1. Introduction
Gulf War Illness (GWI) is a chronic illness with no known cure and affects some 250,000 veterans that have returned from the first Gulf War fought over 25 years ago. GWI is a complex multisymptom disorder that requires long-term treatment and monitoring, which not only places a great financial burden on the patient, but also on the patient’s family, as well on society. Symptoms of GWI include fatigue, musculoskeletal pain, and gastrointestinal and cognitive dysfunctions [
1]. The symptomatology of GWI can call for patients to take a variety of medications, including over-the-counter pain relievers or prescription stimulants to relieve fatigue [
2]. Furthermore, veterans from the Gulf War may be taking an increasing amount of medications as they age in order to combat age-related comorbid conditions, such as hypertension, hyperlipidemia, or diabetes.
Table 1 provides a list of drugs commonly used to treat GWI symptoms.
The combination of medications is increasingly becoming a concern. Pharmaceutical drugs are ‘promiscuous’, with each binding to an average of at least six molecular targets [
3]. These ‘promiscuous’ drugs tend to be smaller in size [
4], and bind to proteins which share similar gene families [
5]. This has the potential to pose serious unintended consequences, as off-target drug interactions can have deleterious side effects, especially when drugs are used in combination. The potential for adverse drug reactions (ADRs) poses a specific problem for the already taxed systems of patients suffering from GWI, who possess a dysregulation of immune signaling tied to stress and sex hormone levels [
6,
7,
8]. These interactions are a normal part of homeostatic regulation, but a ‘promiscuous’ drug could throw this balance off, exacerbating ADRs.
A major hypothesis of GWI pathophysiology suggests the involvement of a neuroinflammatory cascade that was possibly triggered by multiple toxins experienced in the battlefield and then aggravated further by stress [
9,
10,
11], resulting in altered homeostatic regulation. This neuroinflammatory process is consistent with the broad-ranging multitude of GWI symptoms that extend beyond the central nervous system to affect immune and endocrine function. The body’s key stress regulation system, the hypothalamic–pituitary–adrenal (HPA) axis, links the peripheral immune and endocrine systems to the brain and mediates the response to environmental stressors. In support of this hypothesis, HPA axis dysfunction has been reported in GWI [
