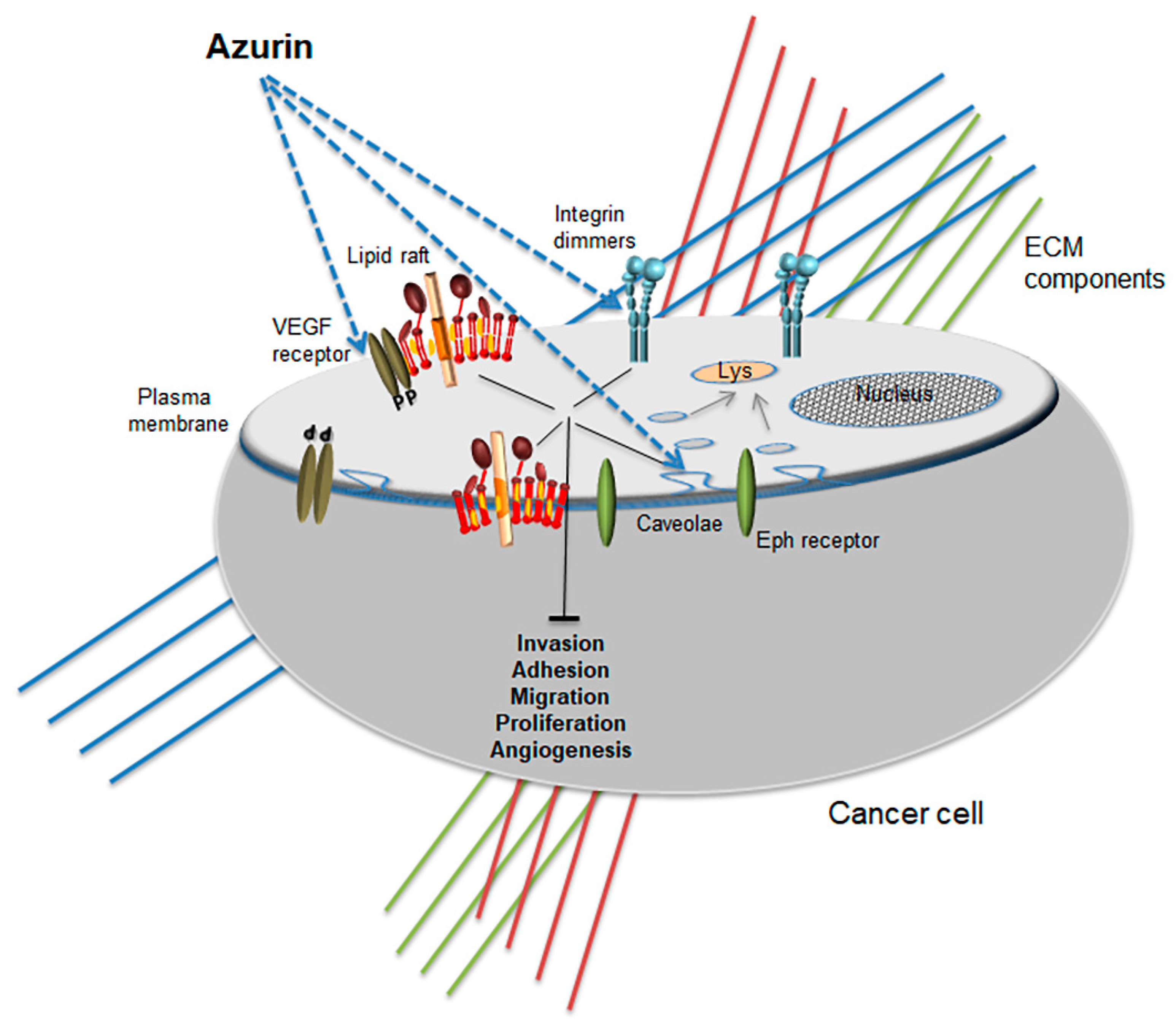
Recently, our group performed a microarray analysis in invasive breast cancer cells overexpressing P-cadherin, a cell-cell adhesion molecule that is aberrantly overexpressed in about 30% of breast cancers [95]. The results evidenced a very significant number of genes with altered expression coding for proteins associated with membrane related processes [93]. Endocytosis and the formation of early endosomes, vesicle mediated processes and membrane remodeling were some of the gene ontology and enriched pathways assigned by the Kegg database that were up-regulated by azurin. On the contrary, focal adhesion, cell motility, biological adhesion and cell surface receptors associated with signal transduction composed most of the genes down-regulated, assigned by the Kegg database [93]. The overexpression of P-cadherin leads to increased invasiveness and it has been associated with the aberrant expression of the α6β4 integrin dimmer which is also related to the hyperactivity of FAK, Src and AKT pro-tumorigenic and proliferative signaling [96]. Treating these cells with azurin led to a down-regulation of this cadherin protein levels, maintaining or even increasing E-cadherin levels, a known tumor suppressor cadherin protein [92]. Furthermore, at the functional level, invasion of the cells through Matrigel is severely impaired, in addition to the decrease of FAK and Src phosphorylation [92]. Indeed, treatments with azurin seem to have led to a global decrease of mechano-sensing associated pathways that drive the invasiveness in these models. Azurin also promoted a deficient adhesion of breast cancer cells to ECM proteins such as collagen, laminin and fibronectin, down-regulating the integrin subunits α6, β4 and β1 and decreases severely the mammosphere-forming efficiency of these cells [93].
