The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis


1
Department of Respiratory Medicine, Graduate School of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
2
Division for Health Service Promotion, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
3
Hastings Center for Pulmonary Research, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
4
Department of Immunology, Genetics and Pathology, Uppsala University, SE-75185 Uppsala, Sweden
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(11), 3611; https://doi.org/10.3390/ijms19113611
Submission received: 26 September 2018
/
Revised: 12 November 2018
/
Accepted: 14 November 2018
/
Published: 15 November 2018
(This article belongs to the Special Issue Inflammation and Cancer 2018)
Abstract
:Idiopathic pulmonary fibrosis (IPF) is a progressive fibrotic lung disease of unknown etiology and dismal prognosis. IPF patients are known to have an increased risk of lung cancer and careful decision-making is required for the treatment of lung cancer associated with IPF. Transforming growth factor (TGF)-β signaling plays a central role in tissue fibrosis and tumorigenesis. TGF-β-mediated pathological changes that occur in IPF lung tissue may promote the process of field cancerization and provide the microenvironment favorable to cancer initiation and progression. This review summarizes the current knowledge related to IPF pathogenesis and explores the molecular mechanisms that underlie the occurrence of lung cancer in the background of IPF, with an emphasis on the multifaceted effects of TGF-β signaling.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease of unknown etiology, and is the most common subtype of idiopathic interstitial pneumonia. The diagnosis of IPF is based on radiological and/or pathological findings, indicative of the morphological pattern of usual interstitial pneumonia (UIP) [1]. The incidence of IPF is estimated to be 3–9 cases per 100,000 person-years [2]. IPF is comprised of sporadic and familial cases, suggesting that genetic factors of variable degree are involved in its pathogenesis [3]. Prognosis for IPF patients is poor with a median survival time of only 2–4 years; however, the clinical course of IPF is highly variable and some patients can survive for long periods of time [4]. Despite recent advances in anti-fibrotic therapies, IPF remains an incurable disease [1,3,4].
Lung cancer is a major comorbidity in IPF patients [5]. A number of studies have demonstrated a strong association between IPF and lung cancer, and epidemiological studies have shown that IPF itself is an independent risk factor for lung cancer [5,6,7]. The reported prevalence of lung cancer among IPF patients is 3–48% [6]. This variance appears to be due to the differences in study populations. A smaller Japanese study calculated the cumulative incidence rates of lung cancer for IPF patients to be 3.3%, 15.4%, and 54.7% at 1, 5, and 10 years, respectively [8]. The survival time of IPF patients with lung cancer is shorter than those without lung cancer [9].
The strong association between IPF and lung cancer indicates that the molecular changes underlying IPF pathogenesis have tumor-promoting effects. Indeed, similar mechanisms have been suggested to be involved in both IPF and lung cancer, including telomere abnormalities, alterations in alveolar epithelial cell features, epithelial–mesenchymal transition (EMT), sustained fibroblast activation, and extracellular matrix (ECM) remodeling [3,4,10,11]. Notably, the anti-fibrotic drug for the treatment of IPF, nintedanib, is also approved as a therapeutic agent for non-small cell lung cancer [12]. Thus, the pathogenic overlap of both diseases may help to better understand the molecular mechanisms involved in IPF and lung cancer, and ultimately may provide clues in developing therapeutic strategies for both diseases.
Transforming growth factor (TGF)-β is a key player in the process of fibrosis and tumorigenesis. TGF-β, as a pleiotropic factor, regulates a variety of biological processes including embryogenesis, cellular differentiation, organogenesis, and immune responses [13]. Under physiological conditions, TGF-β is necessary for lung morphogenesis and homeostasis while aberrant TGF-β signaling has been shown to be central to pulmonary fibrosis and lung cancer progression [11].
In this review, we integrate the current literature of IPF pathogenesis and its relationship with lung cancer, with an emphasis on TGF-β signaling. First, we present an overview of current knowledge about genetic predispositions to IPF and explore its association with lung carcinogenesis. Furthermore, we summarize the recent reports on the genomic features of lung cancer that occurred in the background of IPF. In the latter part of this review, TGF-β signaling is highlighted as a key molecular link between IPF and lung cancer, and we discuss its multifaceted roles in alveolar epithelial cells, fibroblasts, and immune cells.
2. Genetic Predispositions to Pulmonary Fibrosis
Both genetic and environmental factors contribute to the development of IPF. Risk factors for IPF identified through epidemiological studies include ageing, male sex, tobacco smoking, and other environmental exposures [14]. Notably, pulmonary fibrosis tends to occur among smokers in a pedigree of familial pulmonary fibrosis, suggesting that both genetic and environmental factors influence its pathogenesis [15]. Indeed, a significant number of IPF patients are former or current smokers (40–70%) [5,16]. Tobacco smoking is the major risk factor for lung cancer [17], which may confound the association between IPF and lung cancer. However, a population-based cohort study in the United Kingdom demonstrated that the incidence of lung cancer has increased independent of smoking history in IPF patients [5].
In addition to environmental factors, recent genome-wide association studies have identified genetic polymorphisms that are related to an increased risk of IPF; those reported by multiple studies include the regions 5p15 (TERT) and 3q26 (TERC) [18,19]. Telomeres are DNA–protein complexes that protect the ends of chromosomes from degradation and serve to maintain genomic integrity. Shortening of telomeres occurs during each cell division and is implicated in cellular ageing [20]. Telomerase is a ribonucleoprotein complex involved in the addition of repetitive DNA sequences to telomeres and is comprised of telomerase reverse transcriptase (TERT) and telomerase RNA component (TERC).
Besides the telomerase genes, TERT and TERC, multiple studies have identified genetic polymorphisms in the 11p15 region (MUC5B and TOLLIP) related to the risk of IPF [21,22,23], and the genotype of MUC5B and TOLLIP has been shown to be significantly associated with prognosis and therapeutic responses among IPF patients [24,25,26]. It is estimated that the rs35705950 variant in the promoter of MUC5B is the strongest risk factor for developing IPF [27]. Mechanistically, it has been proposed that the rs35705950 variant leads to overexpression of mucin 5B, which may cause abnormal mucociliary clearance and altered immune responses, ultimately leading to chronic alveolar epithelial cell damage [28].
Most convincing evidence of genetic predispositions to IPF comes from pedigrees with familial pulmonary fibrosis. Mutations in surfactant protein C (SFTPC) and surfactant protein A2 (SFTPA2) and their associations with familial pulmonary fibrosis suggest the importance of alveolar epithelial cells in IPF pathogenesis [29,30]. The frequency of SFTPC mutations in familial cases of IPF seems variable depending on ethnicity (2–25%) [31]. In correspondence to the association between TERT or TERC gene polymorphisms and the risk of sporadic IPF, germline TERT or TERC mutations have been found in 8–15% of familial cases of IPF [32,33,34,35]. Two more genes linked to telomere biology are mutated in a subset of familial cases: regulator of telomere elongation helicase 1 (RTEL1) and poly(A)-specific ribonuclease (PARN) [36,37].
