Homology Model and Docking-Based Virtual Screening for Ligands of Human Dyskerin as New Inhibitors of Telomerase for Cancer Treatment

 ,
,
Abstract
:1. Introduction
2. Results
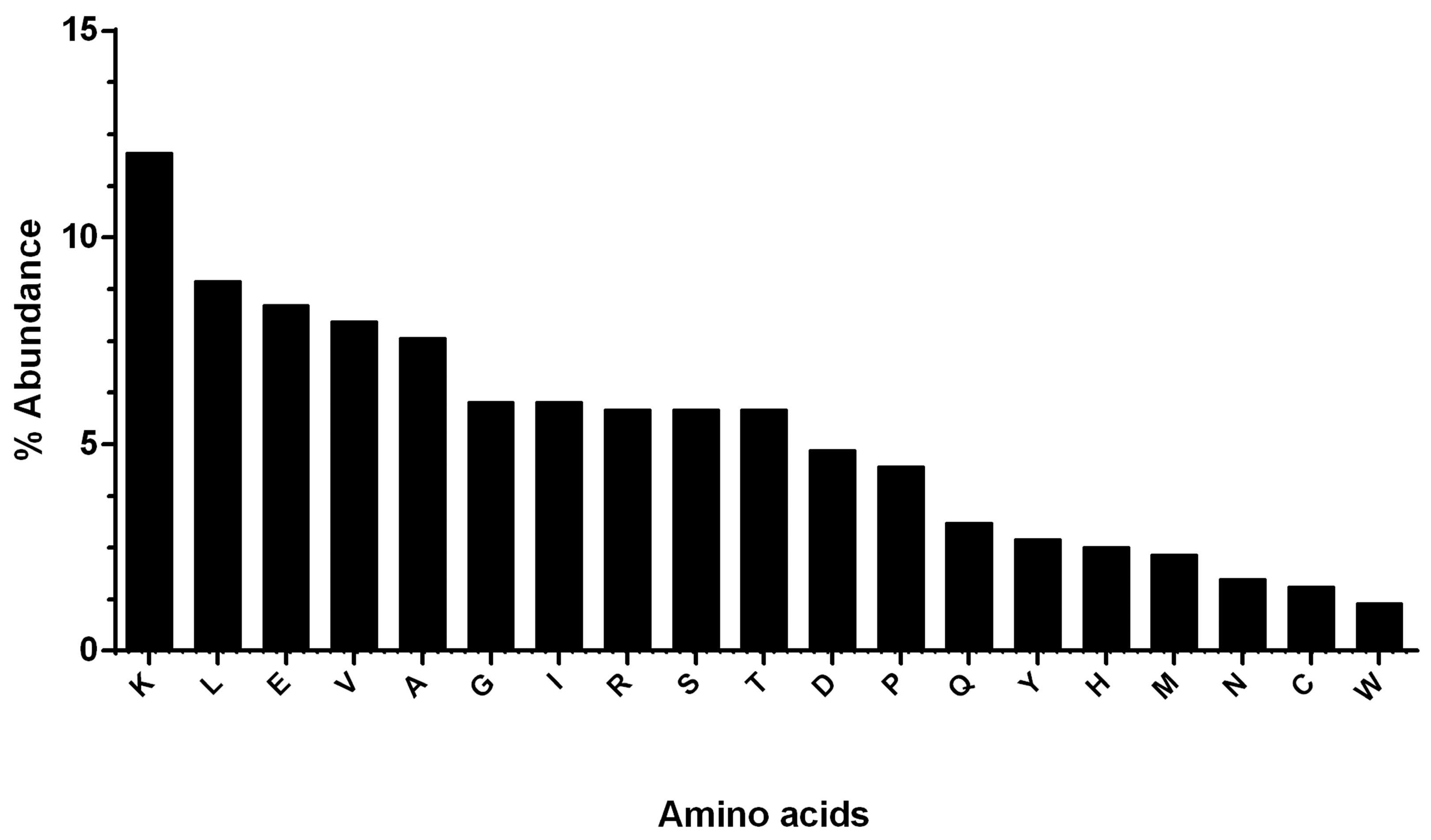
2.1. Physicochemical Properties and the Abundance of aa in hDKC1 Protein
2.2. Prediction of the Two-Dimensional Structure of hDKC1
2.3. Sequence and Secondary Structure Analysis between hDKC1 and Saccharomyces Cerevisiae Dyskerin
2.4. Predicted 3D Homology Model of hDKC1 by I-TASSER
2.5. 3D Structure Validation
2.6. Study of Mutation Stability
2.7. Evaluation and Recognition of Hydrophobic Pockets on hDKC1 Model
2.8. Study and Determination of the Hydrophobic Pocket Containing the Mutation K314
2.9. Docking Based Virtual Screening on the hDKC1 Model by AutoDock Vina
2.10. In Vitro Screening of the Candidate Compounds by Telomerase Activity Assay
3. Discussion
4. Materials and Methods
4.1. Sequence Obtaining and Physicochemical Parameters Analysis of hDKC1
4.2. Secondary Structure Prediction
4.3. Modelling of 3D Structure of hDKC1
4.4. Validation of the Generated Model
4.5. Mutation Stability Analysis
4.6. Hydrophobic Pockets Identification
4.7. Docking Based Virtual Screening
4.8. Cell Line and Culture Conditions
4.9. Determination of Telomerase Activity
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alonso, D.F.; Ripoll, G.V.; Garona, J.; Iannucci, N.B.; Gomez, D.E. Metastasis: Recent discoveries and novel perioperative treatment strategies with particular interest in the hemostatic compound desmopressin. Curr. Pharm. Biotechnol. 2011, 12, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Mengual Gomez, D.L.; Armando, R.G.; Farina, H.G.; Gomez, D.E. Telomerase and telomere: Their structure and dynamics in health and disease. Medicina 2014, 74, 69–76. [Google Scholar] [PubMed]
- Gomez, D.E.; Armando, R.G.; Farina, H.G.; Menna, P.L.; Cerrudo, C.S.; Ghiringhelli, P.D.; Alonso, D.F. Telomere structure and telomerase in health and disease (review). Int. J. Oncol. 2012, 41, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Ashbridge, B.; Orte, A.; Yeoman, J.A.; Kirwan, M.; Vulliamy, T.; Dokal, I.; Klenerman, D.; Balasubramanian, S. Single-molecule analysis of the human telomerase RNA·dyskerin interaction and the effect of dyskeratosis congenita mutations. Biochemistry 2009, 48, 10858–10865. [Google Scholar] [CrossRef] [PubMed]
- Cerrudo, C.S.; Ghiringhelli, P.D.; Gomez, D.E. Protein universe containing a PUA RNA-binding domain. FEBS J. 2014, 281, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Angrisani, A.; Vicidomini, R.; Turano, M.; Furia, M. Human dyskerin: Beyond telomeres. Biol. Chem. 2014, 395, 593–610. [Google Scholar] [CrossRef] [PubMed]
- Garofola, C.; Gross, G.P. Dyskeratosis Congenita; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Dokal, I. Dyskeratosis congenita. In Hematology ASH Education Program; American Society of Hematology: Washington, DC, USA, 2011; Volume 2011, pp. 480–486. [Google Scholar]