12]. It is important to note, however, that activity of the hypothalamic–pituitary–gonadal (HPG) axis is intertwined with that of the HPA axis, as well as the immune system [
13]. For example, the expression of the androgen receptor (AR) has been detected in various immune cell lineages, including neutrophils, mast cells, macrophages, B cells, and T cells [
14,
15], and androgens are known to enhance CD4+, Th1, and CD8+ cell activity [
16].
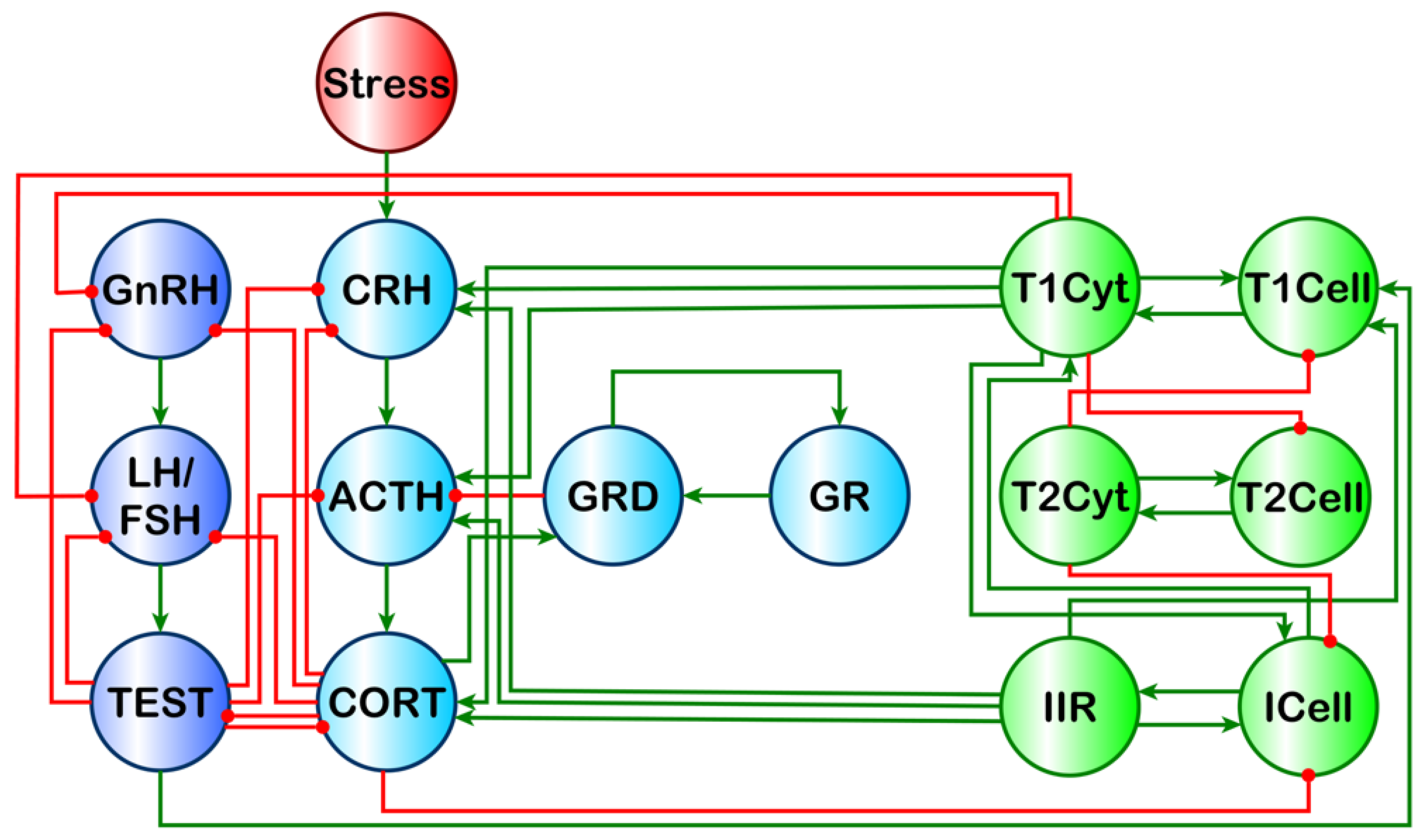
Our previous work aimed to address this issue using a discrete logic model that showed multiple stable homeostatic regulatory behaviors beyond health exist for a simple HPA-HPG-immune network [
7] (
Figure 1). Of these stable regulatory behaviors, the profile of endocrine–immune balance, measured experimentally in a GWI cohort of male subjects, aligned most closely with a state characterized by elevated cortisol, low testosterone, and a shift towards a Th1 immune response. This alignment, however, does not imply that the homeostatic regulatory drive is the sole cause of GWI, but rather suggests that alignment with this alternate stable regulatory behavior acts in part to sustain the chronicity of this complex illness. These alternate homeostatic regulatory regimes are by definition resistant to change and, therefore, could also promote resistance to therapeutic intervention. Thus, these natural regulatory barriers must be compensated for during the design of any treatment avenue to ensure a robust remission from illness. To this end, our simulations predicted that timed treatments inhibiting Th1 cytokines (interleukin (IL)-2, tumor necrosis factor (TNF)-
, or interferon gamma (IFN
)) followed by glucocorticoid receptor (GCR) activity would provide the greatest chance to guide the multiple systems from an altered regulatory state back towards healthy behavior with the minimum number of interventions [
13]. These same simulations also suggested that the addition of an AR agonist may increase the chance of remission.