It is known that mutations in TERT and TERC are related to telomere shortening as a result of impaired telomerase catalytic activity. RTEL1 is required to prevent catastrophic telomere loss and its mutation causes telomere instability [38]. It has been recently shown that PARN is required for post-transcriptional RNA maturation of the TERC gene, further supporting the importance of telomere dysfunction in IPF pathogenesis [39].
Even in the absence of mutations in the telomere-related genes, shortening of telomeres in alveolar epithelial cells is commonly found in IPF patients [34]. In alveolar epithelial type II cells, which are regarded as stem cells in the alveoli [40], telomere shortening results in cellular senescence, stem cell failure, and pulmonary fibrosis in a mouse model [41,42]. Thus, telomere dysfunction through gene–environment interactions appears to be one major pathological mechanism of IPF in general.
Besides the regulatory role of TERT and RTEL1 for telomere function, both genes are also required for DNA replication and DNA repair, ensuring genome integrity [43,44]. This function is of particular importance for alveolar epithelial type II cells that are exposed to a myriad of environmental pollutants and carcinogens. Because alveolar epithelial type II cells are considered to be an origin of lung cancer [45], telomere dysfunction and functional abnormalities of TERT and RTEL1 genes might conceptually connect IPF to lung carcinogenesis.
Apart from telomere dysfunction and shortening involved in IPF pathogenesis, mRNA upregulation or amplification of TERT and TERC genes is frequently found in lung adenocarcinomas and squamous cell carcinomas [46,47]. It is conceivable that TERT and TERC upregulation or amplification leads to increased telomerase activity, thereby avoiding replicative senescence and gaining growth advantage [48]. However, it remains unexplored whether and how telomerase activity is altered during the process of lung carcinogenesis in IPF patients.
3. Genomic Features of Lung Cancer That Occurs in Pulmonary Fibrosis
In recent years, genomic alterations and mutation profiles of lung cancer have been extensively investigated in large cohorts including The Cancer Genome Atlas (TCGA) [46,47]. In general, adenocarcinoma is the most frequent histological subtype of lung cancer (30–40%), followed by squamous cell carcinoma (20–30%) [49]. In contrast, squamous cell carcinoma is the most prevalent subtype (35–40%) of lung cancer that occurs in IPF, and adenocarcinoma accounts for 30–35% of cases [50].
Based on the observation that genomic aberrations and precancerous lesions occur multifocally in a region or an organ, the concept of field cancerization has now been recognized as the priming of normal tissue that precedes the occurrence of cancer [51,52]. A recent report demonstrated that gene expression profiles from bronchial epithelial cells with normal appearance are useful in assessing lung cancer probability [53]. Another report demonstrated that TP53 mutations associated with tobacco smoking are observed in the lungs prior to the development of clinically detectable cancer [54]. These reports suggest that cancer-prone abnormal cells reside in morphologically non-cancerous lung tissues. In IPF lung tissue, progressive bronchiolar proliferation, termed bronchiolization, and squamous metaplasia are frequently found in the fibrotic area [55]. These metaplastic foci have been demonstrated to be associated with atypia and the occurrence of TP53 mutations [56,57]. It is tempting to speculate that these lesions serve as precursors to lung cancer, which may provide an explanation for the higher prevalence of squamous cell carcinoma in IPF patients. Moreover, such a scenario seems to fit with the concept of field cancerization.
Recently, genetic and clinicopathological features of 44 lung adenocarcinomas in the background of UIP, which is the morphological characteristic of IPF, have been analyzed in detail [58]. Compared to lung adenocarcinomas without UIP, adenocarcinomas with comorbid UIP were associated with older age, male gender (93.2%), and smoking history (90.9%). Importantly, adenocarcinomas with UIP showed higher frequencies of invasive mucinous-predominant subtype (29.5% vs. 3.9%) and KRAS mutations (30.2% vs. 8.5%) while EGFR mutations were much less prevalent (2.3% vs. 45.6%) [58]. These observations are consistent with the finding that KRAS mutation-positive lung adenocarcinoma is associated with male sex and smoking history [59].
Recently, it has been demonstrated that MUC5B serves as a marker for the non-terminal respiratory unit (TRU) subtype and the invasive mucinous adenocarcinomas associated with KRAS mutations [60,61]. As mentioned above, MUC5B polymorphism is associated with pulmonary fibrosis [21,22,23], and presumably, mucin 5B is overexpressed in IPF lung tissue. This may be somehow related to the observation that the invasive mucinous-predominant subtype is more frequent in lung adenocarcinomas with UIP [58].
Another study profiled 15 adenocarcinomas and 20 squamous cell carcinomas associated with IPF by sequencing ~500 cancer-related genes [62]. The mean number of somatic mutations was 11.7 per sample with an average of 5.86 mutations/Mb. Interestingly, the frequency of BRAF mutations was high (17.1%), while in this study, KRAS mutation was not found in any sample [62]. EGFR mutation was detected paradoxically in one case of squamous cell carcinoma. This is in contrast with the previous study [58] that showed a higher frequency of KRAS mutations in lung adenocarcinomas associated with UIP. Both studies are based on surgically resected lung cancer samples of East Asian populations, in which the frequencies of KRAS and EGFR mutations are reported to be 8–10% and 40–55%, respectively, in lung adenocarcinomas [59]. Further studies with larger sample size are needed to elucidate oncogenic drivers dominant in lung adenocarcinomas or squamous cell carcinomas that occur in IPF lung tissue.
4. Genome-Wide Methylation Profiles of Pulmonary Fibrosis and Lung Cancer
In recent years, genome-wide methylation profiling studies have identified hypermethylated and hypomethylated genes in lung cancer [63]. In general, DNA hypermethylation of tumor suppressor genes is implicated in lung cancer development. For example, promoter hypermethylation of CDKN2A and RASSF1 leads to perturbed cell cycle regulation, which contributes to lung carcinogenesis. Importantly, hypermethylation of these tumor suppressors has been shown to be associated with smoking behavior in lung cancer patients [64].
In addition, global DNA hypomethylation is also a hallmark of cancer, causing aberrant expression of cancer testis antigens and activation of repetitive elements, such as long interspersed nuclear element-1 (LINE-1), across the whole genome [63,65]. This is clinically important, as transactivation of cancer testis antigens has been postulated to enhance the efficacy of immune therapy. Furthermore, it has been suggested that detection of transactivated repetitive elements might be potentially useful for the diagnosis of lung cancer [63].
There have been few studies describing DNA methylation patterns in the lung tissues of IPF patients [66]. A pioneering study of profiling genome-wide methylation in 94 IPF patients and 67 controls identified 2130 differentially methylated regions, and among them, 738 regions were inversely correlated with gene expression changes [67].
Another study identified 402 CpG islands that were differentially methylated in both IPF lung tissues and lung adenocarcinomas compared with control lungs [68]. Interestingly, IPF lung tissue displayed an intermediate methylation profile of these CpG islands, between normal and cancerous tissues. This observation suggests that DNA methylation patterns in IPF lung tissue may constitute precancerous changes in line with the concept of field cancerization. However, in the same study, IPF lung tissue did not show hypomethylation of LINE-1 repetitive elements, one of the methylation patterns frequently found in lung cancer. This observation suggests that lung cancer DNA methylation profiles are, in some aspects, distinct from those of IPF.
