AMPK: An Epigenetic Landscape Modulator
by
Brendan Gongol
1,2, 
Indah Sari
2,†,
Tiffany Bryant
2,†,
Geraldine Rosete
2,† and
Traci Marin
1,3,* 1
Department of Medicine, University of California, San Diego, CA 92093, USA
2
Department of Cardiopulmonary Sciences, School of Allied Health Professions, Loma Linda University, Loma Linda, CA 92350, USA
3
Department of Health Sciences, Victor Valley College, Victorville, CA 92395, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2018, 19(10), 3238; https://doi.org/10.3390/ijms19103238
Submission received: 26 September 2018
/
Revised: 14 October 2018
/
Accepted: 17 October 2018
/
Published: 19 October 2018
(This article belongs to the Special Issue AMP-Activated Protein Kinase Signalling)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Activated by AMP-dependent and -independent mechanisms, AMP-activated protein kinase (AMPK) plays a central role in the regulation of cellular bioenergetics and cellular survival. AMPK regulates a diverse set of signaling networks that converge to epigenetically mediate transcriptional events. Reversible histone and DNA modifications, such as acetylation and methylation, result in structural chromatin alterations that influence transcriptional machinery access to genomic regulatory elements. The orchestration of these epigenetic events differentiates physiological from pathophysiological phenotypes. AMPK phosphorylation of histones, DNA methyltransferases and histone post-translational modifiers establish AMPK as a key player in epigenetic regulation. This review focuses on the role of AMPK as a mediator of cellular survival through its regulation of chromatin remodeling and the implications this has for health and disease.
1. Introduction
Epigenetic regulation gives rise to a spectrum of cellular phenotypes observed in a single organism independent of primary DNA sequence. Such regulation is hereditable and stable, as occurs in the determination of cell type, but also transient, producing a particular phenotypic outcome to ensure survival [1,2,3]. Influencing gene expression, epigenetics promotes organismal adaption by offering substantial functional variability in response to environmental stimuli [1,2,3]. This regulation occurs, in part, through nucleosomal remodeling as a result of histone, DNA, and DNA-binding protein modifications that include: phosphorylation, acetylation, O-GlcNAcylation, ribosylation, and methylation. Such signature modifications or marks characterize nucleosome remodeling and determine the degree of gene activation or silencing. At a fundamental level, stressors such as nutrient deprivation or heightened physical activity trigger dynamic epigenetic markings that orchestrate adaptive gene regulation to improve survivability [4,5,6,7].
AMP-activated protein kinase (AMPK), a master regulator of energy homeostasis and a key mediator of adaptation and cell survival, is activated by conditions that produce energy deprivation such as hypoxia, exercise, nutrient starvation, and infection [8]. AMPK functions as a heterotrimeric serine/threonine protein kinase composed of a catalytic α-subunit, a scaffolding β-subunit, and a regulatory γ-subunit. Both α and β-subunits exist in two isoforms (α1, α2, and β1, β2), while the regulatory γ-subunit exists in three isoforms (γ1, γ2, γ3). The combination of α, β, and γ isoforms can form 12 different heterotrimeric complexes, each demonstrating unique functions and subcellular or tissue-specific distributions [9,10,11]. Once activated, AMPK phosphorylates targets with well-defined consensus sequences to regulate bioenergetics by modulating metabolic pathways that promote ATP production and limit energy expenditure [12]. Such roles of AMPK in cellular homeostasis are facilitated by gene regulation as a result of chromatin conformational dynamics. To that end, AMPK orchestrates these changes by phosphorylating several histones and proteins involved in nucleosome remodeling that enhance mitochondrial biogenesis and function [13]. Additionally, during acute metabolic stressors, such as fasting or exercise, AMPK associates with chromatin at promoters of genes involved in lipid and glucose metabolism [14].
Chronic AMPK activation plays a role in cellular and organismal inheritance, evidenced by its multi-isoform 2R-ohnologue characteristics, which are often evolutionarily conserved in gene coding regions to support basic survival functions and to increase the possibility for complex tissue diversity and adaptation [15,16,17]. Such adaptation for survival early in life initiates epigenetic programming that correlates with AMPK activation and determines predisposition to disease. For example, insults during development, such as placental insufficiency or maternal metabolic disorders, increase the probability of adverse metabolic disorders in later stages of life, such as diabetes mellitus, metabolic syndrome, insulin resistance, hypertension, vascular disease, and cancer [18]. Furthermore, metabolic disorders, such as maternal obesity, are often associated with reduced AMPK expression or activity and concomitant loss of its epigenetic mechanisms associated with adaptation and survival [19,20]. Although the mechanistic basis for this regulation, as well as its implications for inheritance, is still under exploration, these collective observations point to a fundamentally important role of AMPK as an epigenetic regulator.
2. Histone Modification
Histones are nuclear-localized, primarily positively charged proteins. However, a number of histone splice variants have been described with broad cellular functions and localizations [21,22]. Within the nucleus, histones are packaged into an octamer consisting of two H2A, H2B, H3, and H4 [23]. These octameric histone complexes associate via charge–charge interactions with DNA and make up a nucleosome, which is linked to other nucleosomes by histone H1. Given their tight association with DNA, histones serve as mediators between stress response signaling cascades and the regulation of nucleosomal structure that ultimately influence gene expression and cellular survival [24]. AMPK, both directly and indirectly, regulates the post-translational modification (PTM) status of histones that play a major role in the regulation of nucleosome structure.
2.1. Histone Phosphorylation
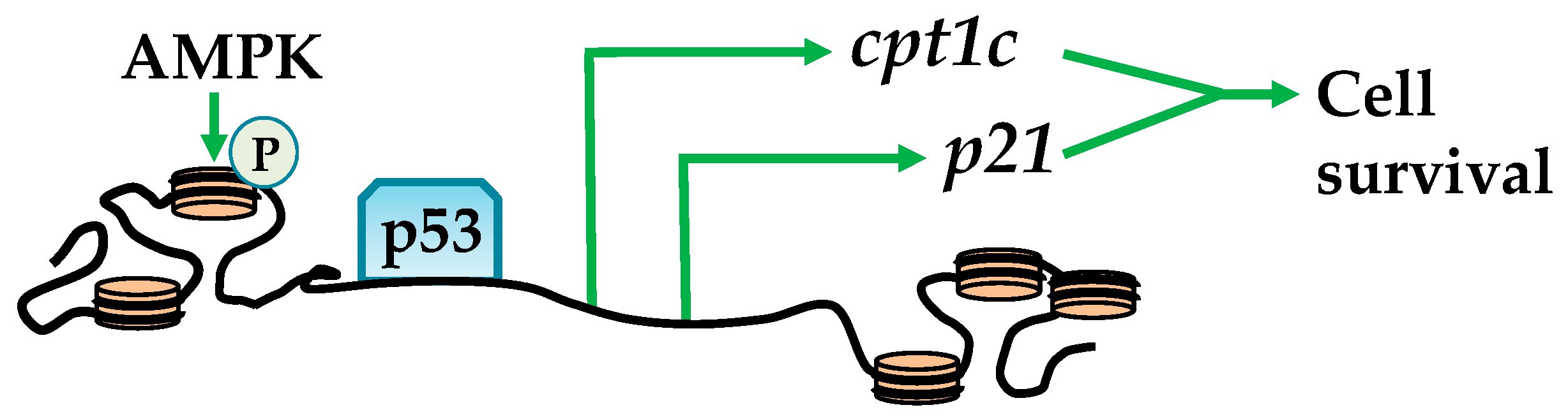
Bioinformatic analysis has identified several histones that contain an AMPK phosphorylation consensus sequence. These include: H1FX, H2AFX, H2AFY, H2AFY2, H2AFZ, H2BFM, and H3F3B [25], suggesting that AMPK plays an important role in epigenetic regulation through histone phosphorylation. Ultimately, such histone phosphorylation promotes cross-talk between epigenetic regulators to facilitate spatio-temporal nucleosome structural changes that influence transcriptional machinery [24]. For example, activation by glucose deprivation or UV radiation results in the colocalization of AMPK and phosphorylating serine 36 in H2B (H2BS36) throughout the carnitine palmitoyltransferase 1C (CPT1C) and cyclin-dependent kinase inhibitor (p21) promoter regions and gene bodies. This exposes TP53 or tumor protein (p53) DNA binding sites and subsequent association with RNA polymerase II (Figure 1) [26,27]. These events enhance CPT1C and p21 expression, which play a role in cellular survival via activating autophagy and transporting long-chain fatty acids to the mitochondria, collectively enhancing β-oxidation and energy production [28,29,30]. Related to cellular survival, charged multivesicular body protein 1B (CHMP1b) is phosphorylated by AMPK, forms a “shell” around nucleosomes enriched with H3 phosphorylation and acetylation, and influences gene transcription [25]. This might play a role in the transition of active and inactive nucleosome regions and potentially heritable epigenetic marks [31].