In non-small cell lung cancer (NCSLC), azurin also decreases the expression of integrin subunit β1 as well as the adhesion to ECM proteins and the signaling pathways associated with increased proliferation such as PI3K/AKT and Src [88]. In this model, overexpression of this integrin is known to control the activity of EGFR. However, pre-treating the cells with azurin prevented the activation of EGFR pathway by its natural ligand EGF. Furthermore, the combination of azurin with EGFR-targeted therapies such as the small chemical inhibitors gefitinib or erlotinib produced a synergistic effect in the inhibition of proliferation stronger than with any of the chemicals alone [88].
At the membrane protein level, azurin also targets the receptor tyrosine kinase Eph receptor B2, competing with its natural ligand ephrin B2 for the binding to the receptor, therefore blocking the proliferative signaling induced by the natural ligand [92]. Later, engineered versions of peptides derived from azurin were conjugated to nicotinamide, increasing the sensitivity to radiotherapy up to 13-fold compared to the nicotinamide alone [93]. Using Surface Plasmon Resonance (SPR) to screen a library of peptides derived from the C-terminal region of azurin that would bind to ephrin receptors EphA2, EphB2 and EphB4, one particular analogue was found to sensitize Lewis lung carcinoma (LCC), both in vitro and in vivo, on solid tumor engraftment models [93]. These experiments increased the potential of this protein or derived peptides to target membrane-associated proteins relevant in different models of cancer.
The natural structure of azurin as an invariant β-sheet sandwich structure (Greek key β-barrel) formed by eight parallel and antiparallel strands and an extended α-helix region situated outside of the barrel, confers on this protein the nature of a scaffold protein. This also seems to give azurin the ability to interfere on multiple cancer–associated signaling events, exhibiting some promiscuity in its targets, unlike rationally designed drugs that target a specific step in a single pathway.
On top of the signaling and functional changes caused by azurin in the different cancer cell models, the cellular biophysical and nanomechanic properties of the plasma membranes were also evaluated. In lung cancer cells treated with azurin the membrane profile was assessed by Atomic Force Microscopy (AFM), in an attempt to explain the interference with cell attachment and response to growth factors [94]. The Young’s modulus (E) of azurin-treated lung cancer cells decreased upon treatment with azurin in about 30% [94]. This result seems to be in accordance with the increased sensitivity towards the EGFR kinase inhibitors, and as described in the previous sections, alterations in membrane stiffness in cancer cells often contribute to the reduced permeability to anticancer drugs. The global membrane order in different cancer cell models was also evaluated recently in three different cancer cell lines (MCF-7, A549 and HT-29) with the environment-sensitive probe Laurdan [97]. In this case, treating cells with azurin decreased the average GP value of plasma membranes, indicating a decrease in the Lo domains suggesting a more fluid membrane state.
Furthermore, a phenylalanine residue of azurin located in the surface hydrophobic patch of the protein is also involved in the entry into cancer cells, mediating interactions with caveolin-1 and GM-1, a ganglioside enriched in lipid rafts pointing to the importance of hydrophobic interactions between azurin and lipid raft resident lipids and proteins [97]. This also correlated well with increased sensitivity to paclitaxel and doxorubicin in the presence of azurin, again suggesting that a part of the anticancer effect of azurin may occur by changing the membrane properties in a way that favors the permeability to anticancer drugs and/or disturbs the pro-tumorigenic signaling of raft resident proteins.
4.2. Induction of Apoptosis
Further experiments also demonstrate that upon entry in cancer cells, azurin triggers apoptosis. Some evidence points to a possible interaction with p53, which would then support apoptosis [88,98,99,100,101]. A peptide derived from azurin, p28, is an amphipathic peptide that adopts an alpha-helical conformation with an overall negative net charge, is now the lead peptide in several pre-clinical and clinical studies towards different cancer types [102,103,104,105,106], based on its pro-apoptotic activity. The entry of this peptide seems to be dependent on the cholesterol levels present in the membrane since its depletion with MβCD strongly impaired its intracellular accumulation [107]. Several chemical inhibitors of different steps in endocytosis showed that p28 entered in cancer cells via caveosome-directed but also caveosome-independent pathways [107], and also that membrane bound glycosaminoglycans are apparently not involved in the uptake of azurin or p28 in the cells but N-glycosated proteins may play a role in the endocytosis of p28 [107]. The p28 peptide has also effects at the membrane since it blocks angiogenesis in endothelial and cancer cells through the modulation of the VEGFR-2 membrane receptor tyrosine kinase [108]. In endothelial HUVEC cells, p28 enters the cells co-localized with caveolin-1, blocking the VEGF and bFGF-induced migration, capillary tube formation and neoangiogenesis in several xenograft models. The down-regulation of VEGFR-2 induces the attenuation of FAK and AKT signaling pathways, namely through the decrease in their phosphorylation, which in turn, affects their signaling activity in the cellular remodeling associated with processes such as adhesion to the extracellular matrix and migration [108].