5. Roles of TGF-β in Alveolar Epithelial Cells
In IPF lung tissue, pathological events include alveolar epithelial regenerative failure (repeated epithelial damage and aberrant repair of the injured epithelium) and sustained fibroproliferative reactions (persistent fibroblast activation, excessive ECM deposition, and the resultant structural alterations) [3,4]. Molecular processes of alveolar epithelial cell damage and defective repair comprise oxidative injury, endoplasmic reticulum stress, mitochondrial dysfunction, cellular senescence, and apoptosis [69,70]. Transforming growth factor (TGF)-β is essential for both epithelial and mesenchymal changes (Figure 1).
Although many other inflammatory signals are involved in IPF pathogenesis, such as interleukin (IL)-1β, IL-13, IL-17, CC chemokine ligand 2 (CCL2), and CXC chemokine ligand 12 (CXCL12) [71], in a clinical setting, anti-inflammatory or immunosuppressive agents, including corticosteroids, did not show clear therapeutic benefits. This supports the hypothesis that IPF is caused by dysregulated epithelial–mesenchymal interactions and is not simply an inflammatory disease.
TGF-β is abundantly expressed in IPF lung tissue, where macrophages and metaplastic alveolar epithelial cells are the major cellular sources of TGF-β [72,73]. In rodent models, ectopic expression of TGF-β in the lungs recapitulates the pathophysiological features of human pulmonary fibrosis, supporting the concept that TGF-β plays a pivotal role in the disease pathogenesis [74,75]. TGF-β induces cytostasis in most epithelial cells and inhibits proliferation of alveolar epithelial cells [76]. Thus, it is conceivable that TGF-β negatively affects epithelial cell regeneration and therefore contributes to IPF pathogenesis.
TGF-β is also known to strongly elicit EMT, a phenotypic change whereby polarized epithelial cells acquire mesenchymal and migratory characteristics [14]. Mechanistically, TGF-β induces the transcriptional repressors, SNAI1, SNAI2, ZEB1, and ZEB2, which subsequently repress cell junctional proteins, in turn, disrupting epithelial cell integrity and apical–basal polarity. Previous studies have demonstrated that TGF-β elicits EMT in alveolar epithelial cells, which may be a part of the altered epithelial features in IPF lung tissue [77]. It remains controversial whether epithelial cells that undergo EMT contribute to mesenchymal cell populations, with conflicting results from studies of cell lineage tracing in mouse models [78].
Single-cell transcriptome analysis is emerging as a powerful method for deciphering heterogeneous cell populations and cellular differentiation processes [79]. Recently, single-cell RNA-sequencing of lung epithelial cells from IPF patients has been performed, which identified three subtypes of epithelial cells associated with IPF. Their transcriptional profiles were distinct from those of alveolar epithelial type II cells, and showed features of conducting airway basal cells or goblet cells, supporting the concept of bronchiolization. Notably, mesenchymal markers and EMT-related molecules were induced in “basal cell” or “indeterminate” subtypes of IPF-derived epithelial cells, indicating the involvement of EMT. In line with this finding, TGF-β signaling pathway genes showed higher expression levels in IPF-derived epithelial cells and pathway analysis revealed aberrant activation of TGF-β signaling [79].
In contrast to normal epithelial cells with organized cell junction and polarity, most cultured cancer cells lack cell polarity and exist in an intermediate cellular status between epithelial and mesenchymal cells, which is termed partial EMT. Progression and completion of EMT processes in cancer cells are associated with the acquisition of malignant cell features including stem cell traits, resistance to apoptosis, cell invasion, and drug resistance [80].
Taken together, it can be postulated that TGF-β-mediated EMT in IPF lung tissue might contribute to malignant transformation. However, further pathological evaluations are required to determine whether EMT is clinically relevant and significantly involved in the fibrotic process of IPF. Moreover, it also needs to be determined whether EMT-related molecular changes are associated with the morphological changes found in IPF, such as alveolar epithelial cell hyperplasia and squamous metaplasia.
6. TGF-β-Mediated Fibroproliferative Reactions in Pulmonary Fibrosis and Lung Cancer
In addition to aberrations in epithelial cells, pathological activation of fibroblasts and excessive ECM accumulation constitute a major hallmark of IPF, leading to irreversible structural alterations and tissue stiffening in the lungs. These changes functionally cause lung volume loss and a reduced vital capacity. The fibrotic thickening of the alveolar wall impairs gas exchange with decreased diffusing capacity and ultimately leads to arterial hypoxemia [3,4].
Activated lung fibroblasts, which conceivably overlap with α-smooth muscle actin (α-SMA)-positive myofibroblasts, are responsible for producing ECM components, such as collagen and laminin [81]. TGF-β potently enhances differentiation from fibroblast to myofibroblast, and promotes the expression of ECM components [11]. Moreover, TGF-β induces the expression of integrins, matrix metalloproteinases, protease inhibitors, and regulators of small GTPases, all of which contribute to tissue remodeling and cell–ECM interactions [82]. TGF-β also stimulates the production of fibrogenic or angiogenic growth factors such as connective tissue growth factor (CTGF), platelet-derived growth factor (PDGF), and vascular endothelial cell growth factor (VEGF).
Corresponding to mesenchymal tissue remodeling in IPF, TGF-β is also central in the development of tumor stroma, which is composed of cancer-associated fibroblasts (CAFs), immune cells, and the ECM. In analogy to IPF, CAFs are α-SMA-positive myofibroblasts activated by TGF-β, and have common features with the myofibroblasts observed in IPF lung tissue [83]. Notably, gene expression profiling analyses have demonstrated that CAFs isolated from lung cancer tissues exhibit increased expression of TGF-β signaling-related genes [84,85].
To compare gene expression profiles from lung cancer stroma samples and the lung tissues of interstitial pneumonia including IPF, we utilized publicly available datasets. The dataset GSE22863 comprises gene expression profiles from normal lung parenchyma and lung tumor stroma while the dataset GSE47460 includes gene expression profiles from control lung tissues and different interstitial lung diseases including IPF. We found that about 10% of genes significantly upregulated in lung cancer stroma were also enriched in lung tissues of interstitial lung diseases compared to normal lung tissues. These commonly upregulated genes included ECM components (COL1A2, COL3A1, and COL5A2) and matrix metalloproteinases (MMP9 and MMP11). Importantly, inferred biological processes of these commonly upregulated genes were mainly related to ECM remodeling and collagen metabolism (Figure 2). These findings support the idea that cancer stroma exhibits molecular features similar to those of interstitial pneumonia or IPF.
The mesenchymal changes in IPF lung tissue, most likely mediated by TGF-β, also influence the repair process of the alveolar epithelium by inducing developmental signals. Particularly, activated fibroblasts produce growth factors for the epithelium such as hepatocyte growth factor (HGF). In IPF patients, HGF levels in serum and bronchoalveolar lavage fluid are significantly higher than in normal controls [86]. Considering the importance of the HGF/c-Met pathway for lung cancer progression [87], an abundance of growth factors in the milieu of IPF lung tissue may facilitate the development of cancer in IPF patients.