2.2. Histone Acetylation
Lysine acetylation of the N-terminal tails of histones disrupts the charge–charge interactions between DNA and histone tails, producing a euchromatin, a relaxed and active, chromatin state. This reaction changes histone–DNA associations, histone–histone associations between adjacent nucleosomes, and histone–regulatory protein interactions [32]. Histone acetylation occurs via transfer of an acetyl group from acetyl coenzyme-A (acetyl-CoA) to the to the ε-ammonium (NH3+) group of lysine catalyzed by histone acetyltransferases (HATs). HAT families are diverse and promote a spectrum of interactions and functions. Histone deacetylation restores the DNA-histone interaction promoting a heterochromatic, a condensed and silent chromatin state [33]. Antagonistically, histone deacetylation, catalyzed by histone deacetylases (HDACs), restores the DNA-histone interaction promoting a euchromatic chromatin state [33]. AMPK regulates the activity of both HATs and HDACs by influencing cofactor or substrate availability through direct phosphorylation.
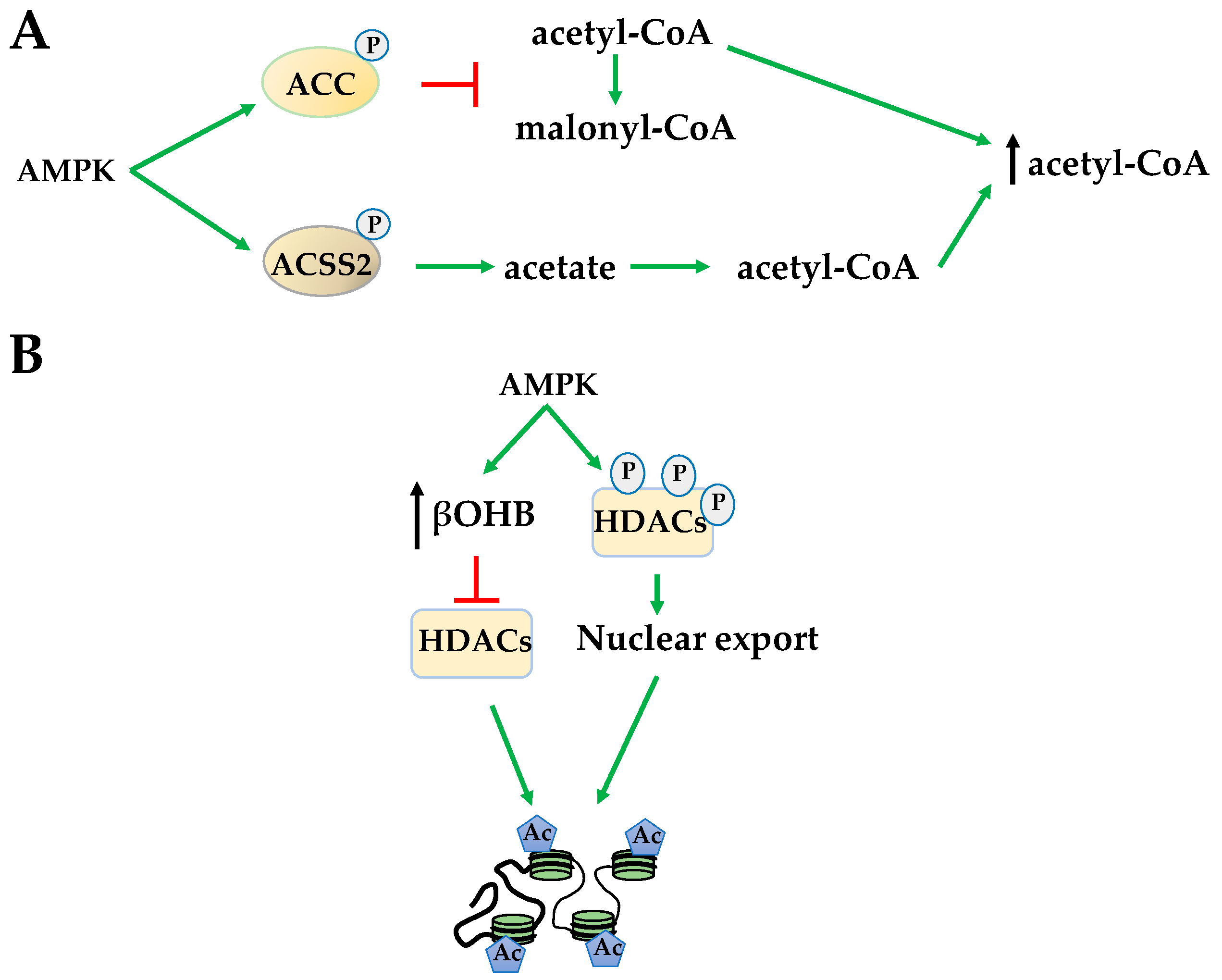
Indirectly, 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) activation of AMPK affects global HAT activity by increasing available acetyl-CoA levels through several mechanisms. AMPK phosphorylates acetyl-CoA carboxylase (ACC) to prevent the conversion of acetyl-CoA to malonyl-CoA increasing available acetyl donating groups for HATs (Figure 2A). AMPK also increases the formation of acetyl-CoA by phosphorylating acetyl-CoA synthetase short-chain family member 2 (ACSS2), causing its nuclear translocation for the conversion of acetate, the byproduct of HDAC histone deacetylation, to acetyl-CoA (Figure 2A). This mechanism has been shown to increase HAT acetylation of H3 at transcription factor EB (TFEB)-responsive promoters to activate genes important for autophagy and lysosomal function [34,35]. Yet, in general, increased acetyl-CoA levels in response to AICAR results in increased H3 acetylation at lysine 14 (H3K14) and H4 acetylation at lysines 5,8,12,16 (H4K5,8,12,16) [36]. AMPK activated by metformin regulates histone acetylation in a metabolic state-dependent manner. For example, metformin reduces H3 acetylation by decreasing bioavailability of mitochondrial acetyl-CoA in breast cancer, bringing acetylation status to normal in promoters of cancer-specific genes [37].
AMPK also regulates substrate availability of HDACs. The class III HDACs, sirtuins (SIRTs), couple deacetylation with nicotinamide adenine dinucleotide (NAD+) hydrolysis to produce O-acetyl-ADP-ribose [38]. AMPK indirectly activates SIRT1, in part, by increasing the NAD+/NADH ratio [38,39]. Once activated, SIRT1 plays a fundamental role in chromatin organization by interacting with and deacetylating a variety of transcription factors and coregulators [40]. However, in contrary to activating the SIRTs, AMPK indirectly inhibits class I and II HDACs by increasing β-hydroxybutyrate (βOHB) during fatty acid oxidation. βOHB acts similarly to the HDAC inhibitor butyrate, increasing global histone acetylation (Figure 2B) [41]. However, in addition to globally inhibiting class I and class II HDACs, AMPK activation also promotes HDACs 4, 5, and 7 hyperphosphorylation and translocation from the nucleus (Figure 2B) [42]. Both of these events increase global histone acetylation. However, despite these global effects, the regulation of gene expression is often promoter- and gene cluster-specific, suggesting the importance of direct phosphorylation events of AMPK on specific HATs and HDACs.