- Zeng, X.L.; Thumati, N.R.; Fleisig, H.B.; Hukezalie, K.R.; Savage, S.A.; Giri, N.; Alter, B.P.; Wong, J.M. The accumulation and not the specific activity of telomerase ribonucleoprotein determines telomere maintenance deficiency in X-linked dyskeratosis congenita. Hum. Mol. Genet. 2012, 21, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Arndt, G.M.; MacKenzie, K.L. New prospects for targeting telomerase beyond the telomere. Nat. Rev. Cancer 2016, 16, 508–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinform. 2009, 10, S8. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, P.; Zhang, L.; Huang, J.; Zhu, K.; Luo, C. Current Strategies and Applications for Precision Drug Design. Front. Pharmacol. 2018, 9, 787. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.K. Editorial: In silico drug design and medicinal chemistry. Curr. Top. Med. Chem. 2015, 15, 971–972. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zhao, J.; Cui, C.; Fu, Z.; Li, X.; Liu, X.; Ding, X.; Tan, X.; Li, F.; Luo, X.; et al. Computational chemical biology and drug design: Facilitating protein structure, function, and modulation studies. Med. Res. Rev. 2018, 38, 914–950. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Comin, M.J.; Hornos, L.; Gonzalez, N.; Defelipe, L.; Turjanski, A.G.; Alonso, D.F.; Gomez, D.E.; Menna, P.L. Preclinical development of novel Rac1-GEF signaling inhibitors using a rational design approach in highly aggressive breast cancer cell lines. Anticancer Agents Med. Chem. 2014, 14, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Duan, J.; Li, D.; Ma, S.; Ye, K. Structure of the Shq1-Cbf5-Nop10-Gar1 complex and implications for H/ACA RNP biogenesis and dyskeratosis congenita. EMBO J. 2011, 30, 5010–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Wang, Z.; Cascio, D.; Feigon, J. Structure and interactions of the CS domain of human H/ACA RNP assembly protein Shq1. J. Mol. Biol. 2015, 427, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Rashid, R.; Liang, B.; Baker, D.L.; Youssef, O.A.; He, Y.; Phipps, K.; Terns, R.M.; Terns, M.P.; Li, H. Crystal structure of a Cbf5-Nop10-Gar1 complex and implications in RNA-guided pseudouridylation and dyskeratosis congenita. Mol. Cell 2006, 21, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ye, K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 2006, 443, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. Ab initio protein structure prediction using chunk-TASSER. Biophys. J. 2007, 93, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995, 23, ii–iv. [Google Scholar] [CrossRef] [PubMed]
- Cozzetto, D.; Kryshtafovych, A.; Tramontano, A. Evaluation of CASP8 model quality predictions. Proteins 2009, 77 (Suppl. 9), 157–166. [Google Scholar] [CrossRef] [Green Version]