Finally, TGF-β signaling is essential in the regulation of immune responses. Its role is evidenced by the systemic inflammation observed in TGF-β1-deficient mice [88,89]. Of particular importance is its involvement in T cell homeostasis and immune-suppressive effects [90]. Cancer progression is dependent on the evasion of immunosurveillance, and immune suppression by TGF-β is an emerging mechanism in tumorigenesis [91,92]. Furthermore, accumulating evidence supports the notion that TGF-β mediates tumor-promoting properties of tumor-associated macrophages and myeloid-derived suppressor cells [93].
A previous study performed immunohistochemistry for immune cell markers on IPF lung tissue and characterized topological patterns of immune cell infiltration. The fibrotic regions had much fewer lymphocytes, monocytes, and macrophages compared to the epithelial dominant areas. This observation suggests that fibrotic lesions in IPF lung tissue might be privileged to escape from immune surveillance. It has been reported that lung cancer might preferentially originate from the fibrotic area as felicitously termed “scar-cinoma” [94]. In support of this notion, lung cancers associated with IPF are frequently found in the lower lobe and peripheral portion where fibrotic changes predominantly occur.
Collectively, IPF lung tissue with abundant TGF-β could represent a microenvironment in which anti-cancer immune surveillance is disrupted, which is a condition favorable to cancer initiation and progression. However, the similarities and differences of immune landscape between IPF and lung cancer tissues remain largely unknown.
7. Clinical Implications and Future Perspectives
The diagnosis of lung cancer in patients with IPF is difficult because radiological findings of cancerous lesions often appear indistinct from the fibrotic or inflammatory changes associated with the histological pattern of UIP. In a clinical setting, special attention is required in therapeutic decision-making for lung cancers complicated with IPF, and standard treatment guidelines for lung cancer are not applicable to patients with IPF. Radiation therapy, whether curative or palliative, is not recommended for IPF patients because of the risk of acute exacerbation. EGFR-tyrosine kinase inhibitors are known to induce fatal acute interstitial pneumonia with a higher frequency among lung cancer patients with prior pulmonary fibrosis (mostly IPF) [95]. For early-stage resectable lung cancers, acute exacerbation of interstitial pneumonia following surgery is often fatal, and it is difficult to estimate the risk of this occurring. It has been reported that acute exacerbation following thoracic surgery may occur in 9% of cases, with a mortality rate of more than 40% [96,97].
Recently, nintedanib and pirfenidone have both been approved as treatment options for IPF [3,4]. Nintedanib is a multi-kinase inhibitor that targets VEGF receptors, PDGF receptors, and fibroblast growth factor receptors. Pirfenidone exerts its effect through downregulation of TGF-β, suppression of fibroblast proliferation, and inhibition of collagen synthesis. These agents slow disease progression [98,99], and recent meta-analyses have demonstrated that these agents have a beneficial impact on survival [100,101].
Importantly, nintedanib has been shown to be effective as a therapeutic agent for non-small cell lung cancer [12]. These clinical observations further suggest the close relationship between IPF and lung cancer at the molecular level.
An obvious question is whether a significant amount of somatic mutations are accumulating in the fibrotic lesion of IPF lung tissue. If so, are they related to the degree of fibrotic changes or precancerous stages? Transcriptome and methylation profiles of lung cancer in IPF patients also remain to be elucidated. Recent studies have suggested that lung cancers of each histological subtype can be further subclassified based on gene expression and methylation patterns, which is related to different responses to cytotoxic agents, molecular targeted therapies, and immune checkpoint inhibitors [102,103]. Given that the mutation burden of lung cancer complicated with IPF is relatively high, as suggested by a recent report [62], immune checkpoint inhibitors might be beneficial. However, interstitial lung disease is one of the immune-related adverse events, which may exacerbate IPF [104], and therefore immune checkpoint blockade should be considered with caution.
The genomic changes that occur during the evolution of lung cancer in IPF lung tissue are still fragmentarily understood, and further studies are necessary to dissect the contributions of telomere dysfunction, smoking, epigenetic changes, and somatic mutations to cancer initiation and progression in IPF patients. In summary, a better understanding of IPF pathogenesis and its association with cancer will aid in the development of more refined diagnostic and therapeutic strategies for both IPF and lung cancer.
Funding
This work was supported by the Takeda Science Foundation, Uehara Memorial Foundation Research Fellowship, and JSPS KAKENHI #18K08170 (A.S.) and #16H02653 (T.N.).
Acknowledgments
We are grateful to Hiromitsu Ohta for his constructive suggestions and critical reading of the manuscript.
Conflicts of Interest
The authors declare that they have no conflict of interests.
References