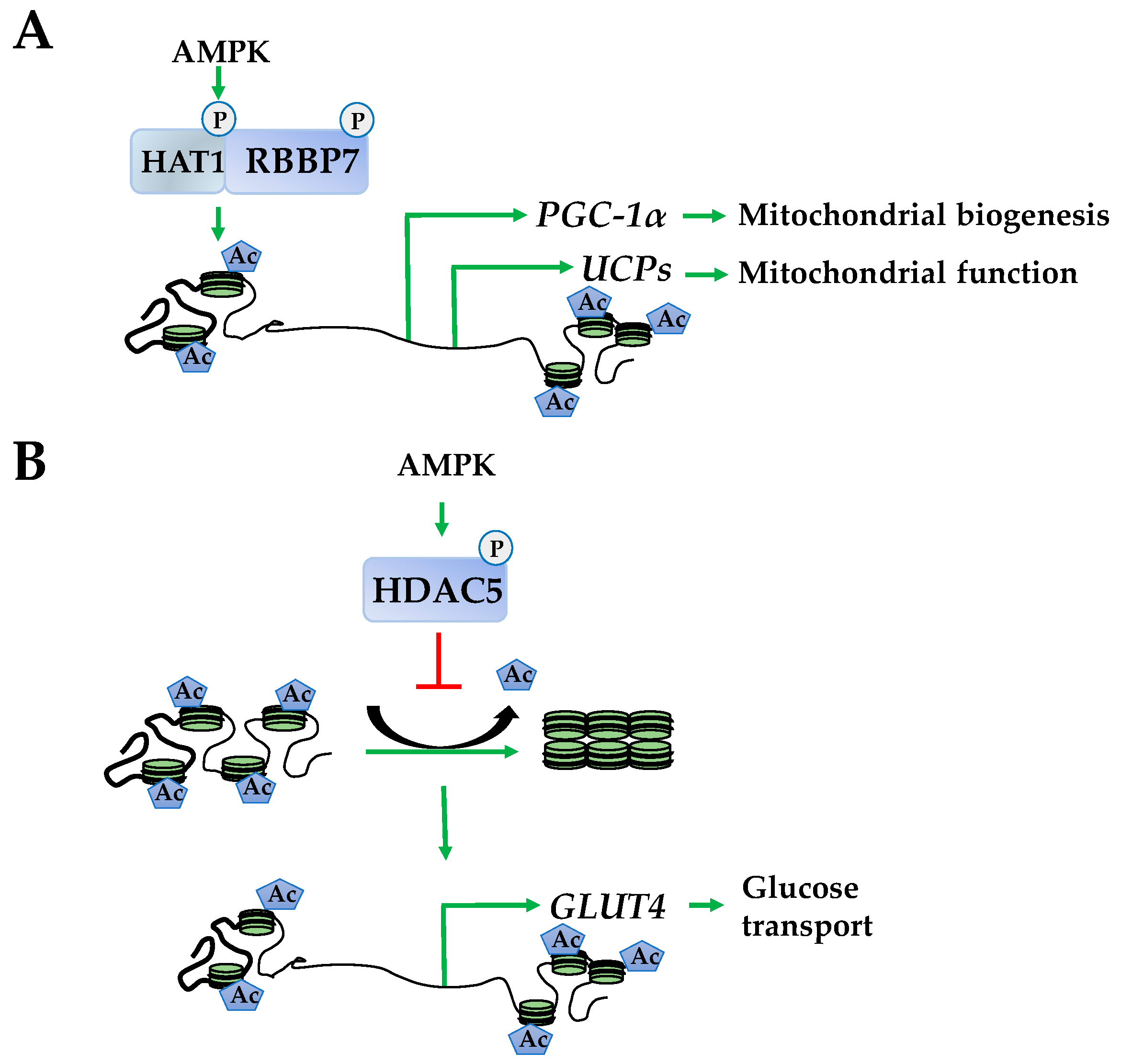
Investigations into epigenetically regulated networks identified HAT1 and retinoblastoma binding protein 7 (RBBP7) as direct targets of AMPK that dimerize following phosphorylation. This results in enhanced euchromatin structure at the promoters of peroxisome proliferator–activated receptor gamma coactivator–1α (PGC-1α), transcription factor A (Tfam), nuclear respiratory factors 1 and 2 (NRF1 and NRF2), and uncoupling proteins 2 and 3 (UCP2 and UCP3). The corresponding induction of these genes produces enhanced mitochondrial function (Figure 3A) [13]. In addition to HAT1 and RBBP7, AMPK phosphorylates HDAC5, promoting its dissociation from the promoters releasing its suppressive effects to increase expression of an array of metabolic genes, including glucose transporter type 4 (GLUT-4) (Figure 3B) [43].
Although less characterized, it is likely that AMPK phosphorylates an array of HATs including transcription regulator family member A (SIN3), CREB binding protein (CREBBP), elongator acetyltransferase complex subunits (ELP) 2, 3, and 4, and K(lysine) acetyltransferases (KAT) 2A, 2B, 6A, 6B, 7, and 8, and HDACs 1–5, 8–9, and 10, 11, as well as SIRTs 2, 3, 4, 5, 6, and 7; all of which contain AMPK consensus sequences and are opportune for future study [13].
2.3. Histone Methylation
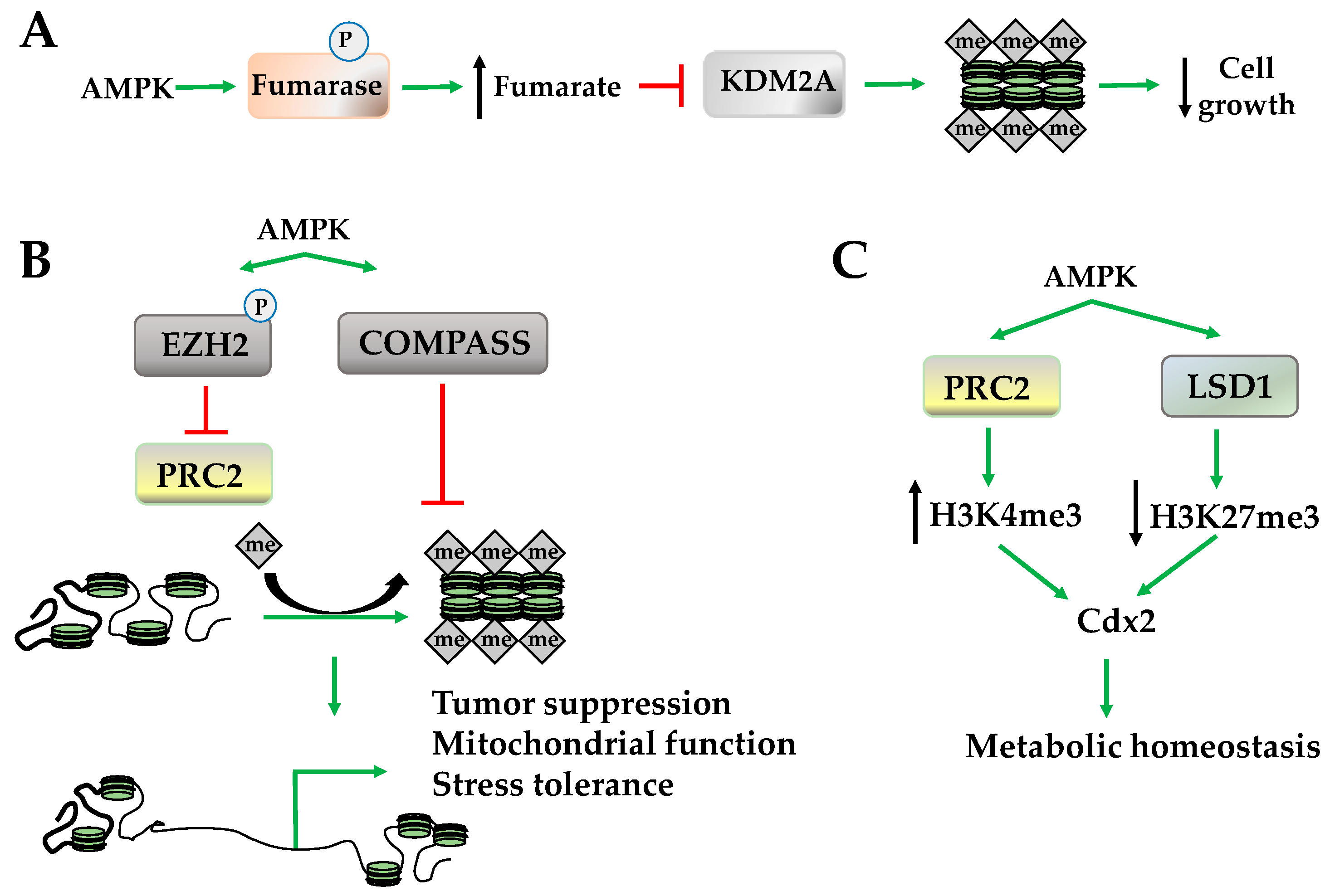
Histone methylation occurs on lysine and arginine, and its effects on gene expression are dependent upon the site and degree of methylation. For example, methylation of H3K4, H3K36, and H3K79 promotes an active euchromatin state; while methylation of H3K9 and H3K27 promotes a silent heterochromatic state [44,45,46]. Both lysine-specific and arginine-specific histone methyltransferases use S-adenosyl methionine (SAM) as cofactors and methyl donor catalyzing the transfer of one, two, or three methyl groups. The demethylases, however, serve a variety of functions and include two primary classes: flavin adenine dinucleotide (FAD)-dependent amine oxidase and Fe(II) and α-ketoglutarate-dependent hydroxylase. The regulation of demethylases via α-ketoglutarate, a tricarboxylic acid (TCA) cycle intermediate, suggests that AMPK may play a role in regulating histone methylation and demethylation because AMPK is activated by conditions where TCA cycle intermediates are depleted and, once activated, these intermediates are restored. For example, enhanced amino acid, fatty acid, or glycogen catabolism by AMPK increases α-ketoglutarate, which, in turn, may increase histone demethylase activity [47]. In addition to restoring α-ketoglutarate levels, AMPK phosphorylates and inhibits fumarase a TCA cycle enzyme that converts fumarate to malate. Subsequently, elevated fumarate levels inhibit lysine-specific demethylase 2A (KDM2A), restoring H3K36 dimethylation (H3K36me2) at promoters of genes mediating cell growth (Figure 4A) [48]. In addition to these indirect effects, AMPK activates the demethyltransferase lysine demethylase 5 (KDM5) and lysine-specific histone demethylase-1 (LSD1) that remove H3K4 trimethylation (H3K4me3) [49]. Although AMPK activates histone demethyltransferase activity, it inhibits several complexes that regulate methyltransferase activity including the methylation complex polycomb repressive complex 2 (PRC2) and the histone methyltransferase-containing COMPASS complex (complex proteins associated with Set1) [20,50]. While the inhibition of PRC2 occurs through the phosphorylation of histone methyl transferase enhancer of zeste homolog 2 (EZH2), which decreases H2K27 monomethylation (H2K27me1), inhibition of the COMPASS complex by AMPK decreases H3K4me3, a marker of transcriptional activation (Figure 4B) [50,51,52]. Globally, inhibition of PRC2 results in the upregulation of tumor suppressor PRC2 target genes and suppression of tumor growth, while COMPASS complex inhibition is a protective mechanism that halts chromatin marking in the presence of nutrient deficiency to promote stress tolerance (Figure 4B) [20,50,51,52].
