HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers

1
State Key Laboratory of Chemical Resource Engineering, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China
2
Key Laboratory of Molecular Pathology, School of basic medical science, Inner Mongolia Medical University, Hohhot 010110, China
3
Key Laboratory of Receptors-Mediated Gene Regulation and Drug Discovery, School of Medicine, Henan University, Kaifeng 475004, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2018, 19(11), 3295; https://doi.org/10.3390/ijms19113295
Submission received: 13 September 2018
/
Revised: 10 October 2018
/
Accepted: 15 October 2018
/
Published: 23 October 2018
(This article belongs to the Special Issue Hepatocyte Growth Factor (HGF))
Abstract
:The HGF/c-MET pathway is active in the development of digestive system cancers, indicating that inhibition of HGF/c-MET signaling may have therapeutic potential. Various HGF/c-MET signaling inhibitors, mainly c-MET inhibitors, have been tested in clinical trials. The observed efficacy and adverse events of some c-MET inhibitors were not very suitable for treating digestive system cancers. The development of new HGF/c-MET inhibitors in preclinical studies may bring promising treatments and synergistic combination (traditional anticancer drugs and c-MET inhibitors) strategies provided anacceptable safety and tolerability. Insights into miRNA biology and miRNA therapeutics have made miRNAs attractive tools to inhibit HGF/c-MET signaling. Recent reports show that several microRNAs participate in inhibiting HGF/c-MET signaling networks through antagonizing c-MET or HGF in digestive system cancers, and the miRNAs-HGF/c-MET axis plays crucial and novel roles for cancer treatment. In the current review, we will discuss recent findings about inhibitors of HGF/c-MET signaling in treating digestive system cancers, and how miRNAs regulate digestive system cancers via mediating HGF/c-MET pathway.
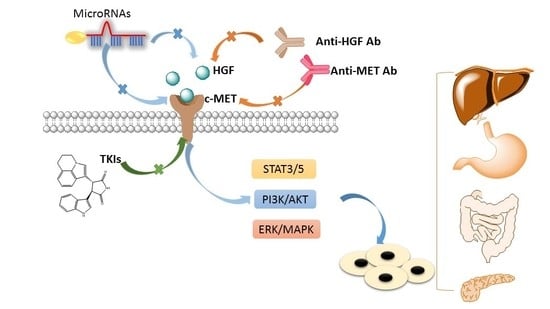
1. Introduction
Digestive tract malignancies, mainly including gastric cancer, hepatocellular carcinoma (HCC), pancreatic cancer, esophageal cancer, and colon and rectal cancer, are highly common cancers and causes of cancer deaths worldwide [1]. Despite surgery, chemotherapy and molecular targeted therapy have been widely used in cancer treatment, median survival for most stage IV digestive cancers is <1 year, with the exception of small bowel and colorectal adenocarcinoma [2]. Thus, it was essential and imperative to explore and identify novel and effective therapeutic targets for the prevention and treatment of digestive system cancers.
The c-mesenchymal–epithelial transition (c-MET), a kinase receptor for hepatocyte growth factor (HGF), is well-known for its roles in driving tumorigenesis [3,4,5]. It is a disulfide-linked heterodimer consisting of a highly glycosylated extracellular α-subunit and a transmembrane β-subunit. Upon binding to the HGF, c-MET triggers dimerization of two subunits, leading to autophosphorylation of tyrosine residues in cytoplasmic domain [4,6,7]. Then, phosphorylation of these tyrosine residues (Tyr1349 and Tyr1356) results in an activated C-terminal docking site, which has been identified to be able to recruit intracellular adaptor proteins [8]. These events trigger several downstream signaling pathways such as phosphoinositide 3-kinase/threonine-protein kinase (PI3K/AKT) pathway, wingless-related integration site (Wnt) pathway, and others [6,9,10]. Moreover, HGF/c-MET induced cell proliferation, migration, survival, invasion, differentiation, and epithelial–mesenchymal transition (EMT), promoting the progression of tumorigenesis [4,11,12]. These common downstream signaling pathways for HGF/c-MET activation has summarized in Figure 1.
The HGF/c-MET receptor tyrosine kinase (RTK) pathway is quiescent in normal tissue, while it is active in various tumors [13]. Growing investigations have confirmed that inhibition of HGF/c-MET signaling is an effective therapeutic strategy in suppressing multiple human cancers, such as non-small cell lung cancer (NSCLC), HCC, gastric cancer, colorectal cancer ovarian cancer, bladder cancer, head and neck cancer, cervical cancer, and some other cancers [4,7,8,14,15,16,17,18,19,20]. In preclinical and clinical trials, it has demonstrated that the inhibitors of c-MET have antitumor activity in treatment of NSCLC. And for EGFR-TKIs (epidermal growth factor receptor-tyrosine kinase inhibitors) resistant and EGFR-TKI naive NSCLC patients, combination use of c-MET inhibitors and EGFR-TKIs (EGFR inhibitors) may be considered as a promising treatment option [20]. In the past years, many c-MET inhibitors have being discovered and developed to suppress tumor [21,22,23,24,25]. For example, Balan et al. found that Honokiol inhibited calcineurin inhibitor-induced renal tumor growth by suppressing c-MET-Ras-HO-1 axis [23]. Therefore, finding effective c-MET inhibitors and understanding the regulation of HGF/c-MET pathway are crucial for further inhibiting prolonged activation of this pathway in human cancers, including digestive system cancers.
In 1993, first microRNA (miRNA) was discovered and identified, then functional studies of miRNA has been expanded in recent years [26,27]. Importantly, the roles of miRNAs in different tumor progression have been reported [28]. Several publications have reported that several miRNAs inhibit digestive system cancer progression through targeting c-MET [29,30,31,32]. For example, Korhan et al. revealed that restoration of miR-181a-5p significantly suppressed hepatocellular carcinoma via directly targeting c-MET [31]. In the future, the miRNAs-HGF/c-MET axis could serve as a potential target for digestive system cancer treatment.
In the current review, the physiological and pathological mechanisms of the aberrant HGF/c-MET in digestive system cancers have been described. Specifically, the new therapies using c-MET inhibitors have been highlighted and some special miRNAs that mediate c-MET in partial digestive system cancers have been summarized.
2. HGF/c-MET Inhibitors in Digestive System Cancers
Multiple inhibitors of HGF/c-MET signaling have shown antitumor activity in the preclinical phase of digestive system cancers [33,34]. However, the clinical efficacy of some inhibitors is limited, and some novel HGF/c-MET signaling inhibitors or medication strategies are being developed for digestive system cancers. Clinically relevant examples and some new potential drugs in preclinical trials are listed in Table 1.
3. Inhibitors of c-MET/HGF Signaling and Hepatocellular Carcinoma (HCC)
HCC is a significant leading cause of cancer-related death in the world [35]. The therapeutic efficacy of conventional chemotherapy is limited, and five-year survival for advanced HCC is <10% [36]. Sorafenib, a multi-kinase inhibitor (Raf serine/threonine kinases and receptor tyrosine kinases), is the only established standard treatment for advanced HCC [37]; the survival benefit from sorafenib is limited and novel effective therapeutic options need to be developed [36].
Tyrosine kinase receptor c-MET, as a mitogenic growth factor for hepatocytes, has been studied HGF/c-MET signaling increases hepatocyte proliferation and tissue remodeling [35,36]. In addition, c-MET is overexpressed at transcriptional and protein levels in HCC [36,38,39]. c-MET inhibitors and multi-kinase inhibitors have been studied for treatment of HCC.
Tepotinib MSC2156119J, highly selective ATP-competitive c-MET inhibitor, has been reported to suppress c-MET-positive HCC tumor in vivo [40]. In Bladt et al.’s research, antitumor effects of MSC2156119J and sorafenib in HCC were compared and evaluated [33]. On the one hand, MSC2156119J effectively inhibits c-MET-positive HCC tumor growth, and also diminished the number of metastatic foci of lung. On the other hand, MSC2156119J monotherapy had superior efficacy compared with sorafenib monotherapy, and the antitumor activity of the combination of MSC2156119J and sorafenib was not superior to MSC2156119J monotherapy In addition, MSC2156119J was also an effective inhibitor for HBV (hepatitis B virus) [33]. Previous reports show that upregulation of HGF is responsible for HBV-induced HCC progression. MSC2156119J is capable of suppressing the HBV-induced HCC possibly through targeting HGF/c-MET signaling [41]. The efficacy of nonclinical studies showed that tepotinib was a promising therapeutic drug in c-MET-positive HCC tumor. In phase Ib trials, tepotinib shows antitumor activity in Asian patients with advanced HCC, including patients with c-MET-positive tumors [42]. Currently, the single-arm phase II trials of tepotinib are continuing in Caucasian HCC patients who are c-MET-positive and sorafenib-insensitive [43]. Overall, tepotinib MSC2156119J shows antitumor activity in nonclinical studies and phase Ib trials in c-MET-positive HCC patients. These results provide evidence for developing a promising antitumor drug for HCC patients.