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.; Fogarty, A.; Hubbard, R.; McKeever, T. Global incidence and mortality of idiopathic pulmonary fibrosis: A systematic review. Eur. Respir. J. 2015, 46, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am. J. Respir. Crit. Care Med. 2000, 161, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir. J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Le Jeune, I.; Gribbin, J.; West, J.; Smith, C.; Cullinan, P.; Hubbard, R. The incidence of cancer in patients with idiopathic pulmonary fibrosis and sarcoidosis in the UK. Respir. Med. 2007, 101, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009, 14, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45, S163–S175. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Nagase, T. Hippo and TGF-β interplay in the lung field. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L756–L767. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Kaiser, R.; Mellemgaard, A.; Douillard, J.Y.; Orlov, S.; Krzakowski, M.; von Pawel, J.; Gottfried, M.; Bondarenko, I.; Liao, M.; et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): A phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014, 15, 143–155. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Dellaripa, P.F.; Lederer, D.J.; Khanna, D.; Young, L.R.; Martinez, F.J. Interstitial lung disease: NHLBI Workshop on the primary prevention of chronic lung diseases. Ann. Am. Thorac. Soc. 2014, 11, S169–S177. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Ren, P.; Avila, N.A.; Chow, C.K.; Franks, T.J.; Travis, W.D.; McCoy, J.P.J.; May, R.M.; Wu, H.P.; Nguyen, D.M.; et al. Early interstitial lung disease in familial pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007, 176, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.; Malvezzi, M.; Negri, E.; La Vecchia, C.; Boffetta, P. Risk factors for lung cancer worldwide. Eur. Respir. J. 2016, 48, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanios, M.; Blackburn, E.H. The telomere syndromes. Nat. Rev. Genet. 2012, 13, 693–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunninghake, G.M.; Hatabu, H.; Okajima, Y.; Gao, W.; Dupuis, J.; Latourelle, J.C.; Nishino, M.; Araki, T.; Zazueta, O.E.; Kurugol, S.; et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N. Engl. J. Med. 2013, 368, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the response to n-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Peljto, A.L.; Zhang, Y.; Fingerlin, T.E.; Ma, S.F.; Garcia, J.G.; Richards, T.J.; Silveira, L.J.; Lindell, K.O.; Steele, M.P.; Loyd, J.E.; et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013, 309, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Evans, C.M.; Fingerlin, T.E.; Schwarz, M.I.; Lynch, D.; Kurche, J.; Warg, L.; Yang, I.V.; Schwartz, D.A. Idiopathic pulmonary fibrosis: A genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol. Rev. 2016, 96, 1567–1591. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.G.; Livraghi-Butrico, A.; Fletcher, A.A.; McElwee, M.M.; Evans, S.E.; Boerner, R.M.; Alexander, S.N.; Bellinghausen, L.K.; Song, A.S.; Petrova, Y.M.; et al. Muc5b is required for airway defence. Nature 2014, 505, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Van Moorsel, C.H.; Van Oosterhout, M.F.; Barlo, N.P.; de Jong, P.A.; Van der Vis, J.J.; Ruven, H.J.; Van Es, H.W.; Van den Bosch, J.M.; Grutters, J.C. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am. J. Respir. Crit. Care Med. 2010, 182, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuan, P.J.; Xing, C.; Cronkhite, J.T.; Torres, F.; Rosenblatt, R.L.; DiMaio, J.M.; Kinch, L.N.; Grishin, N.V.; Garcia, C.K. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am. J. Hum. Genet. 2009, 84, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Kropski, J.A.; Blackwell, T.S.; Loyd, J.E. The genetic basis of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1717–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A.; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronkhite, J.T.; Xing, C.; Raghu, G.; Chin, K.M.; Torres, F.; Rosenblatt, R.L.; Garcia, C.K. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogan, J.D.; Kropski, J.A.; Zhao, M.; Mitchell, D.B.; Rives, L.; Markin, C.; Garnett, E.T.; Montgomery, K.H.; Mason, W.R.; McKean, D.F.; et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2015, 191, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Bertuch, A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016, 13, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.H.; Segal, M.; Boyraz, B.; Guinan, E.; Hofmann, I.; Cahan, P.; Tai, A.K.; Agarwal, S. Poly(A)-specific ribonuclease (PARN) mediates 3’-end maturation of the telomerase RNA component. Nat. Genet. 2015, 47, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannier, J.B.; Sandhu, S.; Petalcorin, M.I.; Wu, X.; Nabi, Z.; Ding, H.; Boulton, S.J. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 2013, 342, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.Y.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The international epidemiology of lung cancer: Latest trends, disparities, and tumor characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef] [PubMed]
- Karampitsakos, T.; Tzilas, V.; Tringidou, R.; Steiropoulos, P.; Aidinis, V.; Papiris, S.A.; Bouros, D.; Tzouvelekis, A. Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2017, 45, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Incze, J.; Vaughan, C.W.J.; Lui, P.; Strong, M.S.; Kulapaditharom, B. Premalignant changes in normal appearing epithelium in patients with squamous cell carcinoma of the upper aerodigestive tract. Am. J. Surg. 1982, 144, 401–405. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.A.; Vachani, A.; Whitney, D.; Elashoff, M.; Porta Smith, K.; Ferguson, J.S.; Parsons, E.; Mitra, N.; Brody, J.; Lenburg, M.E.; et al. A bronchial genomic classifier for the diagnostic evaluation of lung cancer. N. Engl. J. Med. 2015, 373, 243–251. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol. Int. 1999, 49, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Ogura, T.; Yokose, T.; Nagai, K.; Nishiwaki, Y.; Esumi, H. p53 gene alteration in atypical epithelial lesions and carcinoma in patients with idiopathic pulmonary fibrosis. Hum. Pathol. 2001, 32, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Munakata, M.; Ohtsuka, Y.; Nisihara, H.; Nasuhara, Y.; Kamachi-Satoh, A.; Dosaka-Akita, H.; Homma, Y.; Kawakami, Y. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer 2002, 95, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masai, K.; Tsuta, K.; Motoi, N.; Shiraishi, K.; Furuta, K.; Suzuki, S.; Asakura, K.; Nakagawa, K.; Sakurai, H.; Watanabe, S.I.; et al. Clinicopathological, immunohistochemical, and genetic features of primary lung adenocarcinoma occurring in the setting of usual interstitial pneumonia pattern. J. Thorac. Oncol. 