Other examples of protein-based anti-tumor therapies exist in the literature based on human proteins or peptides that act as cell-penetrating peptides (CPP) that also exert a pro-apoptotic effect in cancer cells. Lactoferrin (Lf) and its derived peptide lactoferricin (Lfcin) are extensively studied nutraceutical proteins with demonstrated activity against a broad spectrum um tumor types both as isolated agents as well as in combination with other chemotherapeutics such as tamoxifen [109]. In basal-like breast cancer, their combination resulted in enhanced apoptosis in mouse models of this breast cancer [110]. Another peptide derived from bovine lactoferricin with six amino acids, bLFcin6, demonstrated anti-tumor bioactivity and ability to deliver cargos, such as siRNAs, to the interior of cancer cells through a lipid raft-dependent micropinocytosis mechanism [111].
5. Conclusions and Future Perspectives
In conclusion, the role of the plasma membrane in cancer development and response to therapies is long known; however, only recently has it come to be consistently recognized as a target for the development of new therapies. Most cancer cells alter their lipid biosynthetic pathways in a way that not only favors proliferation and/or resistance to therapies but also induce significant changes in the biophysical properties of the plasma membrane. These alterations impact the interactions established with the vast majority of chemotherapeutics, small chemical compounds, and even biological therapies, protecting most of the times the cells of their activity. Different classes of drugs are therefore arising with the purpose of modulating the biochemical and biophysical features of cancer cells acting directly at the membrane and interacting with lipids and membrane-resident proteins or at the enzymes responsible for sensing and synthesis of membrane lipids. As proteins, azurin and its derived lead peptide p28, in this context, appear to be distinctive in their anticancer activity since through interactions with different plasma membrane raft components in cancer cells they can alter the biophysical profile and attenuate raft-related signaling pathways (Figure 2). The impact of azurin at cancer cells increases membrane fluidity and promotes endocytosis, thus providing opportunities to design new therapeutic strategies and drug-delivery systems based on their activity that may enhance the uptake and effectiveness of other drugs.
Their activity has been demonstrated in different models but it has not been demonstrated yet with more physiological models such as multidrug resistant cells or patient-derived cells, which would undoubtedly strengthen their role as new therapeutics or adjuvants to existent therapies in the most difficult-to-treat and resilient tumors that still represent a huge burden to human health.
Author Contributions
A.M.F. and N.B. conceived and designed the review. N.B. wrote the manuscript in consultation with A.M.F. Both authors read and approved the final manuscript.
Funding
The work presented was supported by scientific project (LISBOA-01-0145-FEDER-030034) financed by the Portuguese Science and Technology Foundation (FCT). Funding received by iBB-Institute for Bioengineering and Biosciences from FCT (UID/BIO/04565/2013) and from Programa Operacional Regional de Lisboa 2020 (Project N. 007317) is acknowledged. FCT also provides a post-doctoral research grant for Nuno Bernardes (SFRH/BPD/98162/2013).
Conflicts of interest
The authors declare no conflict of interest.