Tivantinib ARQ197, a non-ATP competitive selective small-molecular inhibitor, not only targets c-MET, but also targets cyclin B1, the proteasome, and glycogen synthase kinase 3 in cancer [44]. In phase I trials, tivantinib shows antitumor activity via suppressing c-MET signaling, promoting apoptosis and inhibiting angiogenesis. Importantly, the safety and tolerability of this drug is qualified. However, the hematological toxicity (mainly neutropenia) of tivantinib was observed in phase II [45]. In Santoro et al.’s phase II trials, 97 patients with advanced HCC and Child-Pugh A cirrhosis were randomly assigned in tivantinib group (360 mg twice-daily or 240 mg twice-daily, capsule) and placebo group in a 2:1 ratio [46]. The median time to progression was longer in the tivantinib group than in the placebo group. For patients with c-MET-positive, the efficacy is more obvious. Inadequately, 10 patients had neutropenia and eight patients had anemia in the tivantinib group, while no patients had neutropenia or anemia in the placebo group. The most common adverse events were more frequent in patients with higher dose tivantinib, and four patients died of severe neutropenia [46]. The antitumor efficacy of tivantinib is promising, however, the dose should be carefully tested and examined and adverse events should be reduced. Recently, in randomized, double-blind and placebo-controlled phase III study, Rimassa et al. reported that tivantinib (120 mg twice daily, tablet) did not improve overall survival in patients with MET-positive advanced HCC [47]. The negative trial might be attributed to tivantinib formulation and dose or adverse events of tivantinib.
SU11274, another small molecular c-MET inhibitor, may inhibit HCC cell growth via suppressing c-MET activation [48]. However, the study about it is only in vitro, and the data of animal models and clinical studies are necessary to be further studied. C-MET inhibitor combined with sorafenib is a potential strategy for HCC treatment. A novel c-MET inhibitor DE605 plus sorafenib may effectively induce HCC cell apoptosis in vitro and suppress HCC tumor xenografts in vivo. On the one hand, DE605 in combination with sorafenib improves therapeutic efficacy (inhibiting proliferation and inducing apoptosis) for HCC. On the other hand, sorafenib inhibits DE605-mediated ERK (extracellular regulated protein kinases) activation [49]. C-MET inhibitor DE605 combined with VEGF inhibitor sorafenib may enhance efficacy and reduce side effects. Therefore, drug combination may be a better strategy for HCC treatment.
Medicinal peptide LZ8, extracted from the Chinese herbal drug Ganoderma lucidum, has antitumor activity in breast cancer, lung cancer, cervical cancer, and HCC [50,51,52,53]. Unlike other c-MET inhibitors, LZ8 prevents the tumor progression in HCC without c-MET dependent [53]. As a Chinese herbal ingredient, the safety of the drug has been certified, however, it is still required for clinical trials to verify safety and efficacy in HCC patients.
Other c-MET inhibitors, such as cabozantinib capmatinib, golvatinib, and foretinib, also have been reported for HCC treatment [54,55,56,57,58]. All of these c-MET inhibitors show antitumor activity in nonclinical studies in HCC. However, the efficacy and safety of these drugs is unpredictable. Firstly, side effects (liver and bone marrow toxicity, neutropenia, and anemia) caused by c-MET inhibitors have been frequently reported [53]. Secondly, HCC is a heterogeneous disease, individual c-MET-based target therapy has limited benefit for HCC patients, especially for patients with c-MET-negative. Finally, advanced HCC usually is accompanied with fibrosis and cirrhosis, which reduces phase I and phase II metabolic enzyme activity and hepatic drugs clearance [59].
4. Inhibitors of c-MET/HGF Signaling and Gastric Cancer
In gastric cancer, c-MET expression elevation also is a poor overall survival marker, comparing with c-MET-negative tumors [60,61]. Several c-MET inhibitors have been developed as anticancer drugs in gastric cancer treatment.
Tivantinib ARQ197 has been investigated in several clinical trials in different cancers, including NSCLC, HCC and metastatic gastric cancer. In an open-label, multicenter trial phase II, 31 Japanese and Korean patients with metastatic gastric cancer were enrolled; 11 patients achieved disease control [62]. However, the adverse events were still serious.
A Phase I trial of another c-MET inhibitor SAR125844 enrolled 22 Asian patients with gastric cancer; it showed modest antitumor function in two patients with MET-positive gastric cancer, adverse events were common in these patients [63]. Similar to the clinical situation of HCC treatment, most of the c-MET inhibitors have limited benefit for treatment of cancer because of adverse events and c-MET-positive limitation. More and more c-MET inhibitors have been found to have antitumor activity in gastric cancer, but many drug studies have only stayed in nonclinical stage.
Gavine et al. reported that 6% of c-MET genes were amplified and 13% of proteins were overexpressed in Chinese gastric cancer patient tumors [64]. The highly selective c-MET small molecule inhibitor volitinib is capable of inhibiting proliferative activity of gastric cancer cells via avoiding phosphorylation of c-MET, and thus inhibiting the activation of c-MET signaling and downstream pathway (AKT and ERK). Volitinib shows antitumor efficacy in patient-derived tumor xenograft models [64].
Toiyama et al. have shown c-MET was predominantly expressed in the transmembrane area, while HGF was mainly expressed in the cytoplasm of primary gastric cancer cells [65]. Importantly, they confirmed overexpression of HGF and c-MET are correlated with peritoneal dissemination and poor prognosis in gastric cancer. The c-MET inhibitor SU11274 may suppress peritoneal mass in gastric cancer nude xenograft model [65]. In parallel, Yashiro et al. studied the efficacy of the c-Met inhibitor SU11274 in combination with irinotecan and found that they have superior efficacy in suppressing in vivo tumor growth by SP-enriched cell lines (OCUM-2M/SP cells) than either group monotherapy [66]. In terms of mechanisms, SU11274 inhibited c-MET signaling and thus decreased the expression of uridine 50-diphosphate-glucuronosyltransferase 1A1 (UGT1A1), which was related with drug resistance to irinotecan [66]. Desirably, the clinical effects of SU11274 monotherapy and combined SU11274 with irinotecan need to be further explored.
Moreover, other selective small molecule c-Met inhibitors such as KRC-408, KRC-00715, and Simm530, and multitargeted kinase inhibitor T-1840383, suppressed the growth of gastric cell lines and tumor xenograft [21,67,68,69]. Also, KRC-00715 and Simm530 suppressed specifically the proliferation of c-MET-overexpressed gastric cells [21,67]. Consistently, these c-MET inhibitors suppress c-MET phosphorylation and its constitutive downstream effectors AKT and ERK [21,67,68,69].
5. Inhibitors of c-Met/HGF Signaling and Colorectal Cancer (CRC)
CRC is the fourth most common cancer in the world [70]. High expression of c-MET is associated with tumor invasion and lymph node and hepatic metastasis in CRC [71]. Growing data suggest suppression of c-MET activation may inhibit tumor activity for patients with colorectal carcinoma.
It has been mentioned that SU11274 may suppress tumor cell growth in HCC and gastric cancer via specifically inhibiting c-MET phosphorylation, and the antitumor function also been observed in CRC [48,65,66,72,73]. Gao et al. reported that SU11274 showed inhibitory effects on cell proliferation in four colon cancer cell types [73]. In parallel, Gao et al. described SU11274 induced G1-phase arrest and restrained cell survival in vitro, and suppressed the growth of the xenograft tumor in vivo [72]. In these studies, the clinical efficacy and safety of SU11274 is still unclear. So further studies are necessary to explore the effects of SU11274 in colorectal cancer patients.
In addition to SU11274, the clinical effects of tivantinib have been explored in several human cancers, including CRC. In a placebo-controlled and phase 1/2 study, Cathy et al. reported the efficacy of tivantinib combined with irinotecan and cetuximab in patients with KRAS (kirsten rat sarcoma viral oncogene) wild-type metastatic colorectal cancer. However, the progression-free survival has not been significantly improved [74]. Tivantinib is probably not suitable for this CRC subgroup.
Commonly, c-MET is co-present with EGFR in 78% to 80% in CRC [70]. Qiu et al. revealed that Norcantharidin (NCTD) suppressed cell proliferation and induced G2/M phase arrest via decreasing the levels of the total EGFR, the activated EGFR, the total c-MET, and the activated c-MET in colon cancer cells (HCT116 and HT29 cells) [70]. As a c-MET inhibitor, NCTD is the demethylated analogue of Cantharidin and has stronger antitumor activity and much less urological toxicity than Cantharidin [70,75]. Therefore, NCTD may be a promising therapeutic drug and its clinical effect and safety is expected.
Mounting evidence supports the notion that c-MET inhibitors play significant roles in human cancers, including CRC. The reports of HGF inhibitors are relatively rare. Owusu et al. demonstrated that an inhibitor of HGF (SRI 31215) inhibited fibroblast-induced MET activation and DU145 cell EMT and migration. Meaningfully, SRI 31215 overcomes autocrine HGF/MET signaling-mediated the primary resistance to EGFR inhibitors (cetuximab and panitumumab) in colon cancer cells [76]. SRI 31215 might be a useful drug candidate to colorectal cancer patients, in combination with EGFR inhibitors, may improve drug efficiency and overcome drug resistance.
Overall, although the studies of HGF/c-MET inhibitors are still in the preclinical stage, they may serve as a potential direction for CRC treatment.
