Identification of Arbuscular Mycorrhiza Fungi Responsive microRNAs and Their Regulatory Network in Maize


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
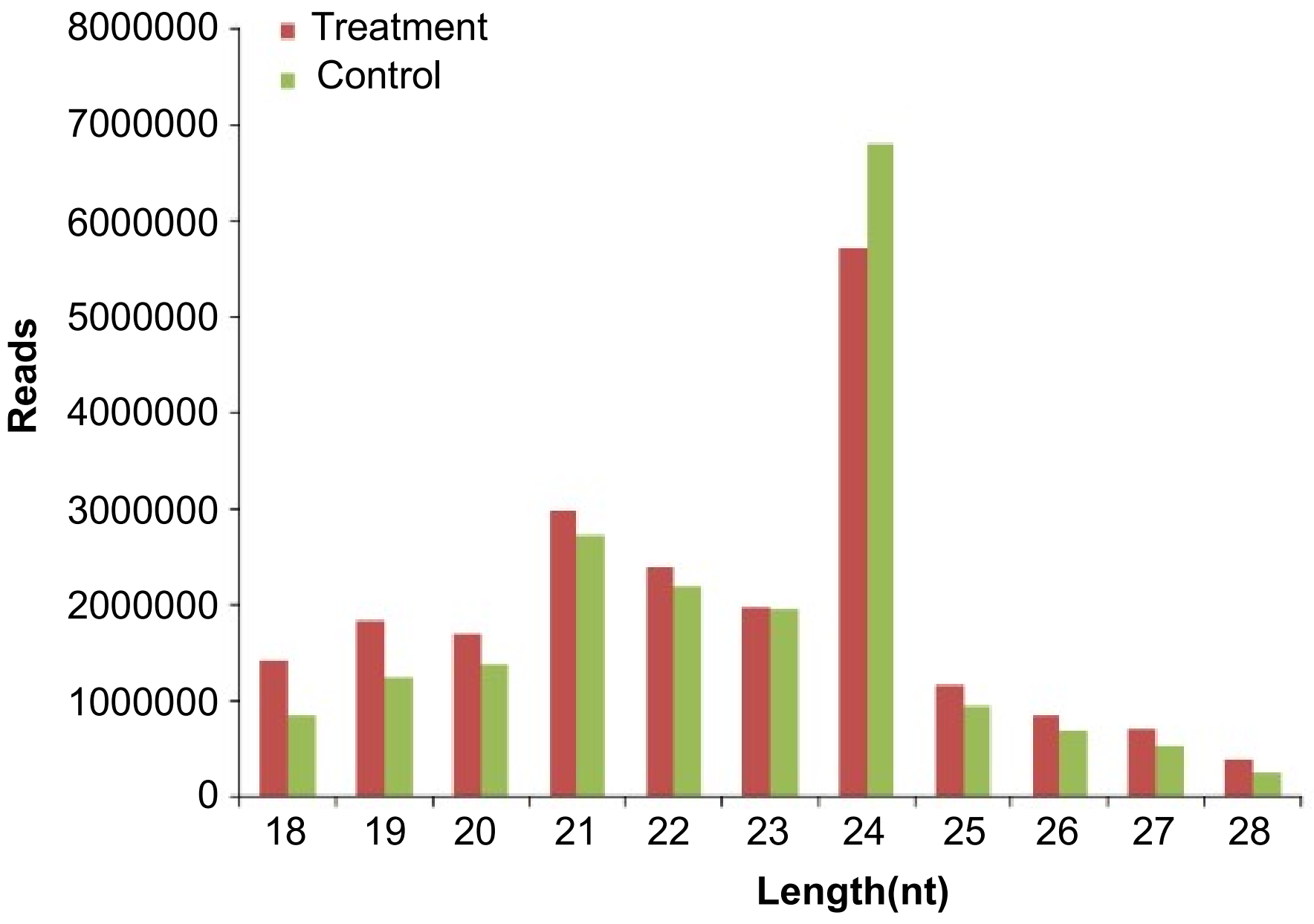
2.1. Sequencing of Small RNAs in Maize Roots
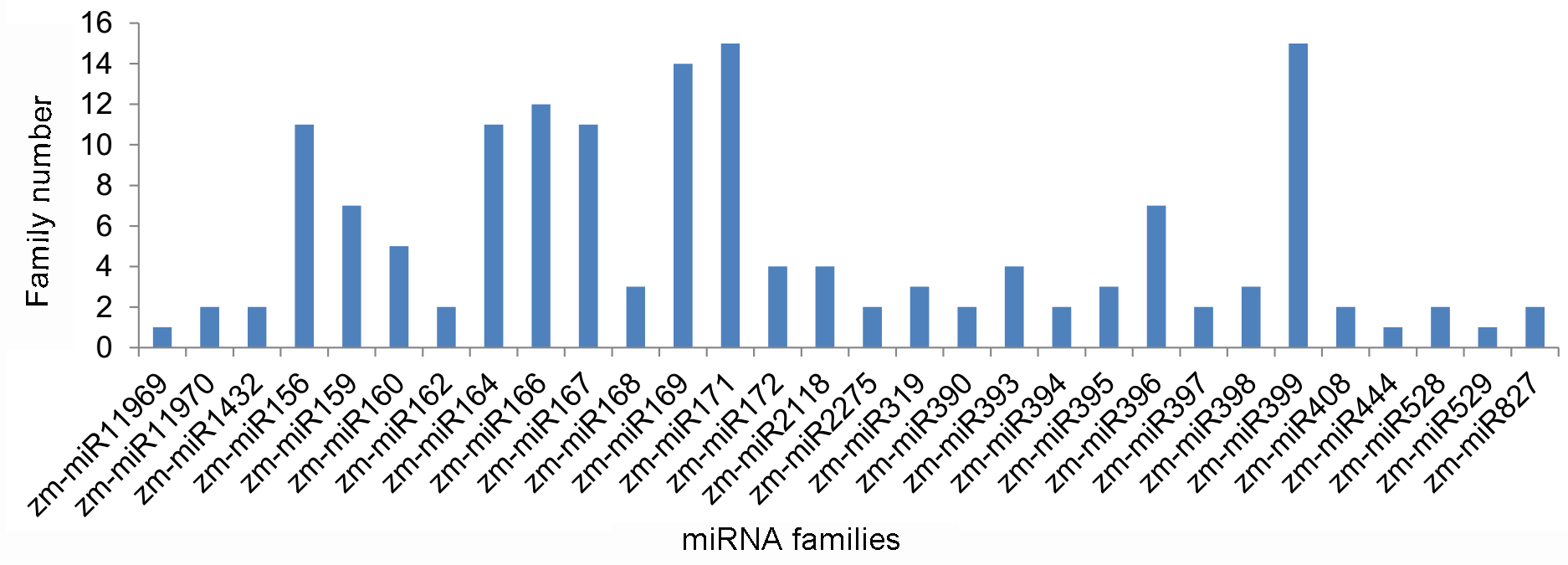
2.2. Identification of Known Maize miRNAs
2.3. Prediction of Novel miRNAs