2016, 11, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Duruisseaux, M.; Antoine, M.; Rabbe, N.; Rodenas, A.; Mc Leer-Florin, A.; Lacave, R.; Poulot, V.; Duchêne, B.; Van Seuningen, I.; Cadranel, J.; et al. Lepidic predominant adenocarcinoma and invasive mucinous adenocarcinoma of the lung exhibit specific mucin expression in relation with oncogenic drivers. Lung Cancer 2017, 109, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, D.H.; Kim, K.B.; Shin, N.; Park, W.Y.; Lee, J.H.; Choi, K.U.; Kim, J.Y.; Lee, C.H.; Sol, M.Y.; et al. MUC5AC and MUC5B enhance the characterization of mucinous adenocarcinomas of the lung and predict poor prognosis. Histopathology 2015, 67, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.A.; Kim, D.; Chun, S.M.; Bae, S.; Song, J.S.; Kim, M.Y.; Koo, H.J.; Song, J.W.; Kim, W.S.; Lee, J.C.; et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J. Pathol. 2018, 244, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Kaczkowski, B.; Ohshima, M.; Matsuzaki, H.; Noguchi, S.; Mikami, Y.; Lizio, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; et al. Integrative CAGE and DNA methylation profiling identify epigenetically regulated genes in NSCLC. Mol. Cancer Res. 2017, 15, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, X.; Hong, Q.; Deng, Z.; Ma, H.; Xin, Y.; Fang, Y.; Ye, H.; Wang, R.; Zhang, C.; et al. Meta-analyses of gene methylation and smoking behavior in non-small cell lung cancer patients. Sci. Rep. 2015, 5, 8897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djureinovic, D.; Hallström, B.M.; Horie, M.; Mattsson, J.S.; La Fleur, L.; Fagerberg, L.; Brunnström, H.; Lindskog, C.; Madjar, K.; Rahnenführer, J.; et al. Profiling cancer testis antigens in non-small-cell lung cancer. JCI Insight 2016, 1, e86837. [Google Scholar] [CrossRef] [PubMed]
- Sanders, Y.Y.; Ambalavanan, N.; Halloran, B.; Zhang, X.; Liu, H.; Crossman, D.K.; Bray, M.; Zhang, K.; Thannickal, V.J.; Hagood, J.S. Altered DNA methylation profile in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Pedersen, B.S.; Rabinovich, E.; Hennessy, C.E.; Davidson, E.J.; Murphy, E.; Guardela, B.J.; Tedrow, J.R.; Zhang, Y.; Singh, M.K.; et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, E.I.; Kapetanaki, M.G.; Steinfeld, I.; Gibson, K.F.; Pandit, K.V.; Yu, G.; Yakhini, Z.; Kaminski, N. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e33770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, W.E.; Cheng, D.S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Ibarra, G.H.; Kaminski, N. Fibrosis: Lessons from OMICS analyses of the human lung. Matrix Biol. 2018, 68, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune mechanisms in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.G.; Cho, S.J.; Kang, M.J.; Chapoval, S.P.; Lee, P.J.; Noble, P.W.; Yehualaeshet, T.; Lu, B.; Flavell, R.A.; Milbrandt, J.; et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J. Exp. Med. 2004, 200, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Mineo-Kuhn, M.M.; Kramer, C.M.; Finkelstein, J.N. Growth factors alter neonatal type II alveolar epithelial cell proliferation. Am. J. Physiol. 1994, 266, L17–L22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, M.; Saito, A.; Mikami, Y.; Ohshima, M.; Morishita, Y.; Nakajima, J.; Kohyama, T.; Nagase, T. Characterization of human lung cancer-associated fibroblasts in three-dimensional in vitro co-culture model. Biochem. Biophys. Res. Commun. 2012, 423, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Suzuki, H.I.; Horie, M.; Ohshima, M.; Morishita, Y.; Abiko, Y.; Nagase, T. An integrated expression profiling reveals target genes of TGF-β and TNF-α possibly mediated by microRNAs in lung cancer cells. PLoS ONE 2013, 8, e56587. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Ostman, A. Exploring the tumour environment: Cancer-associated fibroblasts as targets in cancer therapy. Expert. Opin. Ther. Targets 2005, 9, 1217–1233. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Noguchi, S.; Yamaguchi, Y.; Ohshima, M.; Morishita, Y.; Suzuki, H.I.; Kohyama, T.; Nagase, T. Differential knockdown of TGF-β ligands in a three-dimensional co-culture tumor-stromal interaction model of lung cancer. BMC Cancer 2014, 14, 580. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Yamanouchi, H.; Fujita, J.; Yoshinouchi, T.; Hojo, S.; Kamei, T.; Yamadori, I.; Ohtsuki, Y.; Ueda, N.; Takahara, J. Measurement of hepatocyte growth factor in serum and bronchoalveolar lavage fluid in patients with pulmonary fibrosis. Respir. Med. 1998, 92, 273–278. [Google Scholar] [CrossRef]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T. cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Osterholzer, J.J.; Marazioti, A.; Stathopoulos, G.T. “Scar-cinoma”: Viewing the fibrotic lung mesenchymal cell in the context of cancer biology. Eur. Respir. J. 2016, 47, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Hotta, K.; Kiura, K.; Takigawa, N.; Yoshioka, H.; Harita, S.; Kuyama, S.; Yonei, T.; Fujiwara, K.; Maeda, T.; Aoe, K.; et al. Comparison of the incidence and pattern of interstitial lung disease during erlotinib and gefitinib treatment in Japanese Patients with non-small cell lung cancer: The Okayama Lung Cancer Study Group experience. J. Thorac. Oncol. 2010, 5, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Kishi, K.; Yoshimura, K. A nationwide survey concerning lung surgery for lung cancer associated with idiopathic interstitial pneumonia. Nihon. Kokyuki. Gakkai. Zasshi. 2011, 49, 148–150. [Google Scholar] [PubMed]
- Sato, T.; Teramukai, S.; Kondo, H.; Watanabe, A.; Ebina, M.; Kishi, K.; Fujii, Y.; Mitsudomi, T.; Yoshimura, M.; Maniwa, T.; et al. Impact and predictors of acute exacerbation of interstitial lung diseases after pulmonary resection for lung cancer. J. Thorac. Cardiovasc. Surg. 2014, 147, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.J.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS® trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, N.; Horie, M.; Suzuki, H.I.; Yoshihara, M.; Djureinovic, D.; Persson, J.; runnström, H.; Lindskog, C.; Elfving, H.; Micke, P.; et al. An integrative analysis of transcriptome and epigenome features of ASCL1-positive lung adenocarcinomas. J. Thorac. Oncol. 2018, 13, 1676–1691. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tamiya, M.; Tamiya, A.; Nakahama, K.; Taniguchi, Y.; Shiroyama, T.; Isa, S.I.; Nishino, K.; Kumagai, T.; Kunimasa, K.; et al. Analysis of early death in japanese patients with advanced non-small-cell lung cancer treated with nivolumab. Clin. Lung Cancer 2018, 19, e171–e176. [Google Scholar] [CrossRef] [PubMed]
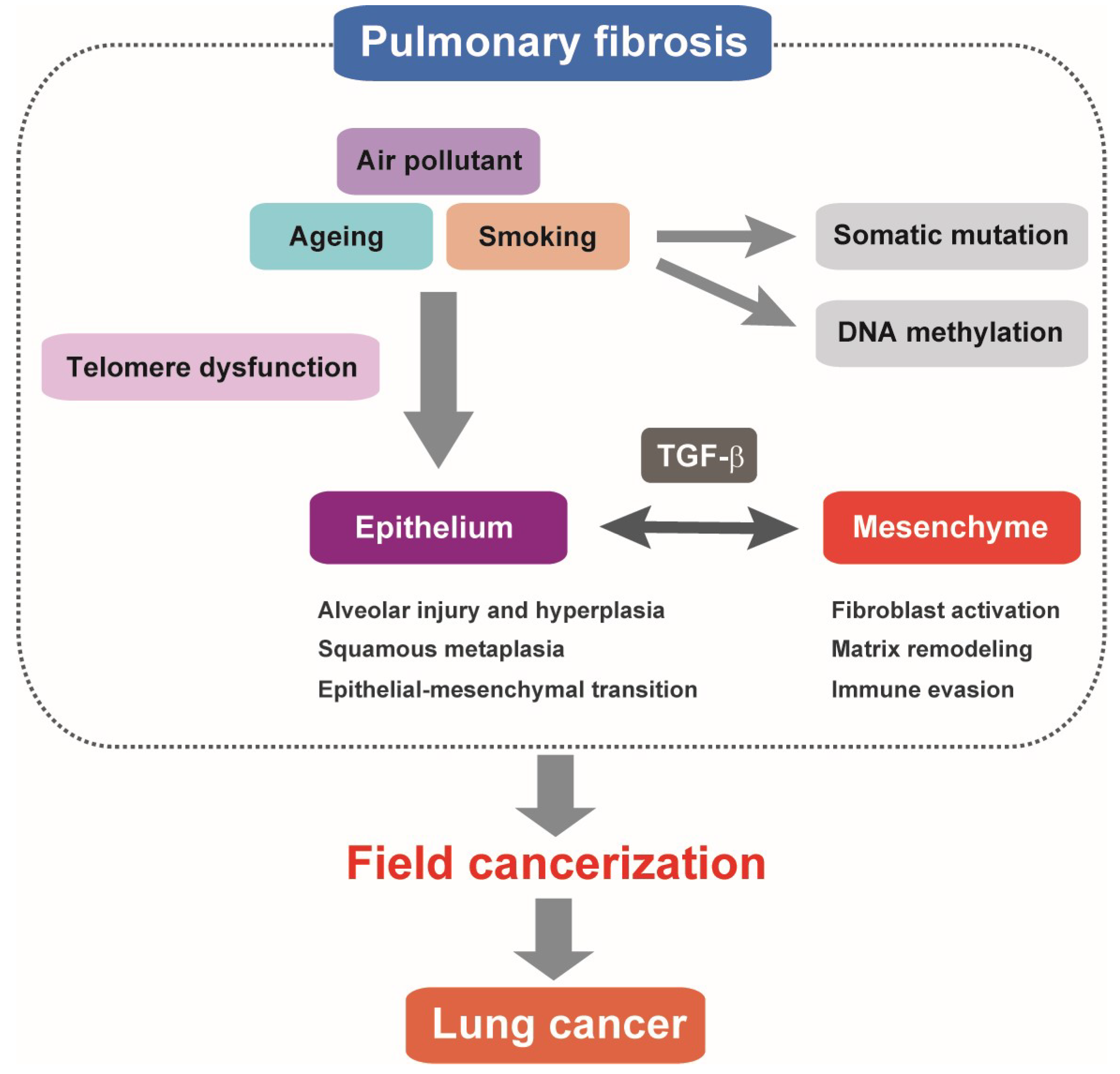
Figure 1.