6. Inhibitors of c-Met/HGF Signaling and Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies in pancreatic cancer [77]. The frequency of c-MET overexpression in PDAC is generally high (80%), and c-MET levels have been considered as an indicator for overall survival and recurrence rates for PDAC patients [78]. Hage et al. reported that c-MET inhibitor cabozantinib increases gemcitabine efficacy and gemcitabine resistance in pancreatic cancer [77]. On the one hand, combination of gemcitabine and cabozantinib exhibits more pronounced antitumor efficacy than monotherapy. On the other hand, cabozantinib overcomes gemcitabine resistance of pancreatic cancer cells by inducing apoptosis and suppressing total c-MET, the phosphorylated c-MET and reprogramming transcription factor SOX2 [77].
Crizotinib, a c-MET/ALK inhibitor, shows antitumor activity in pancreatic cancer cells via suppressing growth and inducing apoptosis. However, Yan et al. considered that antitumor activity of crizotinib was attributed to targeting ALK signaling not c-MET in pancreatic cancer [78]. In parallel, Avan et al. testified synergistic efficacy of crizotinib–gemcitabine and found that PDAC-3-FM-GC tumor growth was significantly inhibited in the combined drugs-treated mouse group compared with that in treated with crizotinib alone group and gemcitabine alone group [79]. Cytidine deaminase activity contributes to inactivation of gemcitabine, and would reduce the therapeutic efficacy. Notably, crizotinib elevated gemcitabine levels in plasma and tissue specimens via inhibiting cytidine deaminase activity [79]. Synergistic combination of crizotinib and gemcitabine may be an attractive therapeutic direction for PDAC but clinical evaluation is necessary for it to be further investigated. In addition, synergistic combination of tivantinib and gemcitabine also show antitumor activity in cells highly expressing c-MET in pancreatic cancer [80].
Further studies will be needed to verify the pharmacodynamics and safety in clinical trials. And synergistic combination of drugs with c-MET inhibitors may be promising therapeutic strategies in treating human cancers, including PDAC.
7. Monoclonal Antibodies against HGF/c-MET in Digestive System Cancers
Several anti-HGF and anti-c-MET monoclonal antibodies direct against extracellular combination of c-MET and HGF have been developed for the inhibition of c-MET-mediated digestive system tumor. Rilotumumab (AMG 102) is a humanized IgG2 monoclonal antibody that selectively binds to HGF [81]. A randomized, placebo-controlled, double-blind phase II clinical trial for patients with advanced gastric or oesophagogastric junction cancer, revealed that rilotumumab plus ECX (epirubicin, cisplatin, and capecitabine) has greater antitumor activity than placebo plus ECX. However, the adverse events were more common in rilotumumab group than that in placebo group [82]. In Catenacci et al.’s investigation, a findings in the clinical phase 3 study (RILOMET-1) showed rilotumumab plus ECX has no effect on improving overall survival for advanced MET-positive gastric or gastro-esophageal junction cancer, and the adverse event frequency was flat with the placebo group [83]. Based on the above research, anti-HGF antibody rilotumumab is not effective on treating of advanced MET-positive gastric cancer.
Onartuzumab is a humanized monoclonal antibody that binds to the c-MET extracellular domain. And onartuzumab has been evaluated in phase III trials in HER2-negative, MET-positive gastroesophageal adenocarcinoma. The combination of first-line mFOLFOX6 (fluorouracil, leucovorin, and oxaliplatin) did not reveal a noteworthy improvement in progression-free survival and overall survival [84]. Similarly, in a randomized double-blind phase II study, Bendell et al. reported that onartuzumab plus mFOLFOX6 and bevacizumab did not significantly improve clinical outcome in metastatic colorectal cancer populations [85].
Furthermore, preclinical studies suggest that anti-c-Met monoclonal antibodies ABT-700 and LY2875358 show antitumor efficacy in solid tumors, including gastric cancer. Both ABT-700 and LY2875358 show preclinical activities via blocking HGF-dependent and HGF-independent MET activation [86,87]. Wang et al. conveyed that ABT-700 showed greater effect on SNU620 cell model in vivo and in vitro than LY2875358 dose [87]. The pharmacodynamics and safety would need to be further validated by clinical trials in the future.
As a consequence, developing anti-HGF and anti-c-MET monoclonal antibodies is still an arduous and demanding task for treatment of digestive cancers.
8. Targeting HGF/c-MET Pathway in Digestive System Cancers by MicroRNAs
Despite advances and improvement in techniques to antagonize HGF/c-MET pathway using small molecule inhibitors, many cancers are unresponsive to these inhibitors due to resistance in clinical research [45]. Therefore, novel approaches are required for treatment of cancers. Developing miRNAs as therapeutic targets is a novel and potential direction for suppression of HGF/c-MET pathway in treatment of digestive system cancers; the miRNAs-HGF/c-MET axis is shown in Figure 2.
9. Targeting HGF/c-MET in Hepatocellular Carcinoma (HCC) by miRNAs
The miRNA dysregulation is causal in many human cancers including HCC [81]. Because of the strong association between HGF/c-MET activation and human cancers, targeting HGF/c-MET by miRNAs may be a promising approach for cancer treatment (Figure 2) [30]. Yang et al. found that miR-26a could directly target HGF to suppress cell viability, migration, and tumor angiogenesis in HCC [88]. Moreover, miR-26a inhibits the production of VEGFA in HCC through antagonizing the HGF-c-MET pathway, and the downstream PI3K/Akt/mTOR, ERK, and VEGFR2 signaling. In this sense, miR-26a may well alleviate tumor angiogenesis of HCC via blocking HGF/c-MET pathway and it might be a novel therapeutic strategy for HCC patients with aberrant HGF and c-MET [88]. Similarly, miR-199a-3p shows antitumor activity by directly targeting HGF, VEGFR1, VEGFA, VEGFR2, and MMP2 in HCC [89]. Ghosh et al. revealed that miR-199a-3p could attenuate HGF/c-MET signaling and thus reduce migration and invasion ability of HCC cells [89]. These reports suggest that targeting miRNAs-HGF axis may be a therapeutic potential for treating HCC.
In parallel, several miRNAs may be tumor-suppressors in HCC via targeting tyrosine kinase receptor c-MET (Figure 2). For example, miR-34a, miR-181b, and miR-198 may decrease migration and invasion of HCC cells by directly targeting c-MET [32,90,91]. Moreover, miR-23b may well suppress HCC cell viability and migration by mediating c-MET and urokinase downmodulation [92]. Yang et al. revealed that miR-122 could decrease cell proliferation and promote apoptosis of HCC cells via targeting c-MET and inhibiting its downstream ERK1/2, STAT3, and Akt/mTOR signaling [93]. Conversely, miR-93 is overexpressed in HCC tumors, and miR-93 level is correlated with c-Met intensity. Ohta et al. verified that miR-93 may bind to the 3’-UTR of PTEN and Cyclin Dependent Kinase Inhibitor 1A (CDKN1A) mRNA and active HGF/c-MET pathway. Meaningfully, sensitivity of HCC cells to sorafenib and tivantinib was attenuated by elevated miR-93 [94]. miR-93, an oncogenic miRNA, may be a novel therapeutic target for HCC. And combination of sorafenib and tivantinib with miR-93 inhibitors or anti-miR-93 drugs might improve drug sensitivity and efficacy of the former. All of these reports reveal the important role of miRNAs-c-MET axis in HCC treatment.
10. miRNAs-HGF/c-MET Axis in Gastric Cancer
miRNA-mediated suppression of oncogene mRNA translation is one of reasons which make miRNAs as interesting candidates in therapeutics. In gastric cancer, both HGF and c-MET contribute and participate in oncogenic pathways [60,61]. Si et al. reported that miR-26a and miR-26b may bind to the 3’-UTR of HGF mRNA, and thus suppress gastric cancer cell viability and migration in vitro and repressed tumor growth and angiogenesis in vivo through inhibiting HGF-VEGF pathway (Figure 2) [95]. In addition, Li et al. identified that miR-16 inhibited gastric cancer cell growth and migration by directly targeting 3’-UTR of HGF mRNA (Figure 2) [96]. Therefore, miRNAs-HGF axis could function as a novel treatment strategy for gastric cancer.
miR-206 levels were decreased in gastric cancer specimens, however, c-MET was overexpressed. Furthermore, the expression of miR-206 correlates inversely with the expression of c-MET in human gastric tumors. In the study of Zheng et al., miR-206 may inhibit cellular proliferation, migration, and invasion, and c-MET, CDK4, p-Akt, p-ERK, and p-Rb were inhibited (Figure 2) [29]. In another report, miR-34a could inhibit EMT in gastric cancer by targeting c-MET and Snail (Figure 2). When miR-34a was downregulated, HGF/c-MET was activated to induce Snail expression. Additionally, miR-34a may directly target Snail. These processes blocked E-cadherin expression and contributed to EMT in gastric cancer [97]. Likewise, miR-1 could also suppress gastric cancer cell growth and migration by targeting MET (Figure 2) [98]. miR-206, miR-34a and miR-1 could suppress gastric cancer by mediating c-MET, the miRNAs-MET pathway could serve as a promising treatment modality in gastric cancer.