However, the orchestration between the methylation and demethylation activities of AMPK is promoter-specific. For example, AMPK recruits PRC2 and LSD1 to the promoter of caudal type homeobox2 (Cdx2), increasing H3K4me3 but not H3K27me3 [53]. This increases CDX2 expression, which transactivates solute carrier family 5 member 8 (SLC5A8) to increase immune function and cell survival under several stress conditions (Figure 4C) [54,55].
2.4. Histone O-GlcNAcylation
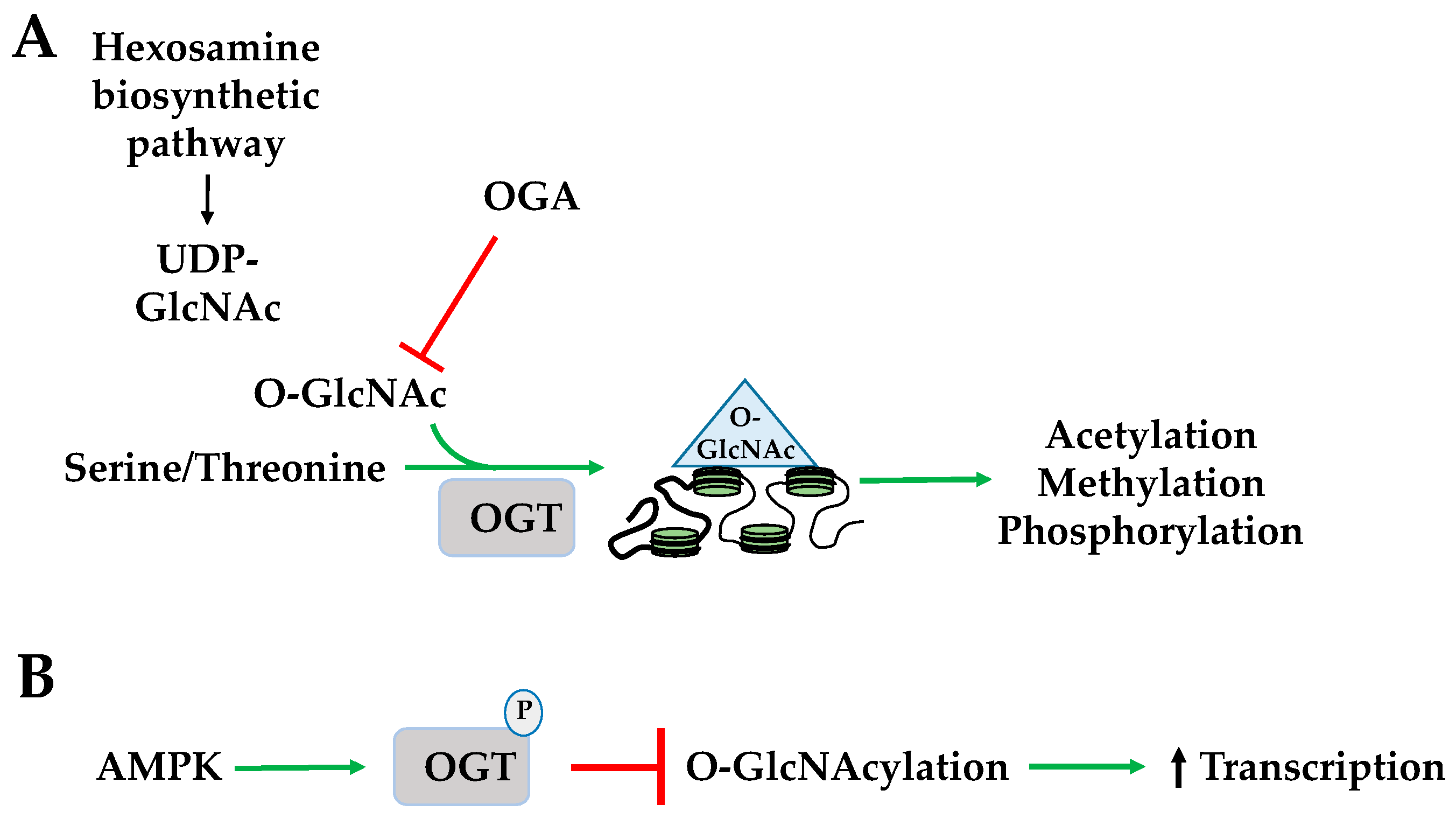
O-GlcNAcylation is the addition of an O-linked N-acetylglucosamine (O-GlcNAc) group to a serine or threonine by O-GlcNAc transferase (OGT). The donor substrate for O-GlcNAcylation, uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), is a product of the hexamine biosynthetic pathway (HBP) in which glucose, fatty acid, amino acid, and ATP metabolism converge implicating its role as a nutrient and stress sensor [56]. O-GlcNAcase (OGA) removes O-GlcNAc through hydrolysis. The dynamics of histone O-GlcNAcylation regulate a variety of activities including other histone PTMs such as acetylation, methylation, and phosphorylation, and is highly sensitive to a spectrum of cellular stressors such as hypoxia, heat shock, and starvation (Figure 5A) [57]. AMPK phosphorylates O-GlcNAc transferase (OGT) promoting its dissociation from chromatin. This inhibits its O-GlcNAcylation of H2BS112 in response to extracellular glucose through the hexosamine biosynthesis pathway (HBP) and promotes H2BK120 mono-ubiquitination and transcriptional activation (Figure 5B) [56].
2.5. Histone Ribosylation
Histone ADP-ribosylation can occur as addition of mono- or poly-ADP-ribosylation units by mono-ADP ribosyltransferases (ARTs) or poly-(ADP-ribose) polymerases (PARPs), respectively. NAD+ serves as the main source for histone ADP-ribosylation, which can occur on many amino acids restructuring chromatin into a euchromatin state [58,59]. AMPK phosphorylates and activates PARP1 [60,61]. Substrates of PARP1 include PARP1 itself and the tail of histones H1, H2A, H2B, H3, and H4, creating a poly(ADP-ribose)ylated mark at transcription start sites in transcriptionally necessary genes during metaphase [62]. Contrarily, when PARP1 is not active as a polymerase, it binds to DNA and promotes a heterochromatin state [59]. This is underscored by AMPK’s phosphorylation of PARP1, causing its dissociation from the B-cell lymphoma protein 6 (Bcl-6) to increase its expression [61].
3. DNA Modification
More abundantly enriched in promoter regions, cytosine-guanine dinucleotides (CpG) are susceptible to methylation at the 5’ cytosine position by a family of DNA methyltransferases (DNMTs). Each DNA methyltransferase member has a slightly different functional role in the global regulation of global DNA methylation. While DNMT 3a and 3b create the methyl “marks” that are carried through mitosis, DNMT1 maintains these methylation marks and regulates the dynamics of DNA modification and nucleosomal remodeling [63]. Following methylation, a number of functional changes occur. Due to its electron donating effects, methylation weakens Watson–Crick base pairing and recruits methyl-CpG binding domain proteins (MBDs), HDACs, and transcriptional repressors that collectively organize chromatin into a heterochromatic, a transcriptionally inactive conformation. These CpG methylations are highly dynamic- and stimulation-dependent. Removal of the CpG methylation requires oxidation of 5-methycytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), which is then converted to 5-formylcytosine (5-fC), and 5-carboxylcytosine (5-caC) followed by complete removal of the functional group by ten-eleven translocation hydroxylases (TETs) [64,65]. Although TETs are required to actively remove CpG methyl groups, the process occurs passively and semi-conservatively during DNA replication [66].