While the order and general targets of this proposed treatment course have been stipulated, the specific pharmaceutical combination remains open. This leaves the clinician with a difficult choice when designing clinical trials based on the previously predicted combination therapy, as determining the allowable drug combinations to be used in such a multi-tiered intervention strategy is a nontrivial task, especially when considering the multitude of comorbid conditions associated with aging. Due to the tight regulation between the hormonal and immune pathways [
18], the identification of drugs that interact with Th1 cytokines, the AR, and the GCR is required in order to avoid ADRs in such clinical trials. Mifepristone is a known potent GCR antagonist which has previously been used to treat Gulf War veterans with chronic multisymptom illness [
19], and the obvious choice for the treatment of Th1 cytokines are monoclonal antibodies which are cytokine-specific [
20,
21] and may be used to treat chronic inflammatory diseases. That being said, monoclonal antibodies also carry significant risks, such as acute anaphylaxis; serum sickness; and the generation of antibodies [
20,
22], which is of concern for GWI. Suramin is a small molecule alternative for inhibiting TNF-
[
23,
24] and IL-2 [
25]; however, its side effect of potentially inducing adrenal insufficiency [
26] makes it an undesirable choice for this population. From a clinical standpoint, it is key to ensure that there are no small molecules that may interfere with such a risky treatment avenue.
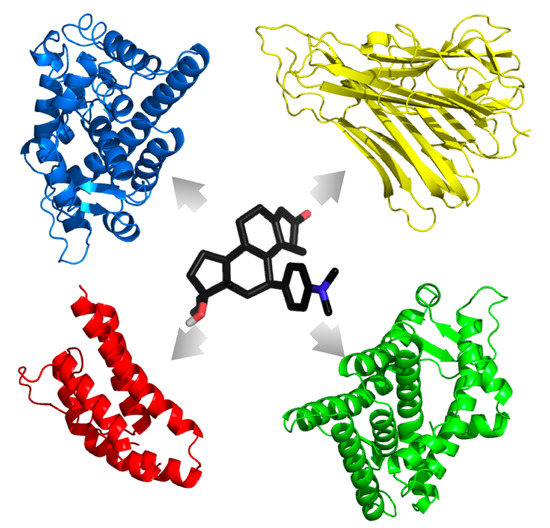
Consistently and reliably predicting the polypharmacologic action of drug agents would be an asset to the healthcare industry for novel drug treatment course development, for the repositioning of United States Food and Drug Administration (FDA)-approved drugs, and for the identification of ADRs. As such, combination treatment design by the clinician should take all precautions to minimize ADRs and off-target interactions, whether for the treatment of a single illness or for the treatment of an illness with comorbid conditions. Here, we characterize FDA-approved drugs commonly used to treat GWI symptoms to find those that have the highest chance of interfering with TNF-
, IL-2, AR, and the GCR in the proposed multidrug GWI treatment course. This was accomplished by performing a consensus docking method using AutoDock 4.2 (AD4) [
27], AutoDock Vina 1.1.2 (VINA) [
28], and Schrödinger’s Glide 2016-4 (GLIDE) [
29] to evaluate potential interactions of 43 FDA-approved small molecule drugs commonly used to treat GWI symptoms (see
Table 1) with multiple crystal structures of the GCR and AR in both agonistic and antagonistic forms, as well as the unbound TNF-
and IL-2 cytokines, representing the stress, male sex, and immune components of our previous models, respectively.
3. Discussion
Nearly 250,000 veterans of the 1990–1991 Persian Gulf War suffer daily with a symptom constellation of fatigue, musculoskeletal pain, and gastrointestinal and cognitive dysfunction collectively known as GWI. While there is no widely accepted biomarker for GWI, and afflicted veterans are commonly diagnosed based only on psychological or psychiatric evaluation, the United States Department of Defense (DoD) is moving to alleviate GWI veterans’ suffering (
http://cdmrp.army.mil/gwirp/default) through the support of basic and applied research, as well as clinical trials evaluating promising treatments. Recent simulation research based on resetting the homeostatic balance has suggested a multi-tiered intervention strategy aimed at inhibiting Th1 cytokines (such as IL-2 or TNF-
) followed by GCR inhibition [
13]. While not a necessity, these same simulations also suggested that the addition of an AR agonist may increase the chance of remission. As clinical trials are currently being developed based on this hypothesized treatment (United States Department of Defense Congressionally Directed Medical Research Program (CDMRP) awards W81XWH-13-2-0085 and GW170044), it is vital for clinicians to be informed about potential interactions with drugs commonly used by veterans with GWI. To this end, the purpose of this work is to identify FDA-approved drugs commonly used to treat symptoms of GWI that may interfere with the proposed Th1 cytokine inhibition, followed by inhibition of the GCR [
13]. This was accomplished by using a virtual screening consensus docking approach with AD4, VINA, and GLIDE to evaluate potential interactions of 43 FDA-approved small molecule drugs with multiple structures of the GCR, AR, TNF-
, and IL-2, representing the stress, male sex, and immune components of our previous models, respectively.