11. miRNAs-HGF/c-MET Axis in CRC
The study about the miRNAs-HGF/c-MET axis is in the initial stage, but it has potential to serve as a new direction for treating CRC. Low miRNA-137 level was confirmed to be correlated with CRC progression by RNA sequencing analysis of 18 colorectal adenoma–carcinoma sequence (ACS) issues. In vitro, miR-137 is capable of inhibiting CRC cell growth, invasion, and colony formation. In an immunodeficient mouse model, miR-137 showed antitumor activity, including inhibiting colorectal tumor progression and hepatic metastasis. c-MET expression is negatively regulated by miR-137, and CRC progression may be suppressed via miR-137-c-MET axis (Figure 2) [99]. miR-137 also can be silenced by Mecp-2 (a DNA methyl-CpG-binding protein)-mediated epigenetic function, contributing to colorectal adenoma–carcinoma sequence and tumor progression. miR-34a has been described as a tumor-suppressor gene in HCC and gastric cancer by targeting c-MET. Consistently, miR-34a could negatively regulate invasion and migration of CRC cells via repressing c-MET (Figure 2) and long intergenic noncoding RNA (GAPLINC) suppresses the function of miR-34a in colorectal cancer [100]. Synthetically, these results provide strong evidence to show the inhibition role of miR-137 and miR-34a in tumor cell migration and invasion in CRC through regulation of c-MET.
12. miRNAs-HGF/c-MET Axis and Pancreatic Cancer
Pancreatic ductal adenocarcinoma ranks among the most lethal cancers, and its incidence is growing [101]. At present, the relationship between pancreatic cancer and miRNAs-HGF/c-MET is being explored. For example, Cao et al. found that miRNA-335-5p could inhibit pancreatic cancer via mediating c-MET (Figure 2) [102]. In detail, miRNA-335-5p may directly target the 3’-UTR of c-MET, and thus inhibit cell growth, migration, and invasion, and promote apoptosis via activating the HGF/c-MET signaling pathway. Moreover, NEAT1, a nuclear-restricted long noncoding RNA, suppresses the microRNA-335-5p/c-MET axis to promote pancreatic cancer malignancy [102]. Consistently, miRNA-335-5p also restrains pancreatic cancer cell tumor growth in an xenograft model [102]. In addition, Tomihara et al. reported c-MET expression was induced by irradiation, while the levels of miR-181b-5p were reduced [91]. On one hand, preoperative chemoradiation therapy might upregulate c-MET expression in remnant PDAC tissues, and irradiation actives HGF/c-MET pathway by enhancing c-MET expression in PDAC cells. On the other hand, irradiation exposure may reduce miR-181b-5p expression and it directly targets the transcription factor ETS1, which binds to the MET promoter and thus actives c-MET. Hence, miR-181b-5p may indirectly suppress the activation of c-MET, and a c-MET inhibitor may be used during preoperative chemoradiation therapy for PDAC patients (Figure 2) [91]. In summary, miRNAs-HGF/c-MET axis would provide more novel treatments for PDAC in the future.
13. miRNAs-HGF/c-MET Axis and Other Cancers
The function of miRNAs-HGF/c-MET axis in several human cancers has also been explored, including NSCLC, renal cancer, breast cancer, bladder cancer, and other cancers [103,104,105,106,107]. For example, miR-199a-3p has been mentioned to play an antitumor role in HCC through targeting c-MET, VEGFA, VEGFR1, VEGFR2, and MMP2. Consistently, miR-199a-3p may inhibit the proliferation of HCC cells via targeting c-MET and antagonizing downstream signals STAT3, mTOR, and ERK1/2 [107]. Similarly, miR-335 has been confirmed to inhibit the migration of cancer cells via binding c-MET mRNA in pancreatic and breast cancer [102,106]. These reports indicate that miRNAs-HGF/c-MET axis is worth developing for multiple cancer treatments.
14. Prospects
The HGF/c-MET pathway is emerging as a therapeutically relevant target in multiple cancers. The discovery and development of c-MET inhibitors has been gradually increasing, including nonselective and selective inhibitors. c-MET inhibitors were diverse for digestive system cancers, however, safety and efficacy have become the main problems in clinical stage studies. New drug developments for c-MET inhibitors offer the potential for better cancer treatment. Clinical trials of these c-MET inhibitors need to be performed to confirm the efficacy and safety. Moreover, insight into the improved safety and efficacy of clinical cancer therapy based on combined medication will provide a new research direction for treating digestive system cancers. In addition, miRNA biology therapy is still in the preclinical stage. Further investigation of the miRNA-HGF/c-MET may supply effective and promising therapy in human cancers, including digestive system cancers.
Author Contributions
H.Z. and Q.F. wrote the manuscript. Y.-D.W. and W.-D.C. edited and revised the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81672433 and No. 81370537) to Yan-Dong Wang, the National Natural Science Foundation of China (Grant No. 81472232 and Grant No. 81270522), Program for Science & Technology Innovation Talents in Universities of Henan Province (HASTIT, Grant No. 13HASTIT024) and Plan for Scientific Innovation Talent of Henan Province to W.-D.C., and the Fundamental Research Funds for the Central Universities (Grant No. PYBZ1803, Grant No. XK1802-8 and Grant No. PYBZ1706) to Yan-Dong Wang.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zuo, C.; Sheng, X.; Ma, M.; Xia, M.; Ouyang, L. ISG15 in the tumorigenesis and treatment of cancer: An emerging role in malignancies of the digestive system. Oncotarget 2016, 7, 74393–74409. [Google Scholar] [CrossRef] [PubMed]
- Ang, C.; Doyle, E.; Branch, A. Bisphosphonates as potential adjuvants for patients with cancers of the digestive system. World J. Gastroenterol. 2016, 22, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Granito, A.; Guidetti, E.; Gramantieri, L. c-MET receptor tyrosine kinase as a molecular target in advanced hepatocellular carcinoma. J. Hepatocell. Carcinoma 2015, 2, 29–38. [Google Scholar] [PubMed]
- Boromand, N.; Hasanzadeh, M.; Sales, S.S.; Farazestanian, M.; Gharib, M.; Fiuji, H.; Behboodi, N.; Ghobadi, N.; Hassanian, S.M.; Ferns, G.A. Clinical and prognostic value of the c-Met/HGF signaling pathway in cervical cancer. J. Cell. Physiol. 2017, 233, 4490–4496. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Lowengrub, J.S. Activation of the HGF/c-Met axis in the tumor microenvironment: A multispecies model. J. Theor. Boil. 2017, 439, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.L.; Lu, D.; Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 2016, 9, 135. [Google Scholar]
- Hu, C.T.; Wu, J.R.; Cheng, C.C.; Wu, W.S. The therapeutic targeting of HGF/c-Met signaling in hepatocellular carcinoma: Alternative approaches. Cancers 2017, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Furge, K.A.; Zhang, Y.W.; Vande Woude, G.F. Met receptor tyrosine kinase: Enhanced signaling through adapter proteins. Oncogene 2000, 19, 5582–5589. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met axis in head and neck cancer. Cancers 2017, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Stanley, A.; Ashrafi, G.H.; Seddon, A.M.; Modjtahedi, H. Synergistic effects of various Her inhibitors in combination with IGF-1R, C-MET and Src targeting agents in breast cancer cell lines. Sci. Rep. 2017, 7, 3964. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.; Kelly, C.; Rauch, J.; Kida, K.; García-Muñoz, A.; Monsefi, N.; Turriziani, B.; Doherty, C.; Mehta, J.P.; Matallanas, D. HGF induces epithelial-to-mesenchymal transition by modulating the mammalian hippo/MST2 and ISG15 pathways. J. Proteome Res. 2014, 13, 2874–2886. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Song, Y.; Qu, J.; Che, X.; Song, N.; Fan, Y.; Wen, T.; Xu, L.; Gong, J.; Wang, X. The chemokine receptor CXCR4 and c-MET cooperatively promote epithelial-mesenchymal transition in gastric cancer cells. Transl. Oncol. 2018, 11, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Alam, S.M.; de Bono, J.S. HGF/c-MET targeted therapeutics: Novel strategies for cancer medicine. Curr. Drug Targets 2011, 12, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A.; Saltotellez, M.; Laurentpuig, P.; Bardelli, A.; Rolfo, C.; Tabernero, J.; Khawaja, H.A.; Lawler, M.; Johnston, P.G.; Schaeybroeck, S.V. Targeting c-MET in gastrointestinal tumours: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2017, 14, 562. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhu, Y.; Liang, Z.; Li, S.; Xu, X.; Wang, X.; Wu, J.; Hu, Z.; Meng, S.; Liu, B. c-Met and CREB1 are involved in miR-433-mediated inhibition of the epithelial–mesenchymal transition in bladder cancer by regulating Akt/GSK-3β/Snail signaling. Cell Death Dis. 2016, 7, e2088. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Yoon, A.; Ryu, J.Y.; Cho, Y.J.; Choi, J.J.; Song, S.Y.; Bang, H.; Lee, J.S.; Cho, W.C.; Choi, C.H. c-MET as a potential therapeutic target in ovarian clear cell carcinoma. Sci. Rep. 2016, 6, 38502. [Google Scholar] [CrossRef] [PubMed]