DNA Methylation
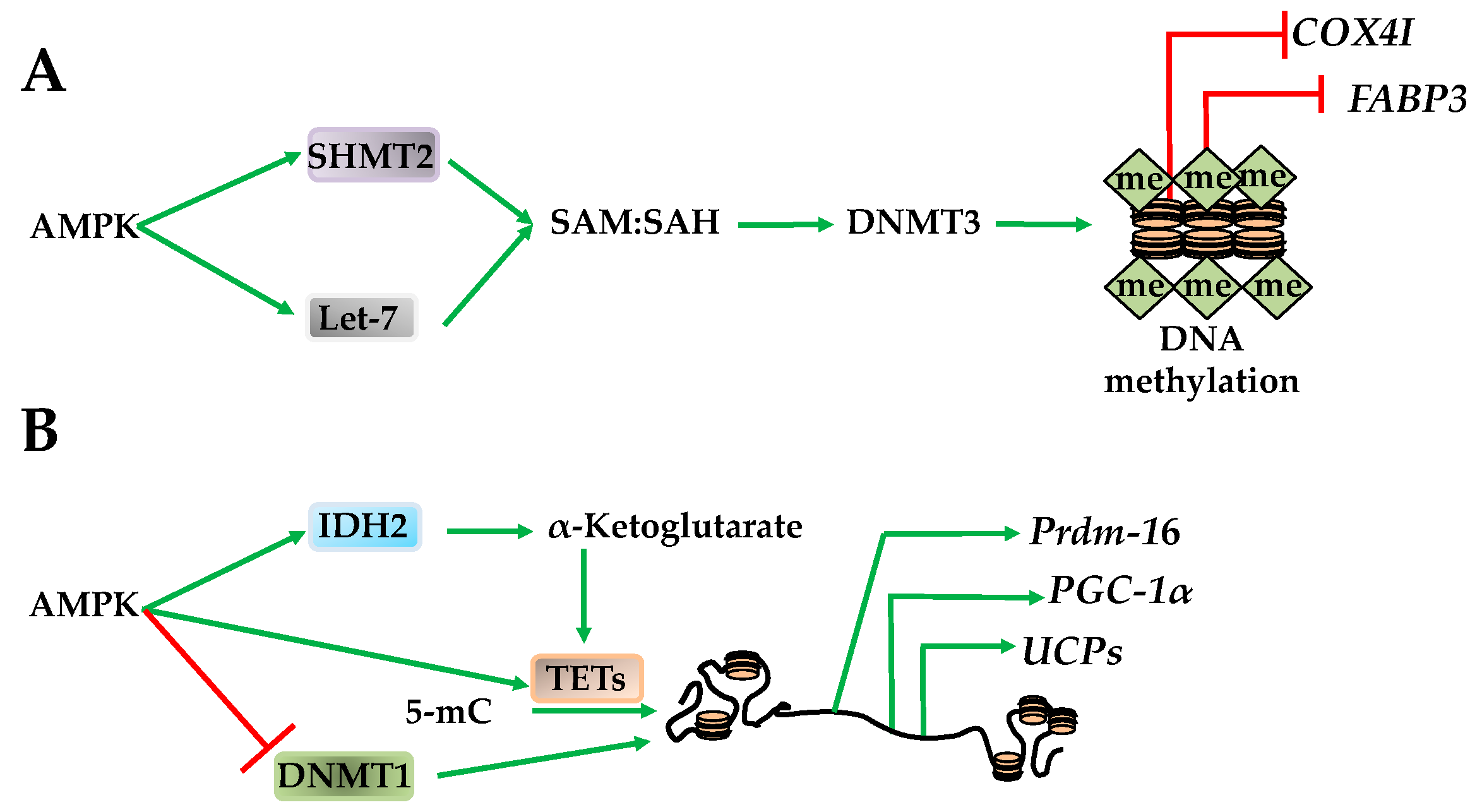
AMPK regulates global methylation by changing the substrates required for DNMT and TET activity. For example, SAM, the methyl donor for DNMT3a and DNMT3b, is converted to S-adenosylhomocysteine (SAH) upon CpG methylation [67]. Therefore, while SAM is required for DNMT3a and DNMT3b activity, SAH has an opposing inhibitory effect [67]. Supporting DNMT3a and DNMT3b activation, AMPK increases mitochondrial function and serine hydroxymethyltransferase 2 (SHMT2). Once activated, SHMT2 facilitates the one carbon transfer from folate to SAH, making SAM, and increasing the SAM/SAH ratio [67]. AMPK also increases the SAM/SAH ratio by transactivating let-7 micro RNA that subsequently degrades H19, relieving a direct inhibitory effect on S-adenosylhomocysteine hydrolase (SAHH). Subsequent activation of SAHH enables DNMT3b activation [68]. Taken together, these studies indicate that AMPK increases the SAM/SAH ratio that has an activating effect on DNMT3a and DNMT3b. These AMPK-dependent shifts in methylation also correlate to metabolic adaptive situations. For example, promoter methylation of cytochrome C oxidase subunit 4I1 (COX4I) and fatty acid binding protein 3 (FABP3) increases during exercise, while peroxisome proliferator-activated receptor δ (PPARδ) promoter methylation increases after fasting (Figure 6A) [69]. However, the overall effect AMPK has on DNA methylation is likely to be stimulation-, tissue-, promoter-, and DNMT isoform-specific. Paralleling its role as an epigenetic metabolic regulator in histone acetylation through HAT1 and RBBP7, AMPK also phosphorylates DNMT1S730, inhibiting methyl CpG in PGC-1α, Tfam, NRF1, NRF2, UCP2 and UCP3 promoters [13]. This results in improved mitochondrial function, a requirement for cell survival [13,70].
In addition to the DNMTs, AMPK also regulates TETs, which, in turn, govern loci-specific CpG methylation. The effect of AMPK on TETs occurs through its regulation of TCA cycle intermediates. For example, AMPK regulates isocitrase dehydrogenase 2 (IDH2) to increase the levels of α-ketoglutarate, an activator of TET1–TET4 resulting in CpG demethylation [47,71]. Activation of TET1–TET4 results in transactivation of the PR domain containing 16 (Prdm16) gene in progenitor cells that promotes brown adipogenesis, therefore supporting AMPK’s role as an epigenetic regulator of metabolic function (Figure 6B) [47].
4. Approaches to Elucidating the AMPK-Modulated Epigenetic Landscape
Although biochemical studies have resolved a number of mechanistic insights into the role AMPK plays in the epigenetic regulation of chromatin structure, the study of AMPK on epigenetic function is still in its infancy. A number of predictive algorithms and studies have identified a potential role of AMPK on a number of epigenetic regulators. However, the role they may play under different physiological stimulations and their tissue specificity are still unknown. Because of the interconnected nature between regulatory cascades, the study of AMPK on epigenetic function in a disease-relevant capacity requires systems biological and bioinformatics approaches.