References
- Peetla, C.; Vijayaraghavalu, S.; Labhasetwar, V. Biophysics of cell membrane lipids in cancer drug resistance: Implications for drug transport and drug delivery with nanoparticles. Adv. Drug Deliv. Rev. 2013, 65, 1686–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V. Membrane-lipid therapy: A historical perspective of membrane-targeted therapies—From lipid bilayer structure to the pathophysiological regulation of cells. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Marco, P.; Marco, C.; Gálvez, X.; Jiménez-López, J.M.; Carrasco, M.P. Alkylphospholipids: An update on molecular mechanisms and clinical relevance. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; ten Hagen, T.L.M. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef]
- Escribá, P.V.; Busquets, X.; Inokuchi, J.; Balogh, G.; Török, Z.; Horváth, I.; Harwood, J.L.; Vígh, L. Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog. Lipid Res. 2015, 59, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [Green Version]
- Azordegan, N.; Fraser, V.; Le, K.; Hillyer, L.M.; Ma, D.W.L.; Fischer, G.; Moghadasian, M.H. Carcinogenesis alters fatty acid profile in breast tissue. Mol. Cell. Biochem. 2013, 374, 223–232. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S. Aberrant lipid metabolism in cancer cells—The role of oncolipid-activated signaling. FEBS J. 2017, 285, 432–443. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug–membrane interaction studies. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz-Jankowska, A.; Augoff, K.; Biernatowska, A.; Podkalicka, J.; Sikorski, A.F. Membrane rafts as a novel target in cancer therapy. Biochim. Biophys. Acta 2014, 1845, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Mouritsen, O.G.; Anderson, R.G.W. Lipid rafts: At a crossroad between cell biology and physics. Nat. Cell Biol. 2007, 9, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; de Kroon, A.I.P.M. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L. The Fluid-Mosaic Model of Membrane Structure: Still relevant to understanding the structure, function and dynamics of biological membranes after more than 40 years. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Stafford, J.H.; Thorpe, P.E. Increased Exposure of Phosphatidylethanolamine on the Surface of Tumor Blood Vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar]
- Zwaal, R.F.A.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Stafford, J.H.; Thorpe, P.E. Increased Exposure of Phosphatidylethanolamine on the Surface of Tumor Vascular Endothelium. Neoplasia 2011, 13, 299-IN2. [Google Scholar] [CrossRef]
- Goñi, F.M. The basic structure and dynamics of cell membranes: An update of the Singer-Nicolson model. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1467–1476. [Google Scholar] [CrossRef]
- London, E. How principles of domain formation in model membranes may explain ambiguities concerning lipid raft formation in cells. Biochim. Biophys. Acta-Mol. Cell Res. 2005, 1746, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Subczynski, W.K.; Wisniewska, A. Physical properties of lipid bilayer membranes: Relevance to membrane biological functions. Acta Biochim. Pol. 2000, 47, 613–625. [Google Scholar] [PubMed]
- Hao, M.; Mukherjee, S.; Sun, Y.; Maxfield, F.R. Effects of Cholesterol Depletion and Increased Lipid Unsaturation on the Properties of Endocytic Membranes. J. Biol. Chem. 2004, 279, 14171–14178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preetha, A.; Banerjee, R.; Huilgol, N. Tensiometric profiles and their modulation by cholesterol: Implications in cervical cancer. Cancer Invest. 2007, 25, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Nabi, I.R. Lipid Rafts, Caveolae, and Their Endocytosis, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2010; Volume 282, ISBN 9780123812568. [Google Scholar]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, P.J.; Wolf, C. The liquid-ordered phase in membranes. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim. Biophys. Acta 2008, 1785, 182–206. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.N.; Goñi, F.M.; Alonso, A. Compartmentalization of Ceramide Signaling: Physical Foundations and Biological Effects. J. Cell. Physiol. 2000, 184, 285–300. [Google Scholar] [CrossRef]
- Quinn, P.J. Lipid-lipid interactions in bilayer membranes: Married couples and casual liaisons. Prog. Lipid Res. 2012, 51, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Tekpli, X.; Holme, J.A.; Sergent, O.; Lagadic-Gossmann, D. Role for membrane remodeling in cell death: Implication for health and disease. Toxicology 2013, 304, 141–157. [Google Scholar] [CrossRef] [PubMed]
- de Laurentiis, A.; Donovan, L.; Arcaro, A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem. J. 2007, 1, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G. Cellular lipidomics. EMBO J. 2005, 24, 3159–3165. [Google Scholar] [CrossRef] [Green Version]