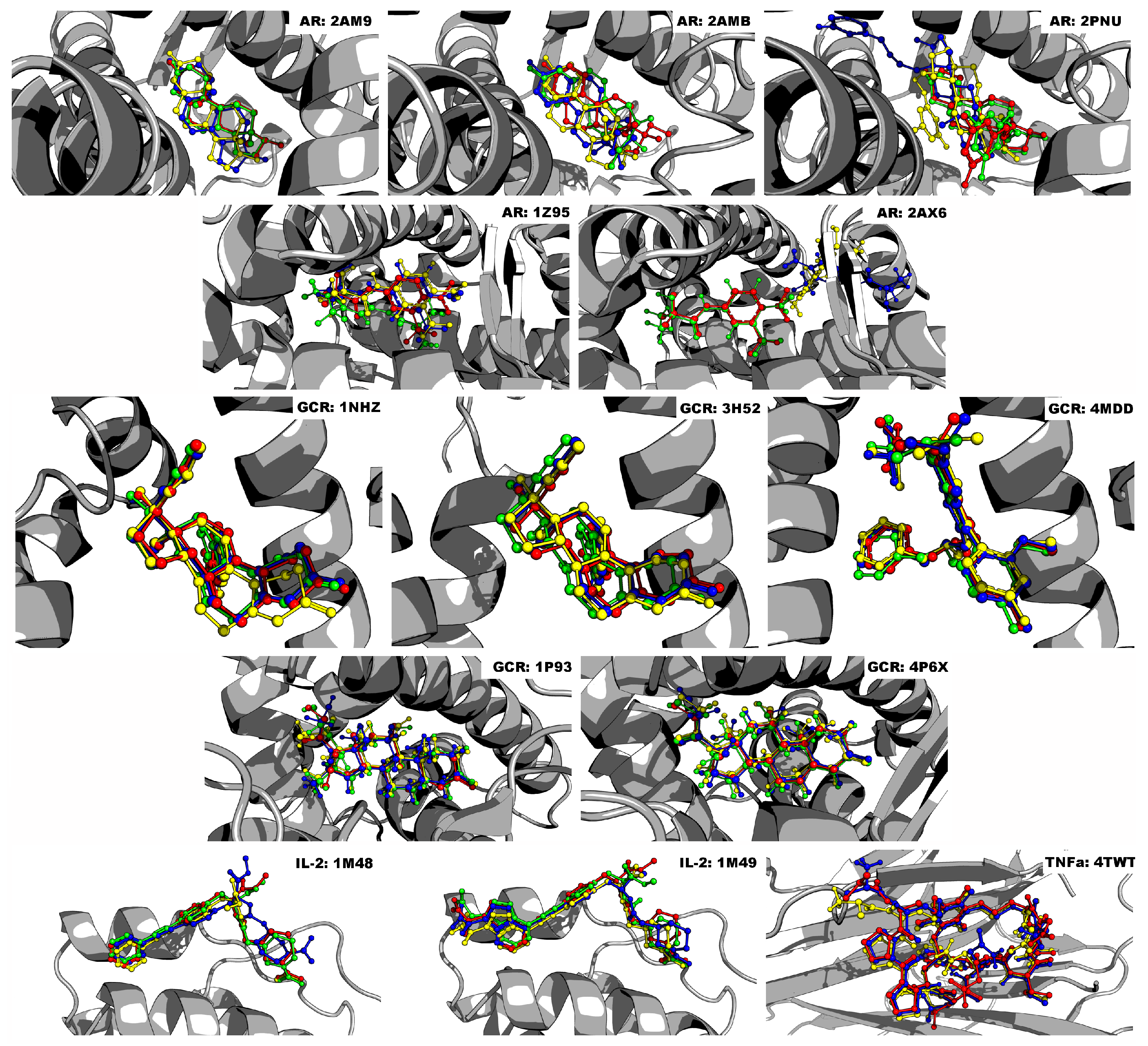
Virtual screening is a standard technique used in the drug discovery pipeline [
30,
36]: drugs are computationally assessed for binding to a specified site on a target based on physicochemical properties in order to determine the ligand’s most likely conformation. The likelihood of the drug–protein interaction is assessed based on the binding energy [
30]. That being said, reliable binding energy prediction can be a challenging and difficult task. Program-specific optimization algorithms and scoring functions can often produce highly variable predictions of likely binding poses and ligand affinities to their targets [
30]. Consensus docking emends this issue by amalgamating numerous scoring algorithms to reduce error and increase prediction accuracy [
30,
37,
38]. Studies have shown that a consensus docking approach is far more reliable and precise than simply using a single docking method [
30,
39,
40]. Houston and Walkinshaw [
30] tested the reliability of such a method, and found that a consensus approach using AD4 and VINA predicted the correct binding pose far more often compared to either of the programs alone. Similarly, other studies have concluded that consensus docking is an improvement over traditional methods for the identification of novel drugs [
41,
42].