Pulmonary fibrosis as field cancerization. Alveolar epithelial cell injury and regeneration failure associated with telomere dysfunction represent a hallmark of pulmonary fibrosis. Pathologically activated TGF-β signaling is involved in altered alveolar epithelial cell features and dysregulated epithelial–mesenchymal interactions. It is postulated that somatic mutations and DNA methylation changes accumulating in pulmonary fibrosis multifocally predispose to lung cancer. TGF-β-mediated fibrotic and immune-suppressive microenvironment in the lung tissue of pulmonary fibrosis may have tumor-promoting features similar to lung cancer stroma.
Figure 1.
Pulmonary fibrosis as field cancerization. Alveolar epithelial cell injury and regeneration failure associated with telomere dysfunction represent a hallmark of pulmonary fibrosis. Pathologically activated TGF-β signaling is involved in altered alveolar epithelial cell features and dysregulated epithelial–mesenchymal interactions. It is postulated that somatic mutations and DNA methylation changes accumulating in pulmonary fibrosis multifocally predispose to lung cancer. TGF-β-mediated fibrotic and immune-suppressive microenvironment in the lung tissue of pulmonary fibrosis may have tumor-promoting features similar to lung cancer stroma.
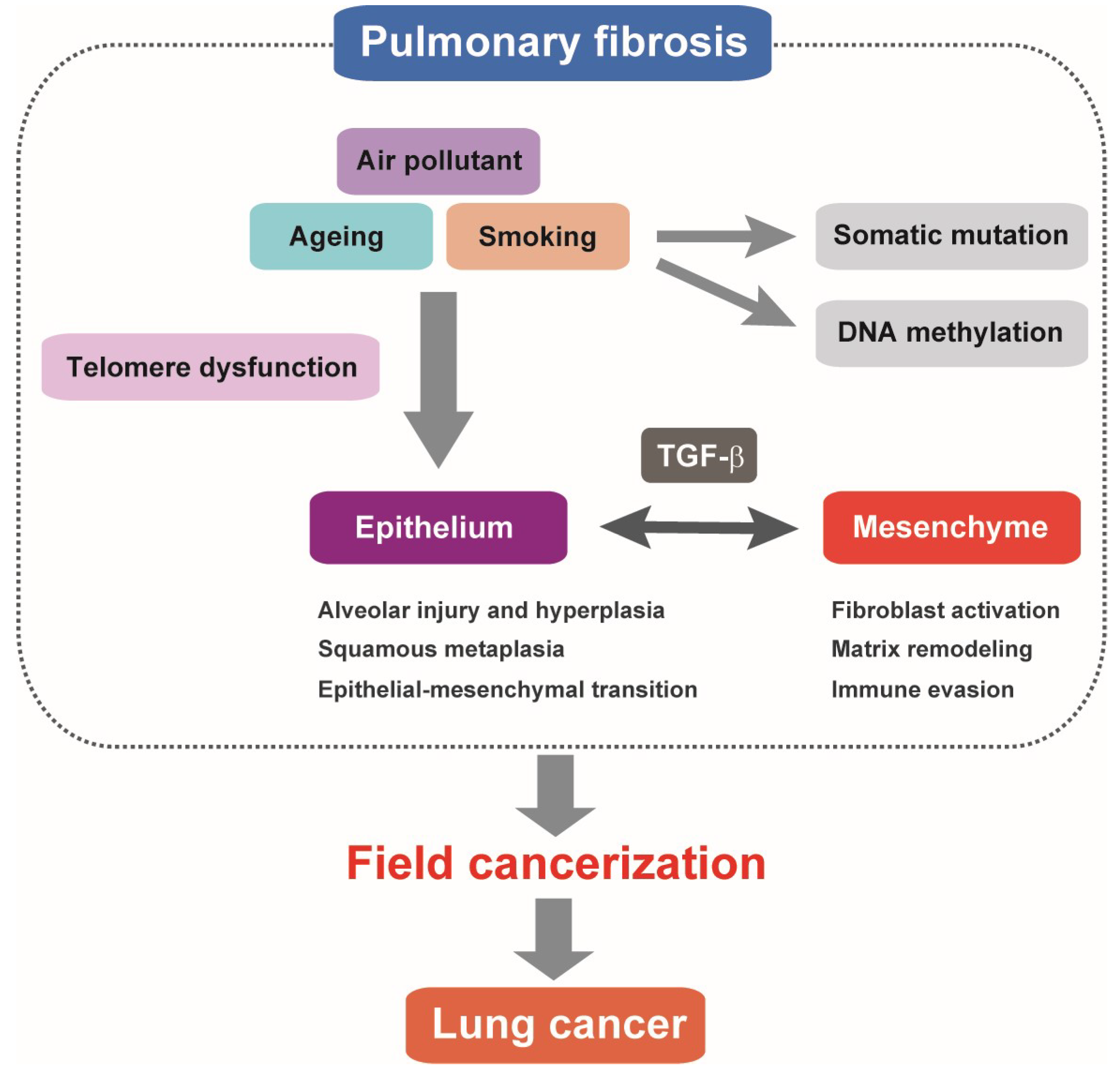
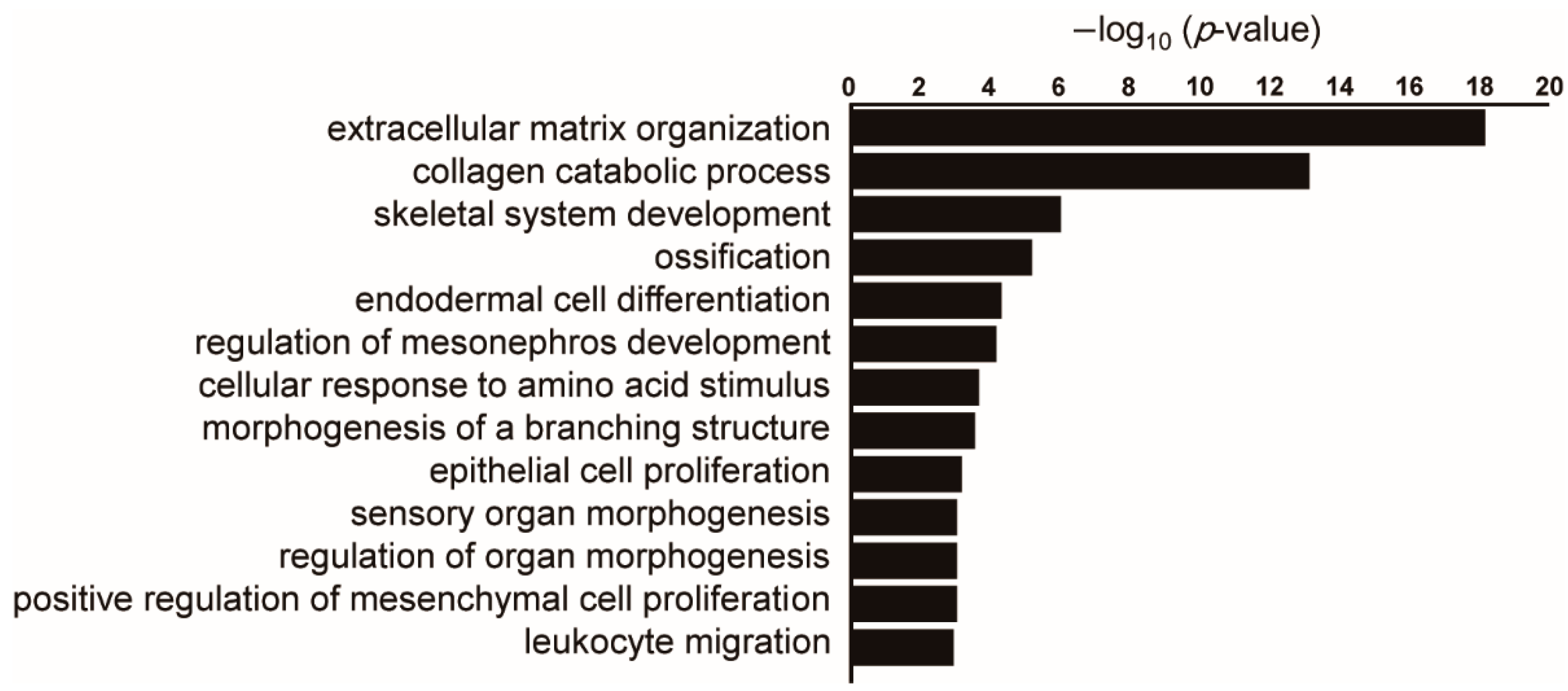
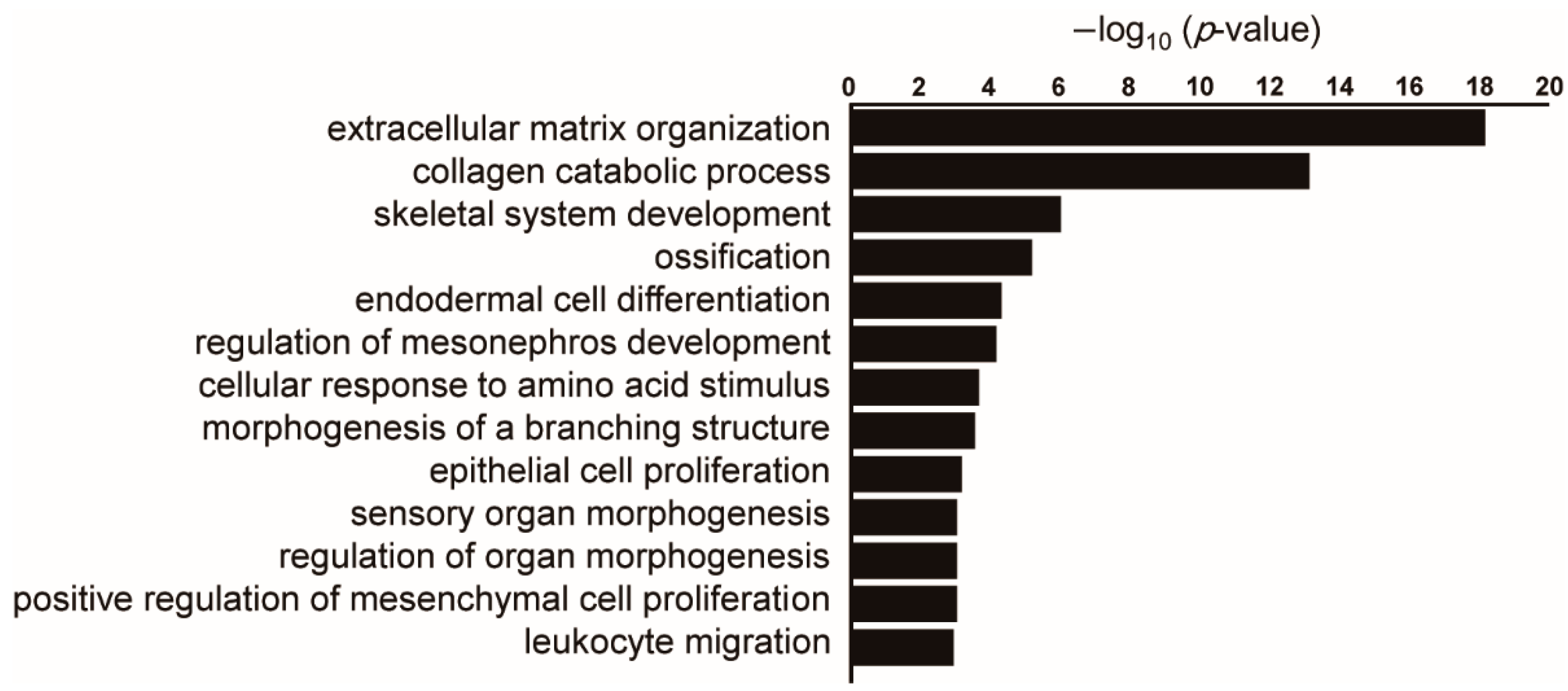
Figure 2.
Pathway analysis of genes commonly upregulated in lung cancer stroma and interstitial pneumonia compared to normal lung tissues. Public datasets of gene expression profiling in lung cancer stroma (GSE22863) and interstitial pneumonia (GSE47460) were compared. When the top 