- Storch, C.H.; Ehehalt, R.; Haefeli, W.E.; Weiss, J. Localization of the Human Breast Cancer Resistance Protein (BCRP/ABCG2) in Lipid Rafts/Caveolae and Modulation of Its Activity by Cholesterol in Vitro. Pharmacology 2007. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Lin, J.; Lu, M.L.; Cells, C.; Solomon, K.R.; Freeman, M.R. Cholesterol-rich Lipid Rafts Mediate Akt-regulated Survival in Prostate Cancer Cells. Cancer Res. 2002, 62, 2227–2231. [Google Scholar] [PubMed]
- Chen, Q.; Pan, Z.; Zhao, M.; Wang, Q.; Qiao, C.; Miao, L.; Ding, X. High cholesterol in lipid rafts reduces the sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.G. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225. [Google Scholar] [CrossRef]
- Parton, R.G.; del Pozo, M.A. Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 2013, 14, 98–112. [Google Scholar] [CrossRef]
- Zeisig, R.; Koklič, T.; Wiesner, B.; Fichtner, I.; Sentjurč, M. Increase in fluidity in the membrane of MT3 breast cancer cells correlates with enhanced cell adhesion in vitro and increased lung metastasis in NOD/SCID mice. Arch. Biochem. Biophys. 2007, 459, 98–106. [Google Scholar] [CrossRef]
- Sok, M.; Sentjurc, M.; Schara, M.; Stare, J.; Rott, T. Cell membrane fluidity and prognosis of lung cancer. Ann. Thorac. Surg. 2002, 73, 1567–1571. [Google Scholar] [CrossRef]
- Ramu, A.; Glaubiger, D.; Magrath, I.T.; Cells, P. Plasma Membrane Lipid Structural Order in Doxorubicin-sensitive and -resistant P388 Cells Plasma Membrane Lipid Structural Order in Doxorubicin-sensitive. Cancer Res. 1983, 43, 5533–5537. [Google Scholar] [PubMed]
- May, G.L.; Wright, L.C.; Dyne, M.; Mackinnon, W.B.; Fox, R.M.; Mountford, C.E. Plasma membrane lipid composition of vinblastine sensitive and resistant human leukaemic lymphoblasts. Int. J. Cancer 1988, 42, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Peetla, C.; Bhave, R.; Vijayaraghavalu, S.; Stine, A.; Kooijman, E.; Labhasetwar, V. Biophysical Characterization of and Doxorubicin Interactions with Membrane Lipids. Mol. Pharm. 2010, 7, 1264–1276. [Google Scholar] [CrossRef]
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Jodoin, J.; Gingras, D.; Béliveau, R. P-glycoprotein is localized in caveolae in resistant cells and in brain capillaries. FEBS Lett. 2000, 466, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, N.; Schumann-Gillett, A.; Mark, A.E.; O’Mara, M.L. Understanding the accumulation of P-glycoprotein substrates within cells: The effect of cholesterol on membrane partitioning. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 776–782. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 1–19. [Google Scholar] [CrossRef]
- Alves, A.C.; Magarkar, A.; Horta, M.; Lima, J.L.F.C.; Bunker, A.; Nunes, C.; Reis, S. Influence of doxorubicin on model cell membrane properties: Insights from in vitro and in silico studies. Sci. Rep. 2017, 7, 6343. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Kartal Yandim, M.; Apohan, E.; Baran, Y. Therapeutic potential of targeting ceramide/glucosylceramide pathway in cancer. Cancer Chemother. Pharmacol. 2013, 71, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Escribá, P.V. Membrane-lipid therapy: A new approach in molecular medicine. Trends Mol. Med. 2006, 12, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Suzui, M.; Weinstein, I.B. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clin. Cancer Res. 2001, 7, 4220–4229. [Google Scholar] [PubMed]
- Shimizu, M.; Deguchi, A.; Lim, J.T.E.; Moriwaki, H.; Kopelovich, L.; Weinstein, I.B. (−)-Epigallocatechin gallate and polyphenon E inhibit growth and activation of the epidermal growth factor receptor and human epidermal growth factor receptor-2 signaling pathways in human colon cancer cells. Clin. cancer Res. 2005, 11, 2735–2746. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Nagao, T.; Ingolfsson, H.I.; Maxfield, F.R.; Andersen, O.S.; Kopelovich, L.; Weinstein, I.B. The inhibitory effect of (−)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in HT29 colon cancer cells. Cancer Res. 2007, 67, 6493–6501. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Wakasaki, T.; Toh, S.; Shimizu, M.; Adachi, S. Chemoprevention of head and neck cancer by green tea extract: EGCG-the role of EGFR signaling and “lipid raft”. J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Tighiouart, M.; Lee, J.E.; Shin, H.J.; Khuri, R.; Yang, C.S.; Chen, Z.G.; Shin, D.M. Synergistic inhibition of head and neck tumor growth by green tea (−)-epigallocatechin-3-gallate and EGFR tyrosine kinase inhibitor. Int. J. Cancer 2008, 123, 1005–1014. [Google Scholar] [CrossRef]
- Eddy, S.F.; Kane, S.E.; Sonenshein, G.E. Trastuzumab-resistant HER2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007, 67, 9018–9023. [Google Scholar] [CrossRef]