- Demkova, L.; Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 2018, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Dong, J.; Wang, M.; Yao, S.; Tian, X.; Cui, X.; Fu, S.; Zhang, S. miR-148a-3p suppresses epithelial ovarian cancer progression primarily by targeting c-Met. Oncol. Lett. 2018, 15, 6131–6136. [Google Scholar] [CrossRef] [PubMed]
- Pasquini, G.; Giaccone, G. C-MET inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.P.; Cho, S.Y.; Ha, J.D.; Jung, H.; Kim, H.R.; Chong, O.L.; Jang, I.Y.; Chong, H.C.; Lee, H.K.; Sang, U.C. Novel c-Met inhibitor suppresses the growth of c-Met-addicted gastric cancer cells. BMC Cancer 2016, 16, 35. [Google Scholar]
- He, C.; Ai, J.; Xing, W.; Chen, Y.; Zhang, H.; Huang, M.; Hu, Y.; Ding, J.; Geng, M. Yhhu3813 is a novel selective inhibitor of c-Met Kinase that inhibits c-Met-dependent neoplastic phenotypes of human cancer cells. Acta Pharmacol. Sin. 2014, 35, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Balan, M.; Chakraborty, S.; Flynn, E.; Zurakowski, D.; Pal, S. Honokiol inhibits c-Met-HO-1 tumor-promoting pathway and its cross-talk with calcineurin inhibitor-mediated renal cancer growth. Sci. Rep. 2017, 7, 5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.S.; Liao, C.H.; Chen, W.S.; Pai, J.T.; Weng, M.S. Shikonin inhibited migration and invasion of human lung cancer cells via suppression of c-Met-mediated epithelial-to-mesenchymal transition. J. Cell. Biochem. 2017, 118, 4639–4651. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ai, J.; Shen, Y.; Zhang, H.; Peng, X.; Huang, M.; Zhang, A.; Ding, J.; Geng, M. SOMCL-863, a novel, selective and orally bioavailable small-molecule c-Met inhibitor, exhibits antitumor activity both in vitro and in vivo. Cancer Lett. 2014, 351, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Lowe, S.W. The microcosmos of cancer. Nature 2012, 482, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203. [Google Scholar] [CrossRef] [PubMed]
- Giglio, S.; Vecchione, A. c-Met and miRs in Cancer. Biomedicines 2015, 3, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Yan, D.; Chen, X.; Huang, H.; Chen, K.; Li, G.; Zhou, L.; Zheng, D.; Tu, L.L.; Dong, X.D. MicroRNA-206: Effective inhibition of gastric cancer progression through the c-Met pathway. PLoS ONE 2015, 10, e0128751. [Google Scholar] [CrossRef] [PubMed]
- Karagonlar, Z.F.; Korhan, P.; Atabey, N. Targeting c-Met in cancer by microRNAs: Potential therapeutic applications in hepatocellular carcinoma. Drug Dev. Res. 2015, 76, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Korhan, P.; Erdal, E.; Atabey, N. MiR-181a-5p is downregulated in hepatocellular carcinoma and suppresses motility, invasion and branching-morphogenesis by directly targeting c-Met. Biochem. Biophys. Res. Commun. 2014, 450, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, R.; Ding, K.; Lobie, P.E.; Zhu, T. miR-198 inhibits migration and invasion of hepatocellular carcinoma cells by targeting the HGF/c-MET pathway. FEBS Lett. 2011, 585, 2229–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bladt, F.; Friesehamim, M.; Ihling, C.; Wilm, C.; Blaukat, A. The c-Met inhibitor MSC2156119J effectively inhibits tumor growth in liver cancer models. Cancers 2014, 6, 1736–1752. [Google Scholar] [CrossRef] [PubMed]
- Bouattour, M.; Raymond, E.; Qin, S.; Cheng, A.L.; Stammberger, U.; Locatelli, G.; Faivre, S. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2017, 67, 1132–1149. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Ding, W.; Dang, H.; Jiang, Y.; Rountree, C.B. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011, 54, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, L.; Muzumdar, M.D.; Zhu, A.X. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Lee, C.R.; Man, S.; Chen, E.; Kerbel, R.S. Effects of sorafenib dose on acquired reversible resistance and toxicity in hepatocellular carcinoma. Cancer Res. 2015, 75, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wu, L.; Zheng, S.; Ding, W.; Teng, L.; Wang, Z.; Ma, Z.; Zhao, W. The clinical value of hepatocyte growth factor and its receptor-c-met for liver cancer patients with hepatectomy. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2006, 38, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Ojima, H.; Tsuda, H.; Hashimoto, J.; Morizane, C.; Ikeda, M.; Ueno, H.; Tamura, K.; Shimada, K.; Kanai, Y.; et al. Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int. J. Clin. Oncol. 2013, 18, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Bladt, F.; Faden, B.; Friesehamim, M.; Knuehl, C.; Wilm, C.; Fittschen, C.; Grädler, U.; Meyring, M.; Dorsch, D.; Jaehrling, F. EMD 1214063 and EMD 1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin. Cancer Res. 2013, 19, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Su, Y.; Dykema, K.; Johnson, J.; Koeman, J.; Giorgi, V.D.; Huang, A.; Schlegel, R.; Essenburg, C.; Liang, K. Overexpression of HGF promotes HBV-induced hepatocellular carcinoma progression and is an effective indicator for Met-targeting therapy. Genes Cancer 2013, 4, 247–260. [Google Scholar]
- Qin, S.; Lim, H.Y.; Ryoo, B.Y.; Li, C.; Xiong, H.; Ihling, C.; Cheng, A.L. 2353 data from a phase Ib/II trial of the oral c-Met inhibitor tepotinib (MSC2156119J) as first-line therapy in Asian patients with advanced hepatocellular carcinoma. Eur. J. Cancer 2015, 51, S452–S453. [Google Scholar] [CrossRef]
- Woo, H.Y.; Yoo, S.Y.; Heo, J. New chemical treatment options in second-line hepatocellular carcinoma: What to do when sorafenib fails? Expert Opin. Pharmacother. 2016, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Remsing Rix, L.L.; Kuenzi, B.M.; Luo, Y.; Remilywood, E.; Kinose, F.; Wright, G.; Li, J.; Koomen, J.M.; Haura, E.B.; Lawrence, H.R. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem. Boil. 2014, 9, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Giglione, P.; Ferrari, A.; Reversi, F.; Liguigli, W.; Imarisio, I.; Ganini, C. Tivantinib (ARQ197) in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2015, 15, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Rimassa, L.; Borbath, I.; Daniele, B.; Salvagni, S.; Van Laethem, J.L.; Van, V.H.; Trojan, J.; Kolligs, F.T.; Weiss, A. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: A randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013, 14, 55–63. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peckradosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Rota, E.C.; Porta, C.; Daniele, B.; Bolondi, L. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- Inagaki, Y.; Qi, F.; Gao, J.; Qu, X.; Hasegawa, K.; Sugawara, Y.; Tang, W.; Kokudo, N. Effect of c-Met inhibitor SU11274 on hepatocellular carcinoma cell growth. Biosci. Trends 2011, 5, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Feng, K.; Zhang, Y.; Li, Z.; Zhou, F.; Dou, H.; Wang, T. Sorafenib and DE605, a novel c-Met inhibitor, synergistically suppress hepatocellular carcinoma. Oncotarget 2015, 6, 12340–12356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínezmontemayor, M.M.; Acevedo, R.R.; Oterofranqui, E.; Cubano, L.A.; Dharmawardhane, S.F. Ganoderma lucidum (Reishi) inhibits cancer cell growth and expression of key molecules in inflammatory breast cancer. Nutr. Cancer 2011, 63, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Hsiao, Y.M.; Hsu, C.P.; Lin, M.Y.; Wang, J.C.H.; Huang, Y.L.; Ko, J.L. Transcriptionally mediated inhibition of telomerase of fungal immunomodulatory protein from Ganoderma tsugae in A549 human lung adenocarcinoma cell line. Mol. Carcinog. 2010, 45, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Yang, S.F.; Chen, G.D.; Han, C.P.; Chen, S.C.; Lin, L.Y.; Ko, J.L. Human nonmetastatic clone 23 type 1 gene suppresses migration of cervical cancer cells and enhances the migration inhibition of fungal immunomodulatory protein from Ganoderma tsugae. Reprod. Sci. 2007, 14, 475. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.R.; Hu, C.T.; You, R.I.; Ma, P.L.; Pan, S.M.; Lee, M.C.; Wu, W.S. Preclinical trials for prevention of tumor progression of hepatocellular carcinoma by LZ-8 targeting c-Met dependent and independent pathways. PLoS ONE 2015, 10, e0114495. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Ong, R.; Soo, K.C. Foretinib demonstrates anti-tumor activity and improves overall survival in preclinical models of hepatocellular carcinoma. Angiogenesis 2012, 15, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Takayuki, N.; Osamu, T.; Atsumi, Y.; Tomohiro, M.; Keiko, T.; Setsuo, F.; Shuji, S.; Makoto, A.; Hiroshi, O. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef] [Green Version]