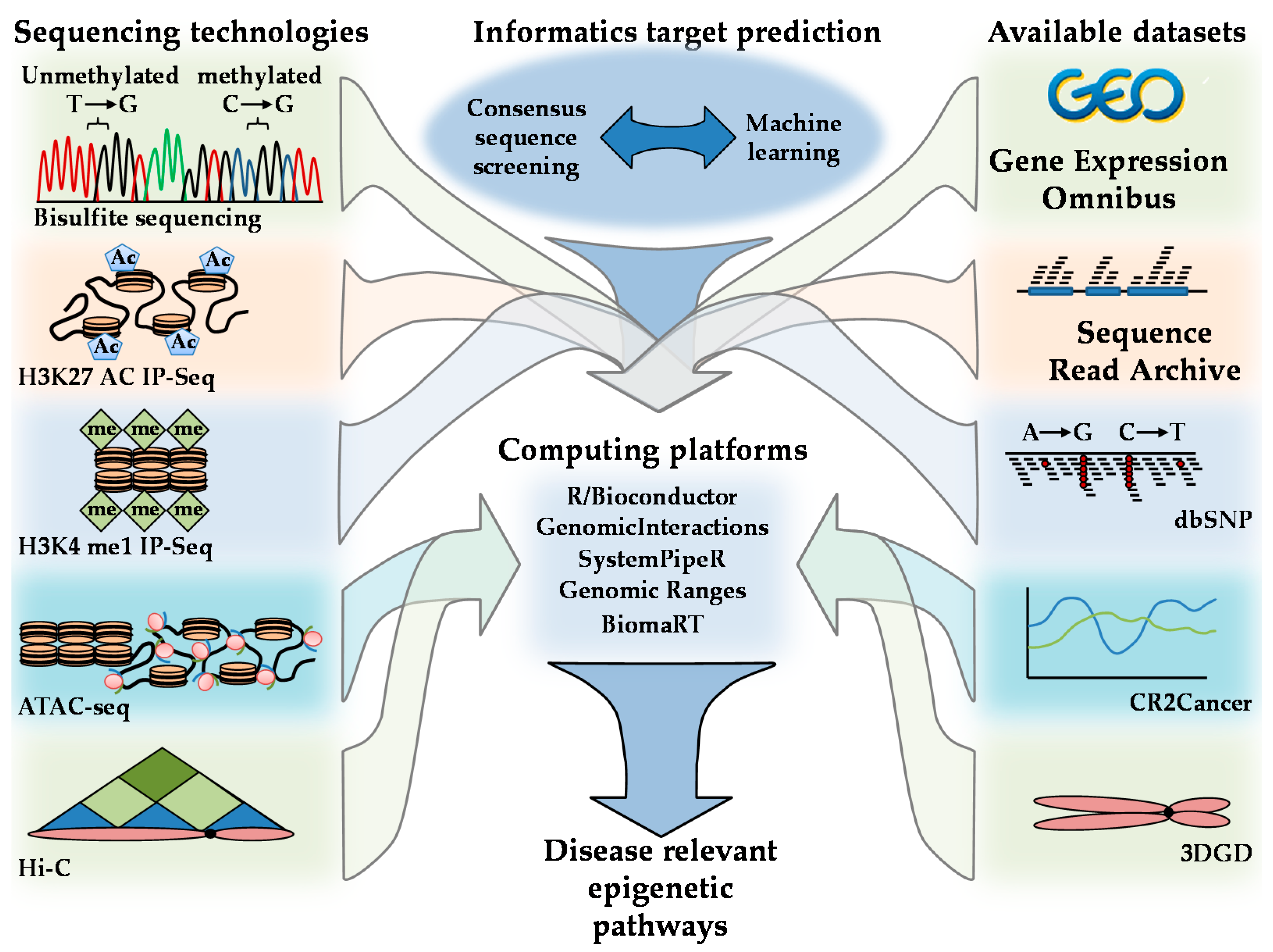
Scientific advances in the computational and Big Data arenas have resulted in novel experimental approaches, databases, and bioinformatics techniques that can be used in the exploration of AMPK epigenetic-regulated signaling pathways. Examples of tools used to explore chromatin characteristics include H3K27 acetylation H3K4 and monomethylation (H3K4me1) immunoprecipitation sequencing (IP-Seq); assay for transposase-accessible chromatin using sequencing (ATAC-seq); assay for transposase-accessible chromatin using sequencing (ATAC-seq); and Hi-C [72]. The relationship between DNA, histone modifications, histone remodeling, followed by Hi-C structural mapping, provides an integrative understanding of how AMPK can influence chromatin architecture. The identification of AMPK-regulated networks can be explored using consensus sequence mapping and machine learning in computing environments such as R/Bioconductor computing [25,73,74,75]. The expression of effected genes and loci and their application of these loci to disease-relevant stimuli can be further explored via cross-referencing with expression profiles housed in the Gene Expression Omnibus (GEO) database, Sequence Read Archive (SRA), Single Nucleotide Polymorphism database (dbSNP), 3D Genome database C (3DGD), and CR2Cancer [76,77,78,79]. Integration of these datasets can provide a comprehensive picture of the influence AMPK has on epigenetic signaling cascades in addition to their genetic loci and disease-specific regulation (Figure 7).
5. Conclusions
AMPK’s role as an epigenetic landscape modulator is underscored by its multifunctional kinase effects that regulate histones and epigenetic enzymes to mediate histone and DNA modifications. AMPK is crucial for cell survival and adaptation as evident by its activation upon nutrient depletion and stressors resulting in histone phosphorylation, acetylation, methylation, O-GlcNAcylation, and ribosylation. Additional, AMPK regulates DNA methylation mediated by influencing the SAM/SAH ratio, TET regulation and inhibition of DNMT1. Technological advances in data analysis continue to reveal the remarkable and diverse roles of AMPK in signaling pathways and epigenetic regulation, providing opportunities to exploit novel therapies relating to health and disease.
Funding
This research received no external funding.
Acknowledgments
We would like to acknowledge David A. Johnson for his thoughtful review of the manuscript and comment to aid in clarity. We would also like to acknowledge John Shyy for his continued support and guidance.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMPK | AMP-activated protein kinase |
| CPT1C | carnitine palmitoyltransferase 1C |
| p21 | cyclin-dependent kinase inhibitor |
| p53 | TP53 or tumor protein |
| HAT | histone deacetylases |
| acetyl-CoA | acetyl coenzyme-A |
| HDAC | histone deacetylases |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| ACC | acetyl-CoA carboxylase |
| ACSS2 | acetyl-CoA synthetase short-chain family member 2 |
| TFEB | transcription factor EB |
| SIRT | sirtuin |
| NAD+ | nicotinamide adenine dinucleotide |
| βOHB | β-hydroxybutyrate |
| RBBP7 | retinoblastoma binding protein 7 |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator-1α |
| Tfam | transcription factor A |
| NRF | nuclear respiratory factors |
| UCP | uncoupling proteins |
| SAM | S-adenosyl methionine |
| GLUT-4 | glucose transporter type 4 |
| KDM2A | lysine-specific demethylase 2A |
| KDM5 | demethyltransferase lysine demethylase 5 |
| LSD1 | lysine-specific histone demethylase-1 |
| PRC2 | polycomb repressive complex 2 |
| COMPASS | complex proteins associated with Set1 |
| EZH2 | enhancer of zeste homolog 2 |
| Cdx2 | caudal type homeobox2 |
| SLC5A8 | solute carrier family 5 member 8 |
| O-GlcNAc | O-linked N-acetylglucosamine |
| OGT | O-GlcNAc transferase |
| UDP-GlcNAc | uridine diphosphate N-acetylglucosamine |
| HBP | hexosamine biosynthesis pathway |
| ART | mono-ADP ribosyltransferase |
| PTM | Post translational modification |
| PARP | poly-(ADP-ribose) polymerase |
| Bcl-6 | B-cell Lymphoma protein 6 |
| CpG | cytosine-guanine dinucleotides |
| DNMT | DNA methyltransferase |
| MBD | methyl-CpG binding domain proteins |
| 5-mC | 5-methycytosine |
| 5-hmC | 5-hydroxymethylcytosine |
| 5-fC | 5-formylcytosine |
| 5-caC | 5-carboxylcytosine |
| TETs | ten-eleven translocation hydroxylases |
| SAH | S-adenosylhomocysteine |
| SHMT2 | serine hydroxymethyltransferase 2 |
| SAHH | S-adenosylhomocysteine hydrolase |
| COX4I | cytochrome C oxidase subunit 4I1 |
| FABP3 | fatty acid binding protein 3 |
| PPARδ | peroxisome proliferator-activated receptor δ |
| TCA | tricarboxylic acid |
| IDH2 | isocitrase dehydrogenase 2 |
| IP-Seq | immunoprecipitation sequencing |
| ATAC-seq | assay for transposase-accessible chromatin using sequencing |
| GEO | gene expression omnibus |
| SRA | sequence read archive |
| dbSNP | single nucleotide polymorphism database |
| 3DGD | 3D genome database |
References
- Trerotola, M.; Relli, V.; Simeone, P.; Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genom. 2015, 28, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Bunkar, N.; Pathak, N.; Lohiya, N.K.; Mishra, P.K. Epigenetics: A key paradigm in reproductive health. Clin. Exp. Reprod. Med. 2016, 43, 59–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, M.J.; Nazor, K.L.; Loring, J.F. Epigenetic regulation of pluripotency and differentiation. Circ. Res. 2014, 115, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Nitert, M.D.; Dayeh, T.; Volkov, P.; Elgzyri, T.; Hall, E.; Nilsson, E.; Yang, B.T.; Lang, S.; Parikh, H.; Wessman, Y.; et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012, 61, 3322–3332. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, G.; Kim, Y.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different gamma subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Vara-Ciruelos, D.; Dandapani, M.; Gray, A.; Egbani, E.O.; Evans, A.M.; Hardie, D.G. Genotoxic Damage Activates the AMPK-α1 Isoform in the Nucleus viaCa2+/CaMKK2 Signaling to Enhance Tumor Cell Survival. Mol. Cancer Res. 2018, 16, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Langendorf, C.G.; Ngoei, K.R.; Scott, J.W.; Ling, N.X.; Issa, S.M.; Gorman, M.A.; Parker, M.W.; Sakamoto, K.; Oakhill, J.S.; Kemp, B.E. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat. Commun. 2016, 7, 10912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, T.L.; Gongol, B.; Zhang, F.; Martin, M.; Johnson, D.A.; Xiao, H.; Wang, Y.; Subramaniam, S.; Chien, S.; Shyy, J.Y. AMPK promotes mitochondrial biogenesis and function by phosphorylating the epigenetic factors DNMT1, RBBP7, and HAT1. Sci. Signal. 2017, 10, eaaf7478. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Mylka, V.; Bougarne, N.; Pawlak, M.; Caron, S.; Hennuyer, N.; Paumelle, R.; De Cauwer, L.; Thommis, J.; Rider, M.H.; et al. Chromatin recruitment of activated AMPK drives fasting response genes co-controlled by GR and PPARα. Nucleic Acids Res. 2016, 44, 10539–10553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, M.; Kawata, M.; McLysaght, A.; Makino, T. Evolution of vertebrate tissues driven by differential modes of gene duplication. DNA Res. 2012, 19, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 2018, 15, 20170774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef]
- Zhu, M.J.; Han, B.; Tong, J.; Ma, C.; Kimzey, J.M.; Underwood, K.R.; Xiao, Y.; Hess, B.W.; Ford, S.P.; Nathanielsz, P.W.; et al. AMP-activated protein kinase signaling pathways are down regulated and skeletal muscle development impaired in fetuses of obese, over-nourished sheep. J. Physiol. 2008, 586, 2651–2664. [Google Scholar] [CrossRef] [PubMed]
- Demoinet, E.; Li, S.; Roy, R. AMPK blocks starvation-inducible transgenerational defects in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2017, 114, E2689–E2698. [Google Scholar] [CrossRef] [PubMed]
- Bönisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.S.; Srebreva, L.N.; Banchev, T.B.; Tasheva, B.T.; Tsanev, R.G. Cytoplasmic pool of histone H1 in mammalian cells. J. Cell Sci. 1990, 96, 461–468. [Google Scholar] [PubMed]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicka, A.; Seiser, C. Sensing core histone phosphorylation- a matter of perfect timing. Biochim. Biophys. Acta 2014, 1839, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.L.; Gongol, B.; Martin, M.; King, S.J.; Smith, L.; Johnson, D.A.; Subramaniam, S.; Chien, S.; Shyy, J.Y. Identification of AMP-activated protein kinase targets by a consensus sequence search of the proteome. BMC Syst. Biol. 2015, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bungard, D.; Fuerth, B.J.; Zeng, P.Y.; Faubert, B.; Maas, N.L.; Viollet, B.; Carling, D.; Thompson, C.B.; Jones, R.G.; Berger, S.L. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 2010, 329, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Roa-Mansergas, X.; Fadó, R.; Atari, M.; Mir, J.F.; Muley, H.; Serra, D.; Casals, N. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci. Rep. 2018, 8, 6997. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I.; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, D.R.; Howard, T.L.; Nyun, T.; Hollenberg, S.M. CHMP1 is a novel nuclear matrix protein affecting chromatin structure and cell-cycle progression. J. Cell Sci. 2001, 114, 2383–2393. [Google Scholar] [PubMed]
- Javaid, N.; Choi, S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Bulusu, V.; Tumanov, S.; Michalopoulou, E.; van den Broek, N.J.; MacKay, G.; Nixon, C.; Dhayade, S.; Schug, Z.T.; Vande Voorde, J.; Blyth, K.; et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017, 18, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol. Cell 2017, 66, 684–697. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Joven, J.; Menendez, J.A. Metformin targets histone acetylation in cancer prone epithelial cells. Cell Cycle. 2016, 15, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK/Snf1 signaling regulates histone acetylation: Impact on gene expression and epigenetic functions. Cell. Signal. 2016, 28, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kraus, W.L. SIRT1-dependent regulation of chromatin and transcription: Linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim. Biophys. Acta 2010, 1804, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Yue, W.; Oppermann, U.; Klose, R. Dynamic protein methylation in chromatin biology. Cell. Mol. Life Sci. 2009, 66, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Feitag, M. Histone Methylation by SET Domain Proteins in Fungi. Annu. Rev. Microbiol. 2017, 71, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Li, C.; Yin, Z.; Wen, J.; Meng, H.; Xue, L.; Wang, J. Histone methylation in DNA repair and clinical practice: New findings during the past 5-years. J. Cancer 2018, 9, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liang, X.; Sun, X.; Zhang, L.; Fu, X.; Rogers, C.; Berim, A.; Zhang, S.; Wang, S.; Wang, B.; et al. AMPK/α-ketoglutarate axis dynamically mediates DNA demethylation in the Prdm16 promoter and brown adipogenesis. Cell Metab. 2016, 24, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, Q.; Li, J.; Hu, B.; Zhao, Q.; Ma, C.; Huang, W.; Zhuo, L.; Fang, H.; Liao, L.; et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat. Cell Biol. 2017, 19, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Shilatifard, A. Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev. Biol. 2010, 339, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Xu, K.; Wei, Y.; Zhang, J.; Han, T.; Fry, C.; Zhang, Z.; Wang, Y.V.; Huang, L.; Yuan, M.; et al. Phosphorylation of EZH2 by AMPK Suppresses PRC2 Methyltransferase Activity and Oncogenic Function. Mol. Cell 2018, 69, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Guo, J.; Zhu, Y.; Huang, Z.; Liu, T.; Cai, J.; Yu, L.; Wang, Z. Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. Int. J. Oncol. 2018, 52, 1899–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloia, L.; Di Stefano, B.; Di Croce, L. Polycomb complexes in stem cells and embryonic development. Development 2013, 140, 2525–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakizaki, F.; Aoki, K.; Miyoshi, H.; Carrasco, N.; Aoki, M.; Taketo, M.M. CDX transcription factors positively regulate expression of solute carrier family 5, member 8 in the colonic epithelium. Gastroenterology 2010, 138, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Gurav, A.; Sivaprakasam, S.; Bhutia, Y.D.; Boettger, T.; Singh, N.; Ganapathy, V. Slc5a8, a Na+-coupled high-affinity transporter for short-chain fatty acids, is a conditional tumour suppressor in colon that protects against colitis and colon cancer under low-fibre dietary conditions. Biochem. J. 2015, 469, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Dehennaut, V.; Leprince, D.; Lefebvre, T. O-GlcNAcylation, an Epigenetic Mark. Focus on the Histone Code, TETFamily Proteins, and Polycomb Group Proteins. Front. Endocrinol. 2014, 5, 155. [Google Scholar] [CrossRef] [PubMed]
- Poirier, G.G.; de Murcia, G.; Jongstra-Bilen, J.; Niedergang, C.; Mandel, P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. USA 1982, 79, 3423–3427. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Zampieri, M.; Caiafa, P. PARP1 orchestrates epigenetic events setting up chromatin domains. Semin. Cell Dev. Biol. 2017, 63, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.W.; Jijon, H.B.; Madsen, K.L. AMP-activated protein kinase is a positive regulator of poly(ADP-ribose) polymerase. Biochem. Biophys. Res. Commun. 2006, 342, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Gongol, B.; Marin, T.; Peng, I.C.; Woo, B.; Martin, M.; King, S.; Sun, W.; Johnson, D.A.; Chien, S.; Shyy, J.Y. AMPKα2 exerts its anti-inflammatory effects through PARP-1 and Bcl-6. Proc. Natl. Acad. Sci. USA 2013, 110, 3161–3166. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, N.; Kossenkov, A.V.; Tulin, A.V. Bookmarking promoters in mitotic chromatin: Poly (ADP-ribose) polymerase-1 as an epigenetic mark. Nucleic Acids Res. 2014, 42, 7028–7038. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.; Qiu, H.; Kim, S.; Jjingo, D.; Hoffman, R.; Kim, C.W.; Jang, I.; Son, D.J.; Kim, D.; Pan, C.; et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J. Clin. Investig. 2014, 124, 3187–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, P.W.; Amouroux, R.; Hajkova, P. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: An emerging complex story. Genomics 2014, 104, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin regulates global DNA methylation via mitochondrial one-carbon metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin alters DNA methylation genome-wide via the H19/SAHH axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Lane, S.C.; Camera, D.M.; Lassiter, D.G.; Areta, J.L.; Bird, S.R.; Yeo, W.K.; Jeacocke, N.A.; Krook, A.; Zierath, J.R.; Burke, L.M.; et al. Effects of sleeping with reduced carbohydrate availability on acute training responses. J. Appl. Physiol. 2015, 119, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, C.M.; Chandel, N.S. Mitochondrial regulation of cell survival and death during low-oxygen conditions. Antioxid. Redox Signal. 2009, 11, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Mortazavi, A. Integrating ChIP-seq with other functional genomics data. Brief. Funct. Genom. 2018, 17, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Tao, W.A. Current technologies to identify protein kinase substrates in high throughput. Front. Biol. 2013, 8, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmston, N.; Ing-Simmons, E.; Perry, M.; Barešić, A.; Lenhard, B. GenomicInteractions: An R/Bioconductor package for manipulating and investigating chromatin interaction data. BMC Genom. 2015, 16, 963. [Google Scholar] [CrossRef] [PubMed]
- Backman, T.W.H.; Girke, T. systemPipeR: NGS workflow and report generation environment. BMC Bioinform. 2016, 20, 388. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P.; Morales, M.E.; White, T.B.; Baddoo, M.; Hedges, D.J.; Servant, G.; Srivastav, S.; Smither, M.E.; Concha, M.; DeHaro, D.L.; et al. A comprehensive approach to expressionof L1 loci. Nucleic Acids Res. 2017, 45, e31. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, M. Searching NCBI’s dbSNP database. Curr. Protoc. Bioinform. 2010. [Google Scholar] [CrossRef]
- Li, C.; Dong, X.; Fan, H.; Wang, C.; Ding, G.; Li, Y. The 3DGD: A database of genome 3D structure. Bioinformatics 2014, 30, 1640–1642. [Google Scholar] [CrossRef] [PubMed]
- Ru, B.; Sun, J.; Tong, Y.; Wong, C.N.; Chandra, A.; Tang, A.T.S.; Chow, L.K.Y.; Wun, W.L.; Levitskaya, Z.; Zhang, J. CR2Cancer: A database for chromatin regulators in human cancer. Nucleic Acids Res. 2018, 46, D918–D924. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
AMPK (AMP-activated protein kinase) promotes cell survival through histone phosphorylation. AMPK phosphorylates H2B to promote chromatin relaxation at tumor protein p53 (p53) recognized promoters and transcription of carnitine palmitoyltransferase 1C (cpt1c) and cyclin-dependent kinase inhibitor (p21) to enhance cell survival.