- Grunt, T.W. Interacting Cancer Machineries: Cell Signaling, Lipid Metabolism, and Epigenetics. Trends Endocrinol. MeTable 2018, 29, 86–98. [Google Scholar] [CrossRef]
- Di Vizio, D.; Adam, R.M.; Kim, J.; Kim, R.; Sotgia, F.; Williams, T.; Demichelis, F.; Solomon, K.R.; Loda, M.; Rubin, M.A.; et al. Caveolin-1 interacts with a lipid raft-associated population of fatty acid synthase. Cell Cycle 2008, 7, 2257–2267. [Google Scholar] [CrossRef] [Green Version]
- Arkenau, H.T.; Voskoboynik, M.; Infante, J.; Brenner, A.; Patel, M.; Borazanci, E.; Falchook, G.; Molife, L.R.; Pant, S.; Dean, E.; et al. Evidence of activity of a new mechanism of action (MoA): A first-in-human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640, as monotherapy or in combination. Eur. J. Cancer 2015, 51, S724. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lan, T.; Hou, J.; Zhang, J.; An, Y.; Tie, L.; Pan, Y.; Liu, J.; Li, X. Atorvastatin sensitizes human non-small cell lung carcinomas to carboplatin via suppression of AKT activation and upregulation of TIMP-1. Int. J. Biochem. Cell Biol. 2012, 44, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Shen, H.-M.; Shui, G.; Wenk, M.R.; Ong, C.-N. Emodin Inhibits Tumor Cell Adhesion through Disruption of the Membrane Lipid Raft-Associated Integrin Signaling Pathway. Cancer Res. 2006, 66, 5807–5815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Shen, H.M.; Ong, C.N. Emodin inhibits tumor cell migration through suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1 pathway. Cell. Mol. Life Sci. 2005, 62, 1167–1175. [Google Scholar] [CrossRef]
- Ok, S.; Kim, S.-M.; Kim, C.; Nam, D.; Shim, B.S.; Kim, S.-H.; Ahn, K.S.; Choi, S.-H.; Ahn, K.S. Emodin inhibits invasion and migration of prostate and lung cancer cells by downregulating the expression of chemokine receptor CXCR4. Immunopharmacol. Immunotoxicol. 2012, 34, 768–778. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid rafts and Fas/CD95 signaling in cancer chemotherapy. Recent Pat. Anticancer Drug Discov. 2011, 6, 274–283. [Google Scholar] [CrossRef]
- Estella-Hermoso de Mendoza, A.; Préat, V.; Mollinedo, F.; Blanco-Prieto, M.J. In vitro and in vivo efficacy of edelfosine-loaded lipid nanoparticles against glioma. J. Control. Release 2011, 156, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; Campanero, M.A.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crul, M.; Rosing, H.; de Klerk, G.J.; Dubbelman, R.; Traiser, M.; Reichert, S.; Knebel, N.G.; Schellens, J.H.M.; Beijnen, J.H.; ten Bokkel Huinink, W.W. Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur. J. Cancer 2002, 38, 1615–1621. [Google Scholar] [CrossRef]
- Vink, S.R.; Schellens, J.H.M.; Van Blitterswijk, W.J.; Verheij, M. Tumor and normal tissue pharmacokinetics of perifosine, an oral anti-cancer alkylphospholipid. Invest. New Drugs 2005, 23, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Fei, H.; Chen, G.; Wang, J.; Wang, F. Perifosine induces cell cycle arrest and apoptosis in human hepatocellular carcinoma cell lines by blockade of Akt phosphorylation. Cytotechnology 2010, 62, 449–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2009, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomé, C.H.; dos Santos, G.A.; Ferreira, G.A.; Scheucher, P.S.; Izumi, C.; Leopoldino, A.M.; Simão, A.M.; Ciancaglini, P.; de Oliveira, K.T.; Chin, A.; et al. Linker for Activation of T-cell Family Member2 (LAT2) a Lipid Raft Adaptor Protein for AKT Signaling, Is an Early Mediator of Alkylphospholipid Anti-leukemic Activity. Mol. Cell. Proteomics 2012, 11, 1898–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomide, A.B.; Thomé, C.H.; Dos Santos, G.A.; Ferreira, G.A.; Faça, V.M.; Rego, E.M.; Greene, L.J.; Stabeli, R.G.; Ciancaglini, P.; Itri, R. Disrupting membrane raft domains by alkylphospholipids. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 1384–1389. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. J. Cell. Biochem. 2004, 91, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Aires, V.; Colin, D.J.; Limagne, E.; Scagliarini, A.; Cotte, A.K.; Ghiringhelli, F. Importance of lipid microdomains, rafts, in absorption, delivery, and biological effects of resveratrol. Ann. N. Y. Acad. Sci. 2013, 1290, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Itoh, M.; Taguchi, Y.; Yamaoka, S.; Umehara, H.; Ichikawa, S.I.; Hirabayashi, Y.; Holleran, W.M.; Okazaki, T. Ceramide reduction and transcriptional up-regulation of glucosylceramide synthase through doxorubicin-activated Sp1 in drug-resistant HL-60/ADR cells. Cancer Res. 2004, 64, 6271–6279. [Google Scholar] [CrossRef] [PubMed]