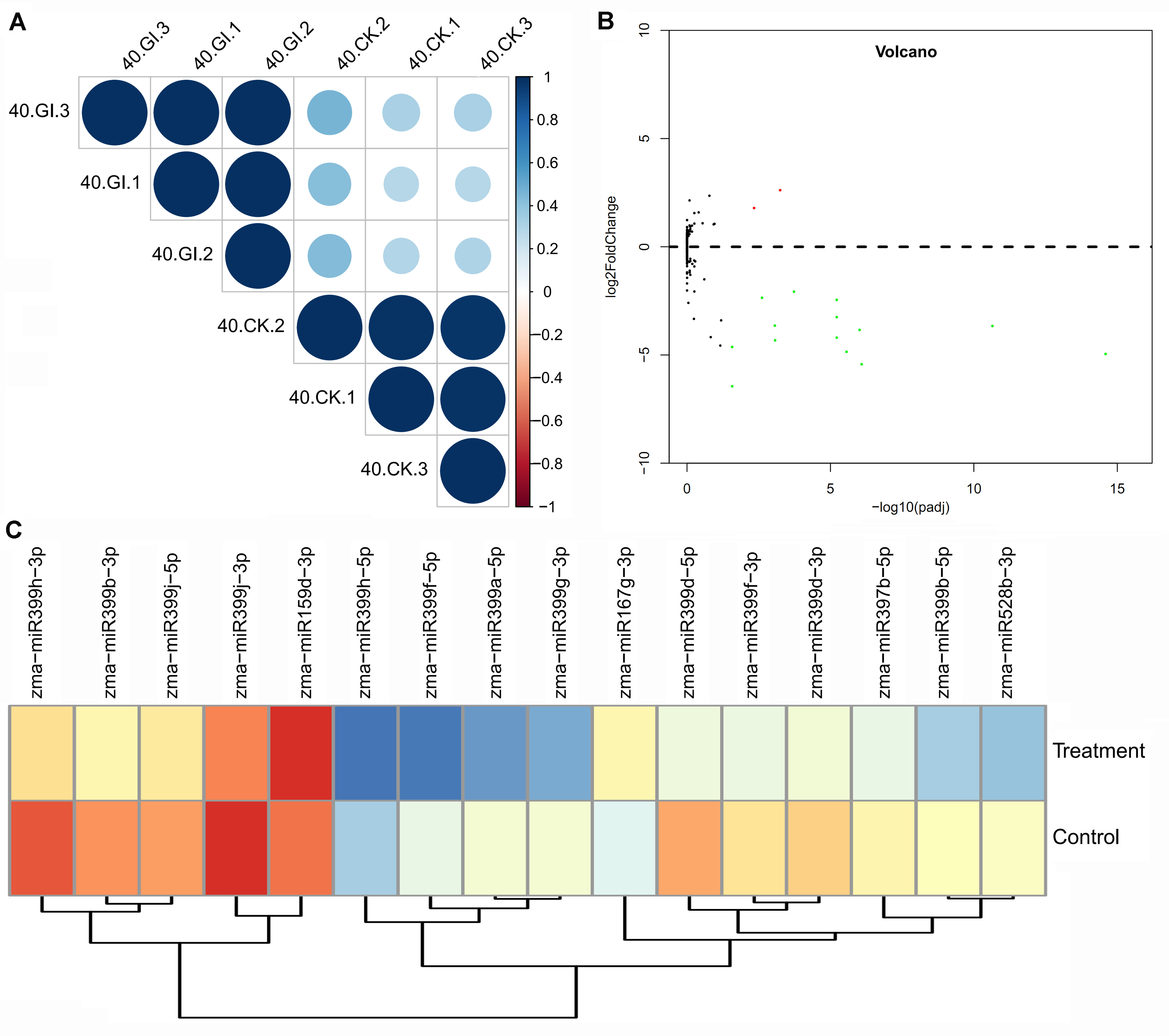
2.4. Differentially Expressed miRNA (DEM) Responsive to Arbuscular Mycorrhiza (AM) Fungus
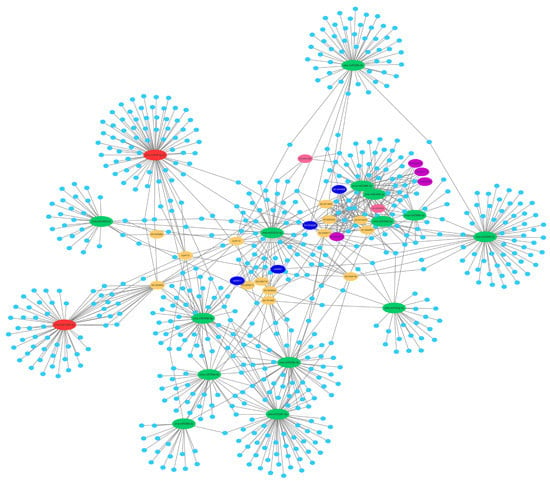