1000 significantly upregulated genes in each dataset were compared, 101 genes were common between these two datasets. Enriched gene ontology terms for biological process were sorted by –log10 (p-value). When there are several similar terms such as “extracellular matrix organization” and “extracellular structure organization”, one term with a lower p-value was selected.
Figure 2.
Pathway analysis of genes commonly upregulated in lung cancer stroma and interstitial pneumonia compared to normal lung tissues. Public datasets of gene expression profiling in lung cancer stroma (GSE22863) and interstitial pneumonia (GSE47460) were compared. When the top 1000 significantly upregulated genes in each dataset were compared, 101 genes were common between these two datasets. Enriched gene ontology terms for biological process were sorted by –log10 (p-value). When there are several similar terms such as “extracellular matrix organization” and “extracellular structure organization”, one term with a lower p-value was selected.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Saito, A.; Horie, M.; Micke, P.; Nagase, T. The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2018, 19, 3611. https://doi.org/10.3390/ijms19113611
AMA Style
Saito A, Horie M, Micke P, Nagase T. The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2018; 19(11):3611. https://doi.org/10.3390/ijms19113611
Chicago/Turabian StyleSaito, Akira, Masafumi Horie, Patrick Micke, and Takahide Nagase. 2018. "The Role of TGF-β Signaling in Lung Cancer Associated with Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 19, no. 11: 3611. https://doi.org/10.3390/ijms19113611
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.