- Perrotta, C.; Cervia, D.; Di Renzo, I.; Moscheni, C.; Bassi, M.T.; Campana, L.; Martelli, C.; Catalani, E.; Giovarelli, M.; Zecchini, S.; et al. Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with syntaxin 4 and acid sphingomyelinase inhibition. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Kok, J.W.; Sietsma, H. Sphingolipid metabolism enzymes as targets for anticancer therapy. Curr. Drug Targets 2004, 5, 375–382. [Google Scholar] [CrossRef]
- Lladó, V.; López, D.J.; Ibarguren, M.; Alonso, M.; Soriano, J.B.; Escribá, P.V.; Busquets, X. Regulation of the cancer cell membrane lipid composition by NaCHOleate: Effects on cell signaling and therapeutical relevance in glioma. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 1619–1627. [Google Scholar] [CrossRef] [PubMed]
- Sparg, S.G.; Light, M.E.; van Staden, J. Biological activities and distribution of plant saponins. J. Ethnopharmacol. 2004, 94, 219–243. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, H.; Niesler, N.; Trautner, A.; Sama, S.; Jerz, G.; Panjideh, H.; Weng, A. Glycosylated Triterpenoids as Endosomal Escape Enhancers in Targeted Tumor Therapies. Biomedicines 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Chen, M.L.; Kimbara, K.; Majumdar, D.; Cunningham, E.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 14098–14103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Kimbara, K.; Das Gupta, T.K.; Chakrabarty, A.M. The Bacterial Redox Protein Azurin Induces Apoptosis in J774 Macrophages through Complex Formation and Stabilization of the Tumor Suppressor Protein p53. Infect. Immun. 2002, 70, 7054–7062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Hiraoka, Y.; Ikehata, M.; Kimbara, K.; Avner, B.S.; Das Gupta, T.K.; Chakrabarty, A.M. Apoptosis or growth arrest: Modulation of tumor suppressor p53’s specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 2004, 101, 4770–4775. [Google Scholar] [CrossRef]
- Punj, V.; Bhattacharyya, S.; Saint-dic, D.; Vasu, C.; Cunningham, E.A.; Graves, J. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene 2004, 23, 2367. [Google Scholar] [CrossRef]
- Bernardes, N.; Ribeiro, A.S.; Abreu, S.; Mota, B.; Matos, R.G.; Arraiano, C.M.; Seruca, R.; Paredes, J.; Fialho, A.M. The bacterial protein azurin impairs invasion and FAK/Src signaling in P-cadherin-overexpressing breast cancer models. PLoS ONE 2013, 19, e69023. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, N.; Ribeiro, A.S.; Abreu, S.; Vieira, A.F.; Carreto, L.; Santos, M.; Seruca, R.; Paredes, J.; Fialho, A.M. High-throughput molecular profiling of a P-cadherin overexpressing breast cancer model reveals new targets for the anti-cancer bacterial protein azurin. Int. J. Biochem. Cell Biol. 2014, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, N.; Abreu, S.; Carvalho, F.A.; Fernandes, F.; Santos, N.C.; Fialho, A.M. Modulation of membrane properties of lung cancer cells by azurin enhances the sensitivity to EGFR-targeted therapy and decreased β1 integrin-mediated adhesion. Cell Cycle 2016, 15, 1415–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albergaria, A.; Ribeiro, A.S.; Vieira, A.F.; Sousa, B.; Nobre, A.R.; Seruca, R.; Schmitt, F.; Paredes, J. P-cadherin role in normal breast development and cancer. Int. J. Dev. Biol. 2011, 55, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, A.F.; Ribeiro, A.S.; Dionísio, M.R.; Sousa, B.; Nobre, A.R.; Albergaria, A.; Santiago-Gómez, A.; Mendes, N.; Gerhard, R.; Schmitt, F.; et al. P-cadherin signals through the laminin receptor α6β4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget 2014, 5, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, N.; Garizo, A.R.; Pinto, S.N.; Caniço, B.; Perdigão, C.; Fernandes, F.; Fialho, A.M. Azurin interaction with the lipid raft components ganglioside GM-1 and caveolin-1 increases membrane fluidity and sensitivity to anti-cancer drugs. Cell Cycle 2018, 17. [Google Scholar] [CrossRef]
- Apiyo, D.; Wittung-Stafshede, P. Unique complex between bacterial azurin and tumor-suppressor protein p53. Biochem. Biophys. Res. Commun. 2005, 332, 965–968. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Probing the interaction between p53 and the bacterial protein azurin by single molecule force spectroscopy. J. Mol. Recognit. 2008, 21, 63–70. [Google Scholar] [CrossRef]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Modeling the interaction between the N-terminal domain of the tumor suppressor p53 and azurin. J. Mol. Recognit. 2009, 22, 215–222. [Google Scholar] [CrossRef]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. P28-Mediated activation of p53 in G2-M phase of the cell cycle enhances the efficacy of DNA damaging and antimitotic chemotherapy. Cancer Res. 2016, 76, 2354–2365. [Google Scholar] [CrossRef]