- O’Neil, B.H.; Bendell, J.C.; Modiano, M.R.; Machiels, J.P.H.; Versola, M.J.; Hodge, J.P.; Sawarna, K.; Tse, N. Phase I/II study of E7050 (golvantinib) in combination with sorafenib in patients (pts) with advanced hepatocellular carcinoma (HCC): Phase I results. J. Clin. Oncol. 2013, 31, 294. [Google Scholar] [CrossRef]
- Bang, Y.J.; Su, W.C.; Nam, D.H.; Lim, W.T.; Bauer, T.M.; Brana, I.; Poon, T.P.; Hong, D.S.; Lin, C.C.; Peng, B. Phase I study of the safety and efficacy of INC280 in patients with advanced MET-dependent solid tumors. J. Clin. Oncol. 2014, 32, 2520. [Google Scholar]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.G.; Götze, O.; Geier, A. Molecular changes in hepatic metabolism and transport in cirrhosis and their functional importance. World J. Gastroenterol. 2016, 22, 72–88. [Google Scholar] [CrossRef] [PubMed]
- Fuse, N.; Kuboki, Y.; Kuwata, T.; Nishina, T.; Kadowaki, S.; Shinozaki, E.; Machida, N.; Yuki, S.; Ooki, A.; Kajiura, S. Prognostic impact of HER2, EGFR, and c-MET status on overall survival of advanced gastric cancer patients. Gastric Cancer 2016, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Metzger, M.L.; Behrens, H.M.; Böger, C.; Haag, J.; Krüger, S.; Röcken, C. MET in gastric cancer—Discarding a 10% cutoff rule. Histopathology 2016, 68, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Muro, K.; Ryu, M.H.; Yasui, H.; Nishina, T.; Ryoo, B.Y.; Kamiya, Y.; Akinaga, S.; Boku, Y.K.S.A.N. A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer. Investig. New Drugs 2014, 32, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Kim, T.M.; Yokota, T.; Goto, M.; Satoh, T.; Ahn, J.H.; Kim, H.S.; Assadourian, S.; Gomez, C.; Harnois, M. Phase I dose-escalation study of the c-Met tyrosine kinase inhibitor SAR125844 in Asian patients with advanced solid tumors, including patients with MET-amplified gastric cancer. Oncotarget 2017, 8, 79546–79555. [Google Scholar] [CrossRef] [PubMed]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol. 2015, 9, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Matushita, K.; Fujikawa, H.; Tanaka, K.; Mohri, Y.; Inoue, Y.; Goel, A.; Kusunoki, M. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. Int. J. Cancer 2012, 130, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Nishii, T.; Hasegawa, T.; Matsuzaki, T.; Morisaki, T.; Fukuoka, T.; Hirakawa, K. A c-Met inhibitor increases the chemosensitivity of cancer stem cells to the irinotecan in gastric carcinoma. Br. J. Cancer 2013, 109, 2619–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhan, Z.; Jiang, X.; Peng, X.; Shen, Y.; Chen, F.; Ji, Y.; Liu, W.; Shi, Y.; Duan, W. Simm530, a novel and highly selective c-Met inhibitor, blocks c-Met-stimulated signaling and neoplastic activities. Oncotarget 2016, 7, 38091–38104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awazu, Y.; Nakamura, K.; Mizutani, A.; Kakoi, Y.; Iwata, H.; Yamasaki, S.; Miyamoto, N.; Imamura, S.; Miki, H.; Hori, A. A novel inhibitor of c-Met and VEGF receptor tyrosine kinases with a broad spectrum of in vivo antitumor activities. Mol. Cancer Ther. 2013, 12, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Jung, K.H.; Park, B.H.; Zheng, H.M.; Lee, H.S.; Choi, M.J.; Yun, J.I.; Kang, N.S.; Lee, J.; Hong, S.S. KRC-408, a novel c-Met inhibitor, suppresses cell proliferation and angiogenesis of gastric cancer. Cancer Lett. 2013, 332, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Wang, S.; Liu, M.; Ma, H.; Zeng, X.; Zhang, M.; Xu, L.; Cui, Y.; Xu, H.; Tang, Y. Norcantharidin inhibits cell growth by suppressing the expression and phosphorylation of both EGFR and c-Met in human colon cancer cells. BMC Cancer 2017, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Dai, G.; Wang, J.; Gao, X.; Zhao, Z.; Duan, Z.; Gu, B.; Yang, W.; Wu, J.; Ju, Y. c-MET inhibition enhances the response of the colorectal cancer cells to irradiation in vitro and in vivo. Oncol. Lett. 2016, 11, 2879. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Bing, X.; Li, M.; Yang, Z.; Li, Y.; Chen, H. Study of critical role of c-Met and its inhibitor SU11274 in colorectal carcinoma. Med. Oncol. 2013, 30, 546. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.H.; Liu, C.; Wei, J.; Feng, Y. Effect of c-Met inhibitor SU11274 on human colon cancer cell growth. Chin. Med. J. 2013, 126, 2705–2709. [Google Scholar] [PubMed]
- Eng, C.; Bessudo, A.; Hart, L.L.; Severtsev, A.; Gladkov, O.; Müller, L.; Kopp, M.V.; Vladimirov, V.; Langdon, R.; Kotiv, B. A randomized, placebo-controlled, phase 1/2 study of tivantinib (ARQ 197) in combination with irinotecan and cetuximab in patients with metastatic colorectal cancer with wild-type KRAS who have received first-line systemic therapy. Int. J. Cancer 2016, 139, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Zhang, X.; Shi, X.; Wei, L.; Zheng, D.; Li, H.; Gao, J.; Li, J.; Hu, Z. NCTD enhances antitumor immunity of GM-CSF prostate cancer cells vaccine by inducing apoptosis of regulatory T cells. Cancer Sci. 2018, 109, 2109. [Google Scholar] [CrossRef] [PubMed]
- Owusu, B.Y.; Bansal, N.; Venukadasula, P.K.M.; Ross, L.J.; Messick, T.E.; Goel, S.; Galemmo, R.A.; Klampfer, L. Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling. Oncotarget 2016, 7, 29492–29506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hage, C.; Rausch, V.; Giese, N.; Giese, T.; Schönsiegel, F.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Herr, I. The novel c-Met inhibitor cabozantinib overcomes gemcitabine resistance and stem cell signaling in pancreatic cancer. Cell Death Dis. 2013, 4, e627. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.H.; Jung, K.H.; Mi, K.S.; Fang, Z.; Kim, S.J.; Ryu, Y.L.; Kim, J.; Kim, M.H.; Hong, S.S. Crizotinib exhibits antitumor activity by targeting alk signaling not c-met in pancreatic cancer. Oncotarget 2014, 5, 9150–9168. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Caretti, V.; Funel, N.; Galvani, E.; Maftouh, M.; Honeywell, R.J.; Lagerweij, T.; Van, T.O.; Campani, D.; Fuchs, D. Crizotinib inhibits metabolic inactivation of gemcitabine in c-Met-driven pancreatic carcinoma. Cancer Res. 2013, 73, 6745–6756. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.D.; Avan, A.; Leon, L.G.; Castiglia, M.; Honeywell, R.; Pauwels, P.; Peeters, M.; Peters, G.; Giovannetti, E. 474ptivantinib-Gemcitabine: Pharmacological rational for a new combination in pancreatic cancer. Ann. Oncol. 2014, 25, iv158. [Google Scholar] [CrossRef]
- Tran, P.; Gooding, W.E.; Villaruz, L.C.; Burns, T.F.; Socinski, M.A.; Tarhini, A.A. Phase I study of rilotumumab (AMG 102), an HGF inhibitor, and erlotinib in patients with advanced non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Iveson, T.; Donehower, R.C.; Davidenko, I.; Tjulandin, S.; Deptala, A.; Harrison, M.; Nirni, S.; Lakshmaiah, K.; Thomas, A.; Jiang, Y. Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: An open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study. Lancet Oncol. 2014, 15, 1007–1018. [Google Scholar] [CrossRef]
- Dvt, C.; Tebbutt, N.C.; Davidenko, I.; Murad, A.M.; Al-Batran, S.E.; Ilson, D.H.; Tjulandin, S.; Gotovkin, E.; Karaszewska, B.; Bondarenko, I. Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1467–1482. [Google Scholar]