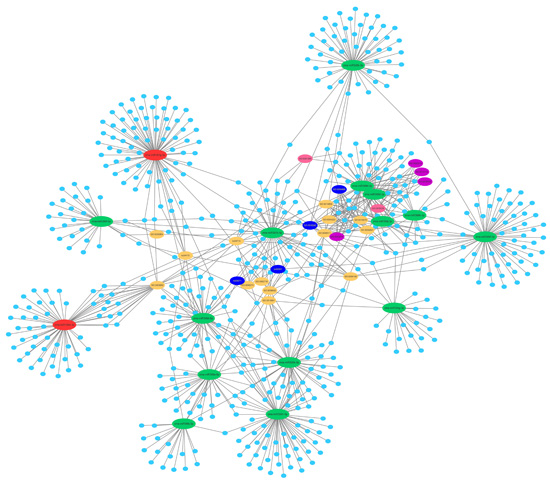
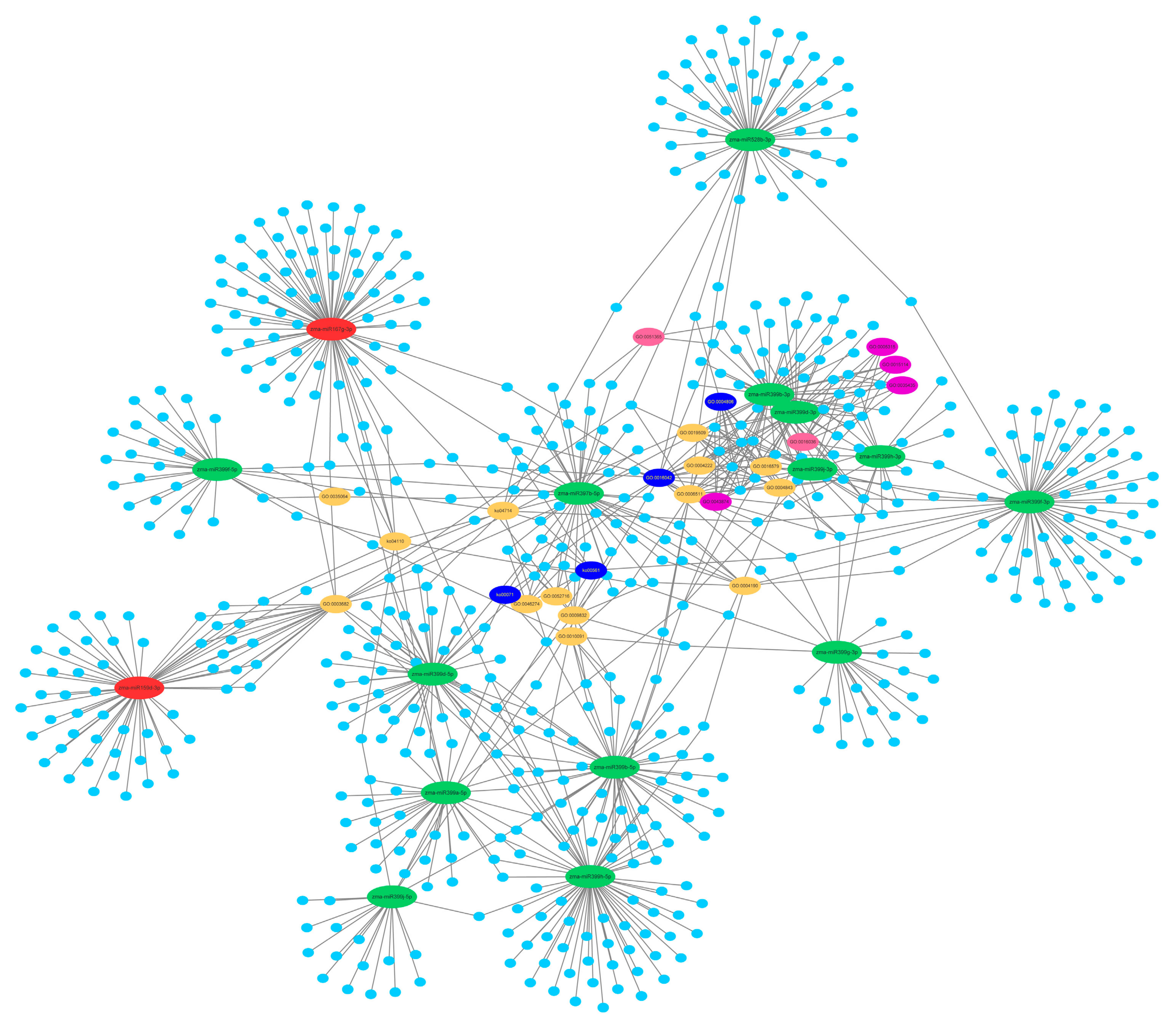
2.5. Prediction of the Differentially Expressed miRNAs Target Genes
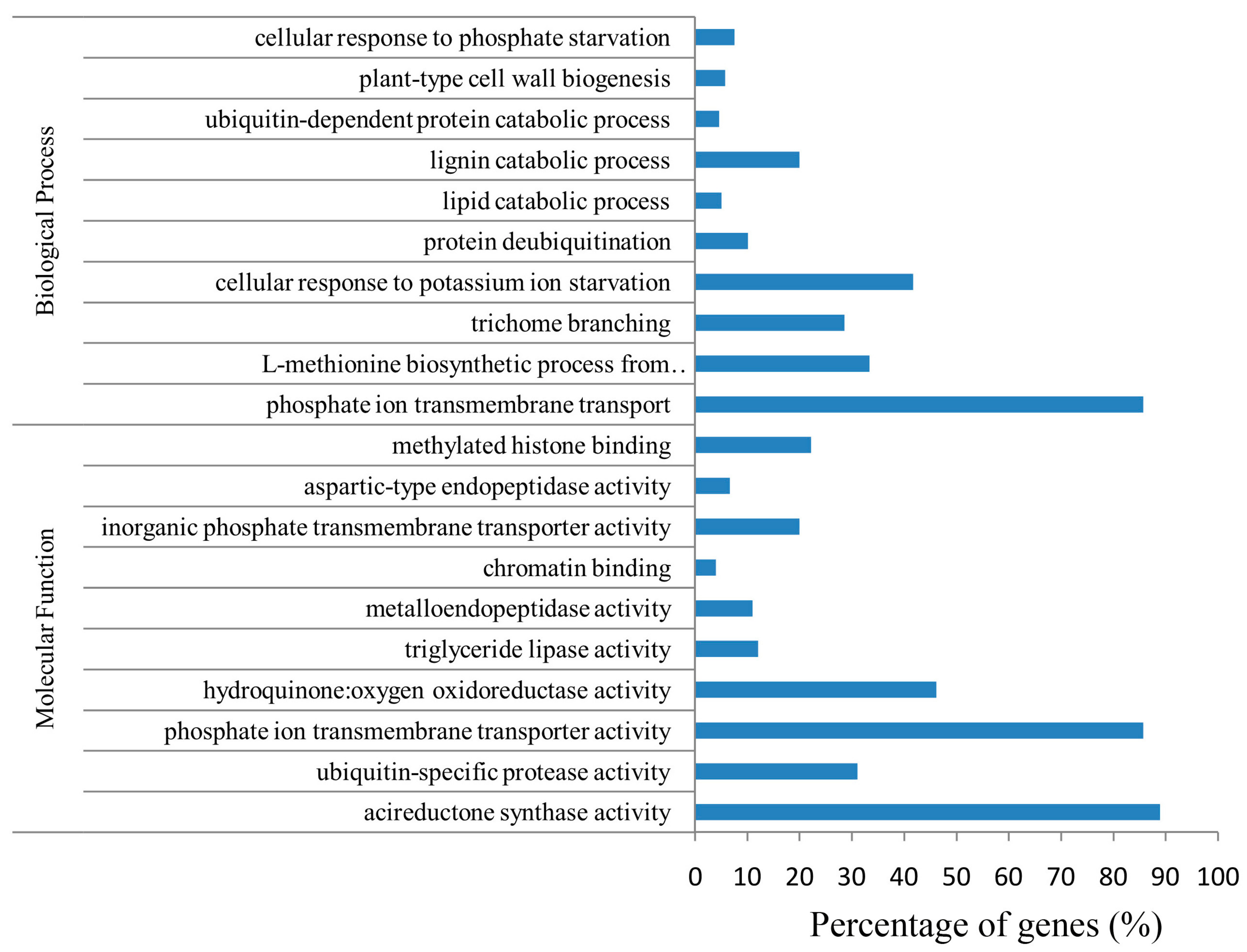