- Mehta, R.R.; Hawthorne, M.; Peng, X.; Shilkaitis, A.; Mehta, R.G.; Beattie, C.W.; Das Gupta, T.K. A 28-amino-acid peptide fragment of the cupredoxin azurin prevents carcinogen-induced mouse mammary lesions. Cancer Prev. Res. 2010, 3, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mehta, R.R.; Lekmine, F.; Christov, K.; King, M.L.; Majumdar, D.; Shilkaitis, A.; Green, A.; Bratescu, L.; Beattie, C.W.; et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol. Cancer Ther. 2009, 8, 2947–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro. Oncol. 2016, 18, 1319–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Gorman, G.S.; Coward, L.U.; Noker, P.E.; Mccormick, D.; Horn, T.L.; Harder, J.B.; Muzzio, M. Preclinical pharmacokinetics, metabolism, and toxicity of azurin-p28 (NSC745104) a peptide inhibitor of p53 ubiquitination. Cancer Chemother. Pharmacol. 2011, 28, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L.; et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef]
- Giansanti, F.; Panella, G.; Leboffe, L.; Antonini, G. Lactoferrin from milk: Nutraceutical and pharmacological properties. Pharmaceuticals 2016, 9, 61. [Google Scholar] [CrossRef]
- Sun, X.; Jiang, R.; Przepiorski, A.; Reddy, S.; Palmano, K.P.; Krissansen, G.W. “Iron-saturated” bovine lactoferrin improves the chemotherapeutic effects of tamoxifen in the treatment of basal-like breast cancer in mice. BMC Cancer 2012, 12, 1–12. [Google Scholar] [CrossRef]
- Fang, B.; Guo, H.Y.; Zhang, M.; Jiang, L.; Ren, F.Z. The six amino acid antimicrobial peptide bLFcin6 penetrates cells and delivers siRNA. FEBS J. 2013, 280, 1007–1017. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of some features of normal and cancer cells depicting lipid composition and extracellular pH (upper panel), and the alterations occurring at the cellular membrane nano/microdomains (lower panel). Metastatic cells with lower cholesterol levels (chol ↓) enhance membrane deformability (lower panel, left) while cells with increased cholesterol levels (chol ↑) enrich membrane in ordered lipid raft domains and rigidity (lower panel, right).
Figure 1.
Schematic representation of some features of normal and cancer cells depicting lipid composition and extracellular pH (upper panel), and the alterations occurring at the cellular membrane nano/microdomains (lower panel). Metastatic cells with lower cholesterol levels (chol ↓) enhance membrane deformability (lower panel, left) while cells with increased cholesterol levels (chol ↑) enrich membrane in ordered lipid raft domains and rigidity (lower panel, right).

Figure 2.
Promiscuous anticancer activity of azurin at the membrane level. Different membrane proteins are targeted by azurin, altering the biophysical profile and attenuating raft-related signaling pathways.
Figure 2.
Promiscuous anticancer activity of azurin at the membrane level. Different membrane proteins are targeted by azurin, altering the biophysical profile and attenuating raft-related signaling pathways.


{kind=link}
{kind=link}
Table 1.
Overview of selected therapeutic agents targeting multiple steps in lipid localization and synthesis to modulate membranes. Chemical structures were obtained from ChemSpider, a free chemical structure database owned by the Royal Society of Chemistry (http://www.chemspider.com/).
Table 1.
Overview of selected therapeutic agents targeting multiple steps in lipid localization and synthesis to modulate membranes. Chemical structures were obtained from ChemSpider, a free chemical structure database owned by the Royal Society of Chemistry (http://www.chemspider.com/).
| Acting Directly at the Membrane | Ref | ||
| i) Lowering cholesterol levels | Cholesterol binding agents: filipin |  | [6] |
| Cholesterol chemical depletion: MβCD |  | [38] | |
| ii) Stabilization of pro-apoptotic domains | ALPs Edelfosine |  | [70,71,72,73,74,75,76,77,78] |
| Miltefosine |  | ||
| perifosine |  | ||
| ODPC |  | ||
| Acting Intracellularly in lipid metabolism pathways | |||
| i) Fatty acid synthesis inhibition | TVB-2640 | unavailable | [62] |
| ii) Statins (inhibition of mevalonate pathway) | Lovastatin |  | [65] |
| Atorvastatin |  | [66] | |
| iii) Cer metabolism (activation of SMase) | Ara-C; |  | [83] |
| Decitabine; |  | [1,6] | |
| glucosylceramide analogue (PDMP) |  | [52,84] | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bernardes, N.; Fialho, A.M. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. Int. J. Mol. Sci. 2018, 19, 3871. https://doi.org/10.3390/ijms19123871
AMA Style
Bernardes N, Fialho AM. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. International Journal of Molecular Sciences. 2018; 19(12):3871. https://doi.org/10.3390/ijms19123871
Chicago/Turabian StyleBernardes, Nuno, and Arsenio M. Fialho. 2018. "Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies" International Journal of Molecular Sciences 19, no. 12: 3871. https://doi.org/10.3390/ijms19123871
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.