Our main goal was to gauge any potential for interaction of FDA-approved drugs commonly used to treat symptoms of GWI with targets of a proposed multi-intervention treatment strategy for GWI, with the goal to inform the clinician of which drugs to avoid. Clinically, in this case, it is better to err on the side of making false positive predictions, rather than false negatives, as it is better to predict an interaction and exclude the use of a drug with no effect than include one that has interactions. This is opposed to standard rational drug discovery, which aims to rule out all false positives for the purpose of lead development, as including a false positive adds to the overall cost of drug testing and design. Here, we followed the latter principle to ensure the reliability of our predictions at the cost of limiting our overall predictions. However, we provide our results so that erring on the side of false positive predictions can be instituted by allowing more inclusive cutoff criteria (see
Supplementary_Tables.xlsx).
Another limiting factor in the standard consensus docking post-processing approach is how to filter results. Filtering is commonly performed by calculating the RMSD between a predicted ligand’s docked pose and the experimental gold standard conformation [
30,
43,
44,
45]. If the two poses differ by more than 2.0 Å, then the ligand is removed from further consideration. This method is only useful when the crystallographic pose is available to compare with, and in the case of consensus docking, the RMSDs between each program’s pose are compared. However, if they differ and no crystallographic pose exists, then it is impossible to know which one is correct [
30]. Other issues of filtering include too lenient or stringent criteria. Garcia-Sosa and Maran [
37] only retained drugs whose binding energy for each docking program was comparable to or lower than the known binder’s energy. This method becomes increasingly exclusive as more docking programs are added. Excessive filtration is especially harmful when important interactions between ligands and targets are overlooked, as in the case of ADRs. For this reason, we opted to include all docking results in the form of a mean binding energy with its standard deviation. The standard deviation provides a measure of consensus across the multiple receptor–program combinations, with a smaller standard deviation indicating better consensus.
The many symptoms of GWI can call for patients to take a variety of medications, such as pain relievers or prescription stimulants to relieve fatigue [
2]. Additionally, patients may be taking medications for common comorbidities, such as hypertension and hyperlipidemia. To evaluate the potential for such medications to interact with the proposed multi-intervention Th1-GCR inhibition strategy, we examined the interaction of medications associated with GWI and common comorbidities on the proposed targets of the combined HPA-HPG-immune model. Common pain relief medications include ibuprofen, acetaminophen, diclofenac, and naproxen, among others [
2,
35]. Atenolol treats high blood pressure or hypertension, clofibrate is a medication for high cholesterol or hyperlipidemia, and carbamazepine is used to prevent and control seizures [
35]. In regard to fatigue and/or cognitive dysfunction, amantadine, modafinil, nimodipine are all drugs commonly used for the treatment of these symptoms [
2].