- Shah, M.A.; Bang, Y.J.; Lordick, F.; Alsina, M.; Chen, M.; Hack, S.P.; Bruey, J.M.; Smith, D.; Mccaffery, I.; Shames, D.S. Effect of fluorouracil, leucovorin, and oxaliplatin with or without onartuzumab in HER2-Negative, MET-positive gastroesophageal adenocarcinoma: The METGastric randomized clinical trial. JAMA Oncol. 2017, 3, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Hochster, H.; Hart, L.L.; Firdaus, I.; Mace, J.R.; Mcfarlane, J.J.; Kozloff, M.; Catenacci, D.; Hsu, J.J.; Hack, S.P. A phase II randomized trial (GO27827) of first-line FOLFOX plus bevacizumab with or without the MET inhibitor onartuzumab in patients with metastatic colorectal cancer. Oncologist 2017, 22, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zeng, W.; Wortinger, M.A.; Yan, S.B.; Cornwell, P.; Peek, V.L.; Stephens, J.R.; Tetreault, J.W.; Xia, J.; Manro, J.R. LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6059–6070. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goetsch, L.; Tucker, L.; Zhang, Q.; Gonzalez, A.; Vaidya, K.S.; Oleksijew, A.; Boghaert, E.; Song, M.; Sokolova, I. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer 2016, 16, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, X.F.; Lu, X.; Jia, H.L.; Liang, L.; Dong, Q.Z.; Ye, Q.H.; Qin, L.X. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology 2014, 59, 1874–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Dasgupta, D.; Ghosh, A.; Roychoudhury, S.; Kumar, D.; Gorain, M.; Butti, R.; Datta, S.; Agarwal, S.; Gupta, S. MiRNA199a-3p suppresses tumor growth, migration, invasion and angiogenesis in hepatocellular carcinoma by targeting VEGFA, VEGFR1, VEGFR2, HGF and MMP2. Cell Death Dis. 2017, 8, e2706. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, H.; Yi, T.; Hu, Z.; Kong, W.; Wu, Y.; Zheng, X. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009, 275, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Tomihara, H.; Yamada, D.; Eguchi, H.; Iwagami, Y.; Noda, T.; Asaoka, T.; Wada, H.; Kawamoto, K.; Gotoh, K.; Takeda, Y. MicroRNA-181b-5p, ETS1, and the c-Met pathway exacerbate the prognosis of pancreatic ductal adenocarcinoma after radiation therapy. Cancer Sci. 2017, 108, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, A.; Sabelli, C.; Moncini, S.; Venturin, M.; Arici, B.; Riva, P.; Portolani, N.; Giulini, S.M.; De, P.G.; Barlati, S. MicroRNA-23b mediates urokinase and c-met downmodulation and a decreased migration of human hepatocellular carcinoma cells. FEBS J. 2009, 276, 2966–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.M.; Gyu, L.C.; Hyun, K.J.; Hyun, K.T.; Min, L.J.; An, J.; Mo, K.K.; Geon, K.S. Gα12overexpressed in hepatocellular carcinoma reduces microRNA-122 expression via HNF4α inactivation, which causes c-Met induction. Oncotarget 2015, 6, 19055–19069. [Google Scholar] [PubMed]
- Ohta, K.; Hoshino, H.; Wang, J.; Ono, S.; Iida, Y.; Hata, K.; Huang, S.K.; Colquhoun, S.; Hoon, D.S. MicroRNA-93 activates c-Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget 2015, 6, 3211–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, Y.; Zhang, H.; Ning, T.; Bai, M.; Wang, Y.; Yang, H.; Wang, X.; Li, J.; Ying, G.; Ba, Y. miR-26a/b inhibit tumor growth and angiogenesis by targeting the HGF-VEGF axis in gastric carcinoma. Cell. Physiol. Biochem. 2017, 42, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Shuang, L.; Zhang, H.; Wang, X.; Qu, Y.; Duan, J.; Rui, L.; Deng, T.; Tao, N.; Le, Z.; Ming, B. Direct targeting of HGF by miR-16 regulates proliferation and migration in gastric cancer. Tumor Biol. 2016, 37, 15175–15183. [Google Scholar]
- Liu, Y.; Sun, M.; Xia, R.; Zhang, E.; Liu, X.; Zhang, Z.; Xu, T.; De, W.; Liu, B.; Wang, Z. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhou, Y.; An, Q.; Li, F.; Li, D.; Zhang, X.; Yu, Z.; Zheng, L.; Duan, Z.; Kan, Q. MicroRNA-1 (miR-1) inhibits gastric cancer cell proliferation and migration by targeting MET. Tumor Boil. 2015, 36, 6715–6723. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cai, S.L.; Li, J.; Qi, Z.P.; Li, X.Q.; Ye, L.C.; Xie, X.F.; Hou, Y.Y.; Yao, L.Q.; Xu, M.D. Mecp2-mediated epigenetic silencing of miR-137 contributes to colorectal adenoma-carcinoma sequence and tumor progression via relieving the suppression of c-Met. Sci. Rep. 2017, 7, 44543. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ouyang, J.; Zhou, D.; Zhong, S.; Wen, M.; Ou, W.; Yu, H.; Jia, L.; Huang, Y. Long noncoding RNA GAPLINC promotes cells migration and invasion in colorectal cancer cell by regulating miR-34a/c-MET signal pathway. Dig. Dis. Sci. 2018, 63, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.S.; Coburn, N.; Liu, N.; Porter, J.M.; Seung, S.J.; Cheung, M.C.; Goyert, N.; Leighl, N.B.; Hoch, J.S.; Trudeau, M.E. A population-based study of the epidemiology of pancreatic cancer: A brief report. Curr. Oncol. 2015, 22, e478. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Zhang, Y.; Yang, J.; He, S.; Li, M.; Yan, S.; Chen, Y.; Qu, C.; Xu, L. NEAT1 regulates pancreatic cancer cell growth, invasion and migration though mircroRNA-335-5p/c-met axis. Am. J. Cancer Res. 2016, 6, 2361. [Google Scholar] [PubMed]
- Luo, W.; Huang, B.; Li, Z.; Li, H.; Sun, L.; Zhang, Q.; Qiu, X.; Wang, E. MicroRNA-449a is downregulated in non-small cell lung cancer and inhibits migration and invasion by targeting c-Met. PLoS ONE 2013, 8, e64759. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Lin, Y.; Chen, H.; Mao, Y.; Wu, J.; Zhu, Y.; Xu, X.; Xu, X.; Li, S.; Zheng, X.; et al. MicroRNA-101 suppresses motility of bladder cancer cells by targeting c-Met. Biochem. Biophys. Res. Commun. 2013, 435, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, H.; Lin, Y.; Hu, Z.; Mao, Y.; Wu, J.; Xu, X.; Zhu, Y.; Li, S.; Zheng, X. MicroRNA-409-3p inhibits migration and invasion of bladder cancer cells via targeting c-Met. Mol. Cells 2013, 36, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Zeng, F.; Wu, J.Y.; Li, H.Y.; Fan, J.J.; Mai, L.; Zhang, J.; Ma, D.M.; Li, Y.; Song, F.Z. MiR-335 inhibits migration of breast cancer cells through targeting oncoprotein c-Met. Tumor Boil. J. Int. Soc. Oncodev. Boil. Med. 2015, 36, 2875–2883. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dong, B.; Zhang, J.; Kong, W.; Chen, Y.; Xue, W.; Liu, D.; Huang, Y. miR-199a-3p inhibits hepatocyte growth factor/c-Met signaling in renal cancer carcinoma. Tumor Boil. 2014, 35, 5833–5843. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The schematic diagram of c-mesenchymal–epithelial transition (c-MET) activation signaling pathways. Activated c-MET binds adaptor molecules GRB-1 and GRB-2, and then they recruit Ras/Raf, SH2 containing protein tyrosine phosphatase (Shp2), STAT3/5, and PI3K signals. In addition, activation of other receptor tyrosine kinases (RTKs) signaling molecules (HER2, VEGFR, EGFR, and ALK) may cross-talk with the c-MET, further recruiting PI3K, ERK, and STAT3/5 signals. These downstream signals are involved in regulating cell morphogenesis, survival, proliferation, and invasion. Inhibition of the HGF/c-MET pathway can be achieved through c-MET kinase inhibitors (TKIs), anti-HGF monoclonal antibodies (Anti-HGF Ab), and anti-MET monoclonal antibodies (Anti-MET Ab). HGF, hepatocyte growth factor; ALK, anaplastic lymphoma kinase; JAK, Janus kinase; STAT3/5, signal transducers and activators of transcription 3/signal transducers and activators of transcription 5; HER2, human epidermal growth factor receptor 2; VEGFR, vascular endothelial growth factor receptor 2; EGFR, epidermal growth factor receptor; P, phosphorylation; GRB, growth factor receptor-bound protein; SOS, son of sevenless; Ras, GTPases and key transducers of receptor tyrosine kinase; Raf, raf kinase, effector of Ras; PI3K, phosphatidylinositide 3-kinases; AKT, protein kinase B; ERK, extracellular regulated protein kinases; MAPK, mitogen-activated protein kinase.
Figure 1.
The schematic diagram of c-mesenchymal–epithelial transition (c-MET) activation signaling pathways. Activated c-MET binds adaptor molecules GRB-1 and GRB-2, and then they recruit Ras/Raf, SH2 containing protein tyrosine phosphatase (Shp2), STAT3/5, and PI3K signals. In addition, activation of other receptor tyrosine kinases (RTKs) signaling molecules (HER2, VEGFR, EGFR, and ALK) may cross-talk with the c-MET, further recruiting PI3K, ERK, and STAT3/5 signals. These downstream signals are involved in regulating cell morphogenesis, survival, proliferation, and invasion. Inhibition of the HGF/c-MET pathway can be achieved through c-MET kinase inhibitors (TKIs), anti-HGF monoclonal antibodies (Anti-HGF Ab), and anti-MET monoclonal antibodies (Anti-MET Ab). HGF, hepatocyte growth factor; ALK, anaplastic lymphoma kinase; JAK, Janus kinase; STAT3/5, signal transducers and activators of transcription 3/signal transducers and activators of transcription 5; HER2, human epidermal growth factor receptor 2; VEGFR, vascular endothelial growth factor receptor 2; EGFR, epidermal growth factor receptor; P, phosphorylation; GRB, growth factor receptor-bound protein; SOS, son of sevenless; Ras, GTPases and key transducers of receptor tyrosine kinase; Raf, raf kinase, effector of Ras; PI3K, phosphatidylinositide 3-kinases; AKT, protein kinase B; ERK, extracellular regulated protein kinases; MAPK, mitogen-activated protein kinase.