- Abdelmonsef, A.H.; Dulapalli, R.; Dasari, T.; Padmarao, L.S.; Mukkera, T.; Vuruputuri, U. Identification of Novel Antagonists for Rab38 Protein by Homology Modeling and Virtual Screening. Comb. Chem. High Throughput Screen. 2016, 19, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, S.; Jamil, F.; Ali, A.; Sehgal, S.A. Comparative Modeling, Molecular Docking, and Revealing of Potential Binding Pockets of RASSF2; a Candidate Cancer Gene. Interdiscip. Sci. 2017, 9, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Rout, S.; Mahapatra, R.K. In silico screening of novel inhibitors of M17 Leucine Amino Peptidase (LAP) of Plasmodium vivax as therapeutic candidate. Biomed. Pharmacother. 2016, 82, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Rashidieh, B.; Madani, Z.; Azam, M.K.; Maklavani, S.K.; Akbari, N.R.; Tavakoli, S.; Rigi, G. Molecular docking based virtual screening of compounds for inhibiting sortase A in L. monocytogenes. Bioinformation 2015, 11, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Aruleba, R.T.; Adekiya, T.A.; Oyinloye, B.E.; Kappo, A.P. Structural Studies of Predicted Ligand Binding Sites and Molecular Docking Analysis of Slc2a4 as a Therapeutic Target for the Treatment of Cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Adekiya, T.A.; Aruleba, R.T.; Khanyile, S.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Structural Analysis and Epitope Prediction of MHC Class-1-Chain Related Protein-A for Cancer Vaccine Development. Vaccines (Basel) 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.T.; Meier, U.T. RNA-guided isomerization of uridine to pseudouridine--pseudouridylation. RNA Biol. 2014, 11, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.A. Dyskeratosis Congenita. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Halim, S.A.; Khan, S.; Khan, A.; Wadood, A.; Mabood, F.; Hussain, J.; Al-Harrasi, A. Targeting Dengue Virus NS-3 Helicase by Ligand based Pharmacophore Modeling and Structure based Virtual Screening. Front. Chem. 2017, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Gonzalez, N.; Maggio, J.; Menna, P.L.; Gomez, D.E. Rho GTPases as therapeutic targets in cancer (Review). Int. J. Oncol. 2017, 51, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Barakat, K.H.; Tuszynski, J.A. Computational Characterization of Small Molecules Binding to the Human XPF Active Site and Virtual Screening to Identify Potential New DNA Repair Inhibitors Targeting the ERCC1-XPF Endonuclease. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Cheng, T.; Li, Q.; Zhou, Z.; Wang, Y.; Bryant, S.H. Structure-based virtual screening for drug discovery: A problem-centric review. AAPS J. 2012, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Ishida, T.; Ikeda, K.; Mochizuki, M.; Teramoto, R.; Taguchi, Y.H.; Iwadate, M.; Umeyama, H.; Ramakrishnan, C.; Thangakani, A.M.; et al. An iterative compound screening contest method for identifying target protein inhibitors using the tyrosine-protein kinase Yes. Sci. Rep. 2017, 7, 12038. [Google Scholar] [CrossRef] [PubMed]