In regard to the proposed multi-intervention Th1-GCR inhibition strategy, mifepristone was only found to interact with the GCR in antagonist form, as expected, confirming that it is a reliable, selective GCR antagonist. Testosterone, a suggested supplement to the strategy [
13], was found to bind most strongly to the AR in agonist form, as expected, but also showed potential interaction with the AR in antagonist form, as well as the GCR in agonist form. The former may be due to the poorer performance of the AR antagonist predictions, as evidenced by its relatively low PPV. However, since the binding energy of testosterone to the AR in agonist form is much lower than to the antagonist form, this also suggests that if bound to the antagonist form, it may induce a structural change to the agonist form, consistent with expectations. The interaction of testosterone with the GCR in the agonist form, however, cannot be so easily explained. The GCR agonist prediction showed a high PPV of 94.57%, leaving a small chance that the prediction is a false positive. However, it must also be noted that there is evidence that testosterone interferes with dexamethasone binding to the glucocorticoid receptor [
46]; however, this is only a weak interaction with the GCR, which is unable to form all the amino acid contacts necessary to yield a stable, transcriptionally active GCR conformation [
47]. Our predictions may be reflecting this weak interaction.
In regards to the 43 commonly used medications to treat GWI symptoms, our results suggest that 10 show real potential to interfere with the proposed Th1-GCR multi-inhibition strategy. These include trazadone, buspirone, and carbamazepine with putative interference of AR agonist activity, and buspirone, proxetine, trazodone, nefazodone, sertraline, valaciclovir, rofecoxib, gabapentin, and baclofen with putative interference of TNF-
activity. Use of these drugs should be considered exclusionary criteria for any trial of the Th1-GCR multi-inhibition strategy. Due to the low PPV for the AR antagonist, the potential interactions of the 43 drugs were not considered reliable. However, as naproxen, diclofenac, ibuprofen, and clofibrate were predicted binders to the AR in antagonist form and have been shown to have AR antagonizing effects [
35], use of these medications should also be considered as exclusionary criteria in any trial, especially those using a testosterone supplement. While no drugs were found to be statistically comparable to mifepristone binding to the GCR, or with suramin to IL-2, both mifepristone and testosterone were shown to have putative interference with suramin’s effect on TNF-
. As such, it is also not advised to combine suramin with mifepristone or testosterone in the proposed Th1-GCR multi-inhibition strategy. Interference with the AR or TNF-
activity poses a greater concern for such a treatment strategy. The majority of the drugs predicted to have a putative interference with the multi-intervention strategy include medications to treat anxiety and depression. As there has been shown to be a link between inflammation, anxiety, and depression [
48], excluding individuals taking these medications from the multi-intervention Th1-GCR inhibition strategy would be prudent. However, it must be noted that the predictions presented in this work have not been experimentally validated. While docking studies such as these can provide useful information about drug–target interaction, they must be experimentally verified to ensure the accuracy of the predictions made. That being said, our goal here is to provide clinicians moving ahead with the proposed Th1-GCR multi-inhibition strategy a gauge for potential drug interactions. It is better to err on the side of caution in this regard.
Our study focused on the prime targets involved in a putative multi-tiered intervention strategy aimed at inhibiting Th1 cytokines (such as IL-2 or TNF-
) followed by GCR inhibition as a first-round assessment of inclusion/exclusion criteria for clinical trials. However, the study is by no means comprehensive. The targets chosen here only represent only a fraction of the HPA-HPG-immune systems. There are many more enzymes, receptors, and targets involved, including multiple immune targets, such as other cytokines and chemokines; other steroid receptors, such as estrogen receptors and mineralcorticoid receptors; as well as other nuclear receptor family members, such as thyroids, PPAR-
, and liver-X, to name a few. The methods used in this work are not immutable, and may be expanded upon. Docking programs, crystal structures, and drug databases may be substituted or used in addition to our chosen set to account for additional targets, as well as agonist/agonistic forms of the receptors involved. There are no limits to the number of docking programs used; in fact, increasing the number of programs decreases the variance. Our post-processing protocol removes only the most extreme values, such that ligands are ordered based on their average energy. The modularity of the pipeline allows us to investigate a multitude of targets that each implicate numerous pathways without ignoring off-target effects. In this vein, we provide our full dataset in the XLSX file format (see
Supplementary_Tables.xlsx) and encourage others to continuously add results from additional docking programs, crystal structures, and additional drugs. This information is not illness-specific and can serve to benefit a multitude of complex diseases by mapping each drug’s interactivity.

 ,
, 



{kind=link}
{kind=link}
{kind=link}