Figure 2.
Targeting HGF/c-MET pathway in digestive system cancers by microRNAs. (A) targeting HGF/c-MET in HCC by miRNAs; (B) miRNAs-HGF/c-MET axis in gastric cancer; (C) miRNAs-HGF/c-MET axis in CRC; (D) miRNAs-HGF/c-MET axis and pancreatic cancer. Abbreviations: EMT, epithelial–mesenchymal transition; ACS, adenoma–carcinoma sequence. ![Ijms 19 03295 i016]() directly target;
directly target; ![Ijms 19 03295 i017]() induction.
induction.
 directly target;
directly target;  induction.
induction.
Figure 2.
Targeting HGF/c-MET pathway in digestive system cancers by microRNAs. (A) targeting HGF/c-MET in HCC by miRNAs; (B) miRNAs-HGF/c-MET axis in gastric cancer; (C) miRNAs-HGF/c-MET axis in CRC; (D) miRNAs-HGF/c-MET axis and pancreatic cancer. Abbreviations: EMT, epithelial–mesenchymal transition; ACS, adenoma–carcinoma sequence. ![Ijms 19 03295 i016]() directly target;
directly target; ![Ijms 19 03295 i017]() induction.
induction.
directly target; induction.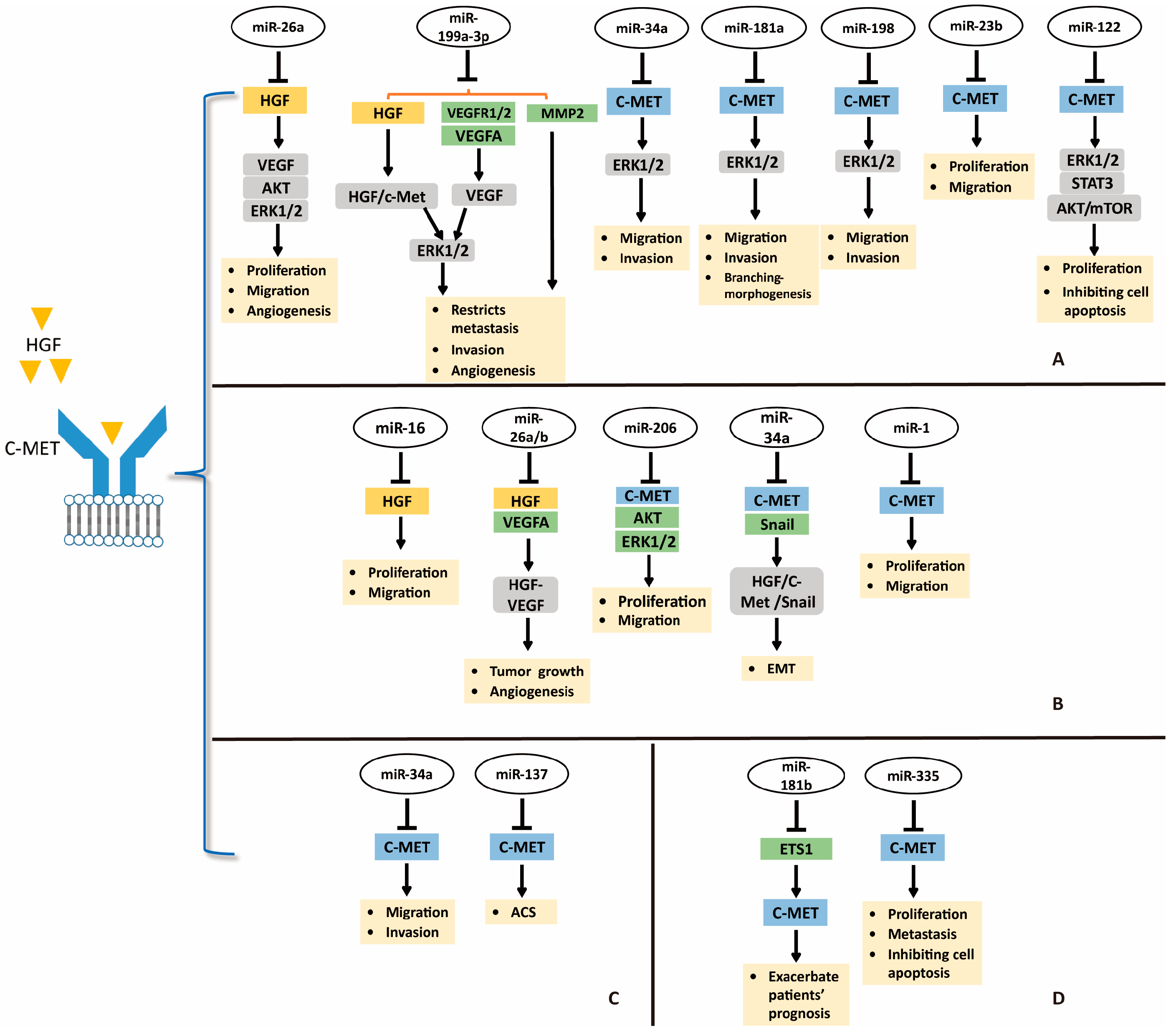

{kind=link}
{kind=link}
{kind=link}
Table 1.
Properties of c-MET inhibitors for digestive system cancer treatment.
| Type | Agent | Structure | Target(s) of Inhibitor | Patient Population |
|---|---|---|---|---|
| ATP-competitive small-molecular inhibitor | Tepotinib (MSC2156119J) |  | c-MET | Advanced HCC [33] |
| ATP-competitive small-molecular inhibitor | SU11274 |  | c-MET | Advanced HCC [48] Gastric cancer [65,66] CRC [72,73] |
| Non-ATP competitive selective small-molecular inhibitor | Tivantinib (ARQ 197) | 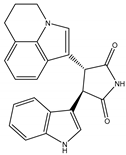 | c-MET Gsk 3 Cyclin B1, Proteasome | Cirrhotic patients with HCC [38,45,46,47] |
| c-Met (DE605) | Metastatic gastric cancer [62] | |||
| Pancreatic cancer [80] | ||||
| Combination | Sorafenib and DE605 |  (DE605) | HCC [49] | |
| Medicinal peptide | LZ8 |  | c-MET, ERK, AKT, and JNK | HCC [53] |
| ATP competitive selective c-Met small molecule inhibitor | Volitinib | 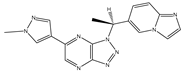 | c-MET | Gastric cancer [64] |
| Selective c-MET small molecule inhibitor | SAR125844 | 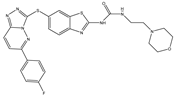 | c-MET | MET-amplified gastric cancer [63] |
| ATP-competitive selective c-Met inhibitor | KRC-408 |  | c-MET | Gastric cancer [69] |
| Selective c-Met inhibitor | KRC-00715 |  | c-MET | Gastric cancer [21] |
| ATP competitive selective c-Met small molecule inhibitor | Simm530 |  | c-MET | Gastric cancer [67] |
| ATP-competitive multitargeted kinase inhibitor | T-1840383 | 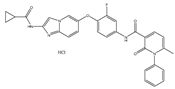 | VEGFRs c-MET | Gastric cancer [68] |
| Synthetic compound | Norcantharidin | 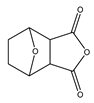 | EGFR c-MET | CRC [70] |
| Triplex inhibitors | SRI 31215 |  | Matriptase, Hepsin, and HGFA | CRC [76] |
| Combination (ATP-competitive small molecule crizotinib and Gemcitabine) | Crizotinib and Gemcitabine | 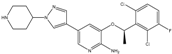 (Crizotinib) | c-MET/ALK | PDAC [78,79] |
| Nonselective oral multi-kinase inhibitor | Cabozantinib | 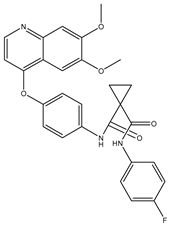 | c-MET and VEGFR-2 | HCC [58] Pancreatic cancer [77]. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, H.; Feng, Q.; Chen, W.-D.; Wang, Y.-D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. Int. J. Mol. Sci. 2018, 19, 3295. https://doi.org/10.3390/ijms19113295
AMA Style
Zhang H, Feng Q, Chen W-D, Wang Y-D. HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers. International Journal of Molecular Sciences. 2018; 19(11):3295. https://doi.org/10.3390/ijms19113295
Chicago/Turabian StyleZhang, Hongli, Qingqing Feng, Wei-Dong Chen, and Yan-Dong Wang. 2018. "HGF/c-MET: A Promising Therapeutic Target in the Digestive System Cancers" International Journal of Molecular Sciences 19, no. 11: 3295. https://doi.org/10.3390/ijms19113295
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.