- Kolosenko, I.; Yu, Y.; Busker, S.; Dyczynski, M.; Liu, J.; Haraldsson, M.; Palm Apergi, C.; Helleday, T.; Tamm, K.P.; Page, B.D.G.; et al. Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLoS ONE 2017, 12, e0178844. [Google Scholar] [CrossRef] [PubMed]
- Billones, J.B.; Carrillo, M.C.; Organo, V.G.; Sy, J.B.; Clavio, N.A.; Macalino, S.J.; Emnacen, I.A.; Lee, A.P.; Ko, P.K.; Concepcion, G.P. In silico discovery and in vitro activity of inhibitors against Mycobacterium tuberculosis 7,8-diaminopelargonic acid synthase (Mtb BioA). Drug Des. Dev. Ther. 2017, 11, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Brender, J.R.; Zhang, J.; Zhang, Y. Protein structure prediction provides comparable performance to crystallographic structures in docking-based virtual screening. Methods 2015, 71, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazi, H.; Saleh, E.; El-Awady, R. In-silico screening for DNA-dependent protein kinase (DNA-PK) inhibitors: Combined homology modeling, docking, molecular dynamic study followed by biological investigation. Biomed. Pharmacother. 2016, 83, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Kumar Deokar, H.; Barch, H.P.; Buolamwini, J.K. Homology Modeling of Human Concentrative Nucleoside Transporters (hCNTs) and Validation by Virtual Screening and Experimental Testing to Identify Novel hCNT1 Inhibitors. Drug Des. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Dehzangi, A.; Lyons, J.; Paliwal, K.; Sharma, A.; Wang, J.; Sattar, A.; Zhou, Y.; Yang, Y. Highly accurate sequence-based prediction of half-sphere exposures of amino acid residues in proteins. Bioinformatics 2016, 32, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ye, Y.; Sang, P.; Yin, Y.; Hu, W.; Wang, J.; Zhang, C.; Li, D.; Wan, W.; Li, R.; et al. Effect of R119G Mutation on Human P5CR1 Dynamic Property and Enzymatic Activity. Biomed. Res. Int. 2017, 2017, 4184106. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Wege, H.; Chui, M.S.; Le, H.T.; Tran, J.M.; Zern, M.A. SYBR Green real-time telomeric repeat amplification protocol for the rapid quantification of telomerase activity. Nucleic Acids Res. 2003, 31, e3. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hDKC1 Model | C-Score | TM Score | RMSD (Å) |
|---|---|---|---|
| Homology Model | −0.09 | 0.70 ± 0.12 | 7.1 ± 4.2 |
| Ab initio model | −2.32 | 0.44 ± 0.14 | 13.0 ± 4.2 |
| Relevance (%) | Volume (Å3) | Residue Number | |
|---|---|---|---|
| 1 | 100 | 615 | 96, 101, 102, 103, 122, 123, 124, 125, 126, 128, 129, 130, 131, 132, 153, 187, 188, 246, 248 |
| 2 | 94 | 203 | 299, 300, 301, 314, 315, 317, 320, 322, 323, 350, 354, 355, 358, 360, 361, 362, 363 |
| 3 | 79 | 284 | 81, 85, 88, 89, 138, 289, 290, 341, 342, 370, 371, 372, 375 |
| 4 | 69 | 926 | 259, 260, 261, 265, 272, 280, 281, 284, 285, 287 |
| 5 | 69 | 289 | 141, 142, 144, 145, 293, 296, 332, 333, 334, 344, 345, 346, 367, 368, 370 |