Figure 1.
AMPK (AMP-activated protein kinase) promotes cell survival through histone phosphorylation. AMPK phosphorylates H2B to promote chromatin relaxation at tumor protein p53 (p53) recognized promoters and transcription of carnitine palmitoyltransferase 1C (cpt1c) and cyclin-dependent kinase inhibitor (p21) to enhance cell survival.

Figure 2.
AMPK activation increases acetyl-CoA and promotes histone acetylation. (A) Through phosphorylation, AMPK inhibits acetyl-CoA carboxylase (ACC) while activating acetyl-CoA synthetase short-chain family member 2 (ACSS2) to increase acetyl-CoA availability for acetylation; (B) AMPK increases β-hydroxybutyrate (βOHB) to inhibit histone deacetylases (HDACs) and promotes HDAC nuclear export via hyperphosphorylation. Both of these events increase histone acetylation.
Figure 2.
AMPK activation increases acetyl-CoA and promotes histone acetylation. (A) Through phosphorylation, AMPK inhibits acetyl-CoA carboxylase (ACC) while activating acetyl-CoA synthetase short-chain family member 2 (ACSS2) to increase acetyl-CoA availability for acetylation; (B) AMPK increases β-hydroxybutyrate (βOHB) to inhibit histone deacetylases (HDACs) and promotes HDAC nuclear export via hyperphosphorylation. Both of these events increase histone acetylation.

Figure 3.
AMPK inhibits histone deacetylase activity to promote histone acetylation. (A) AMPK phosphorylates histone acetylase1 and retinoblastoma binding protein 7 (RBBP7) to form a complex that acetylates histones at promoters of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), uncoupling proteins (UCPs) to increase mitochondrial biogenesis and function; (B) AMPK phosphorylates and inhibits HDAC5 enhancing acetylation at the glucose transporter type 4 (GLUT-4) promoter increasing its transcription.
Figure 3.
AMPK inhibits histone deacetylase activity to promote histone acetylation. (A) AMPK phosphorylates histone acetylase1 and retinoblastoma binding protein 7 (RBBP7) to form a complex that acetylates histones at promoters of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), uncoupling proteins (UCPs) to increase mitochondrial biogenesis and function; (B) AMPK phosphorylates and inhibits HDAC5 enhancing acetylation at the glucose transporter type 4 (GLUT-4) promoter increasing its transcription.

Figure 4.
AMPK regulates histone methylation. (A) AMPK phosphorylates fumarase increase fumarate, which inhibits lysine-specific demethylase 2A (KDM2A), increasing histone methylation at cell growth; (B) AMPK decreases histone methylation through inhibition of polycomb repressive complex 2 (PRC2) and complex proteins associated with set1 (COMPASS complex); (C) AMPK modulates histone 3 methylations status to increase caudal type homeobox2 [cell differentiation]) (Cdx2) expression and metabolic homeostasis. LSD1: lysine-specific histone demethylase-1; EZH2: zeste homolog 2.
Figure 4.
AMPK regulates histone methylation. (A) AMPK phosphorylates fumarase increase fumarate, which inhibits lysine-specific demethylase 2A (KDM2A), increasing histone methylation at cell growth; (B) AMPK decreases histone methylation through inhibition of polycomb repressive complex 2 (PRC2) and complex proteins associated with set1 (COMPASS complex); (C) AMPK modulates histone 3 methylations status to increase caudal type homeobox2 [cell differentiation]) (Cdx2) expression and metabolic homeostasis. LSD1: lysine-specific histone demethylase-1; EZH2: zeste homolog 2.

Figure 5.
AMPK regulates histone O-GlcNAcylation and ADP-ribosylation. (A) O-GlcNAc transferase adds O-GlcNAc to a serine or threonine to produce O-GlcNAcylation. Nutrient and stress sensor stimulates hexosamine biosynthetic pathway (HBP) to create UDP-GlcNAc, which is a donor substrate for O-GlcNAcylation. Histone O-GlcNAcylation regulates acetylation, methylation, and phosphorylation; (B) AMPK phosphorylates O-GlcNAc transferase and inhibits O-GlcNAcylation to increase transcription. OGA: O-GlcNAcase; OGT: O-GlcNAc transferase.
Figure 5.
AMPK regulates histone O-GlcNAcylation and ADP-ribosylation. (A) O-GlcNAc transferase adds O-GlcNAc to a serine or threonine to produce O-GlcNAcylation. Nutrient and stress sensor stimulates hexosamine biosynthetic pathway (HBP) to create UDP-GlcNAc, which is a donor substrate for O-GlcNAcylation. Histone O-GlcNAcylation regulates acetylation, methylation, and phosphorylation; (B) AMPK phosphorylates O-GlcNAc transferase and inhibits O-GlcNAcylation to increase transcription. OGA: O-GlcNAcase; OGT: O-GlcNAc transferase.

Figure 6.
AMPK regulates DNA methylation. (A) AMPK activates serine hydroxymethyltransferase 2 (SHMT2) and Let-7 to increase SAM:SAH ratio providing substrate for DNA methyltransferase 3 (DNMT3) methylation of promoters such as COX4I and FABP3; (B) AMPK directly regulates isocitrase dehydrogenase 2 (IDH2) to yield α-ketoglutarates to promote ten-eleven translocation hydroxylases (TETs) activation. AMPK likely also directly regulates TETs catalyzing the conversion of 5-mc to unmethylated cytosine while inhibiting DNA methyltransferase 1 (DNMT1). These actions result in decreased promoter methylation of PR domain containing 16 (Prdm16), PGC-1α, and the uncoupler proteins.
Figure 6.
AMPK regulates DNA methylation. (A) AMPK activates serine hydroxymethyltransferase 2 (SHMT2) and Let-7 to increase SAM:SAH ratio providing substrate for DNA methyltransferase 3 (DNMT3) methylation of promoters such as COX4I and FABP3; (B) AMPK directly regulates isocitrase dehydrogenase 2 (IDH2) to yield α-ketoglutarates to promote ten-eleven translocation hydroxylases (TETs) activation. AMPK likely also directly regulates TETs catalyzing the conversion of 5-mc to unmethylated cytosine while inhibiting DNA methyltransferase 1 (DNMT1). These actions result in decreased promoter methylation of PR domain containing 16 (Prdm16), PGC-1α, and the uncoupler proteins.

Figure 7.
Methods for resolving disease-relevant AMPK-regulated epigenetic networks. Developed sequencing technologies are listed on the left-hand figure panels. Publicly available data repositories are listed on the right. Computing platforms R and relevant Bioconductor packages including Genomic interactions, SystemPipeR, Genomic Ranges, and BioMart can be used to integrate sequencing technologies, publicly available datasets, and AMPK target predictions as illustrated in the center panels.
Figure 7.
Methods for resolving disease-relevant AMPK-regulated epigenetic networks. Developed sequencing technologies are listed on the left-hand figure panels. Publicly available data repositories are listed on the right. Computing platforms R and relevant Bioconductor packages including Genomic interactions, SystemPipeR, Genomic Ranges, and BioMart can be used to integrate sequencing technologies, publicly available datasets, and AMPK target predictions as illustrated in the center panels.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gongol, B.; Sari, I.; Bryant, T.; Rosete, G.; Marin, T. AMPK: An Epigenetic Landscape Modulator. Int. J. Mol. Sci. 2018, 19, 3238. https://doi.org/10.3390/ijms19103238
AMA Style
Gongol B, Sari I, Bryant T, Rosete G, Marin T. AMPK: An Epigenetic Landscape Modulator. International Journal of Molecular Sciences. 2018; 19(10):3238. https://doi.org/10.3390/ijms19103238
Chicago/Turabian StyleGongol, Brendan, Indah Sari, Tiffany Bryant, Geraldine Rosete, and Traci Marin. 2018. "AMPK: An Epigenetic Landscape Modulator" International Journal of Molecular Sciences 19, no. 10: 3238. https://doi.org/10.3390/ijms19103238
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.