| 6 | 64 | 810 | 74, 75, 76, 80, 81, 82, 85, 88, 341, 372, 375 |
| 7 | 60 | 208 | 54, 91, 93, 254, 258, 259, 261, 284, 285, 287, 288, 289, 291, 292, |
| 8 | 60 | 444 | 70, 71, 72, 303, 304, 307, 376, 377, 378, 379, 380, |
| 9 | 58 | 100 | 157, 169, 170, 173, 204, 206, 207, 214, 215, 216, 233 |
| 10 | 57 | 367 | 98, 127, 129, 154, 155, 156, 215, 245, 246, 247, 249, 256 |
| 11 | 57 | 994 | 183, 184, 185, 186, 192, 227, 228, 231, 241, 242, 243 |
| 12 | 54 | 525 | 54, 55, 57, 58, 294, 295, 296, 297, 298, 324 |
| 13 | 47 | 580 | 301, 302, 304, 305, 308, 309, 313, 314, 315, 316, 318, 319, 378 |
| 14 | 47 | 158 | 53, 54, 55, 56, 77, 78, 79, 80, 290, 339, 340 |
| 15 | 44 | 92 | 83, 86, 87, 273, 278, 279, 282, 283 |
| 16 | 42 | 146 | 176, 180, 181, 182, 197, 199, 218, 220, 224, 225, 226, 228, 229, 232 |
| 17 | 40 | 295 | 103, 119, 120, 121, 122, 123, 124, 143, 147, 151, 222, 223 |
| 18 | 37 | 268 | 98, 125, 126, 127, 128, 187, 227, 243, 244, 245, 246 |
| 19 | 33 | 274 | 156, 206, 207, 208, 211, 213, 214, 215 |
| 20 | 32 | 425 | 83, 84, 87, 113, 114, 115, 137, 270, 273, 274, 282 |
| 21 | 28 | 374 | 138, 141, 310, 311, 369, 371, 372, 373, 374 |
| Name | Compound | Docking Energy (kcal/mol) | SD | Name | Compound | Docking Energy (kcal/mol) | SD |
|---|---|---|---|---|---|---|---|
| E1 |  | −7.20 | 0.21 | E6 |  | −6.73 | 0.21 |
| E2 |  | −7.11 | 0.08 | E7 |  | −6.69 | 0.11 |
| E3 |  | 7.04 | 0.06 | E8 |  | −6.59 | 0.25 |
| E4 |  | −6.93 | 0.13 | E9 |  | −6.54 | 0.09 |
| E5 |  | −6.81 | 0.17 | E10 |  | −6.52 | 0.45 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armando, R.G.; Mengual Gómez, D.L.; Juritz, E.I.; Lorenzano Menna, P.; Gomez, D.E. Homology Model and Docking-Based Virtual Screening for Ligands of Human Dyskerin as New Inhibitors of Telomerase for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 3216. https://doi.org/10.3390/ijms19103216
Armando RG, Mengual Gómez DL, Juritz EI, Lorenzano Menna P, Gomez DE. Homology Model and Docking-Based Virtual Screening for Ligands of Human Dyskerin as New Inhibitors of Telomerase for Cancer Treatment. International Journal of Molecular Sciences. 2018; 19(10):3216. https://doi.org/10.3390/ijms19103216
Chicago/Turabian StyleArmando, Romina Gabriela, Diego Luis Mengual Gómez, Ezequiel Ivan Juritz, Pablo Lorenzano Menna, and Daniel Eduardo Gomez. 2018. "Homology Model and Docking-Based Virtual Screening for Ligands of Human Dyskerin as New Inhibitors of Telomerase for Cancer Treatment" International Journal of Molecular Sciences 19, no. 10: 3216. https://doi.org/10.3390/ijms19103216
APA StyleArmando, R. G., Mengual Gómez, D. L., Juritz, E. I., Lorenzano Menna, P., & Gomez, D. E. (2018). Homology Model and Docking-Based Virtual Screening for Ligands of Human Dyskerin as New Inhibitors of Telomerase for Cancer Treatment. International Journal of Molecular Sciences, 19(10), 3216. https://doi.org/10.3390/ijms19103216