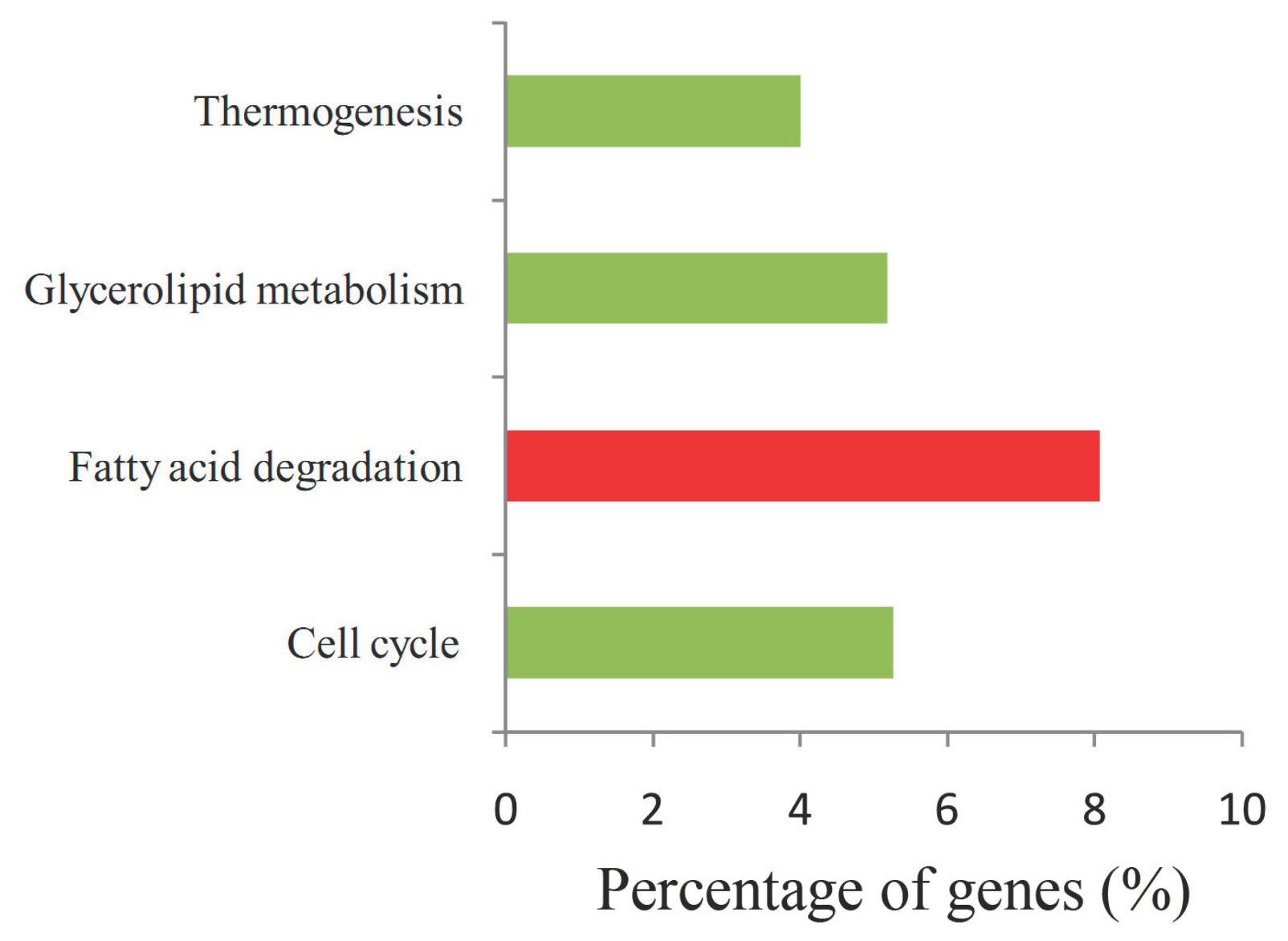
2.6. Function and Enrichment Analyses of the Target Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Detection of Mycorrhizal Colonization
4.3. RNA Extraction, Small RNA Library Preparation and Sequencing
4.4. Identification of Known miRNAs and Novel miRNAs
4.5. Identification of AM Fungus Responsive miRNAs
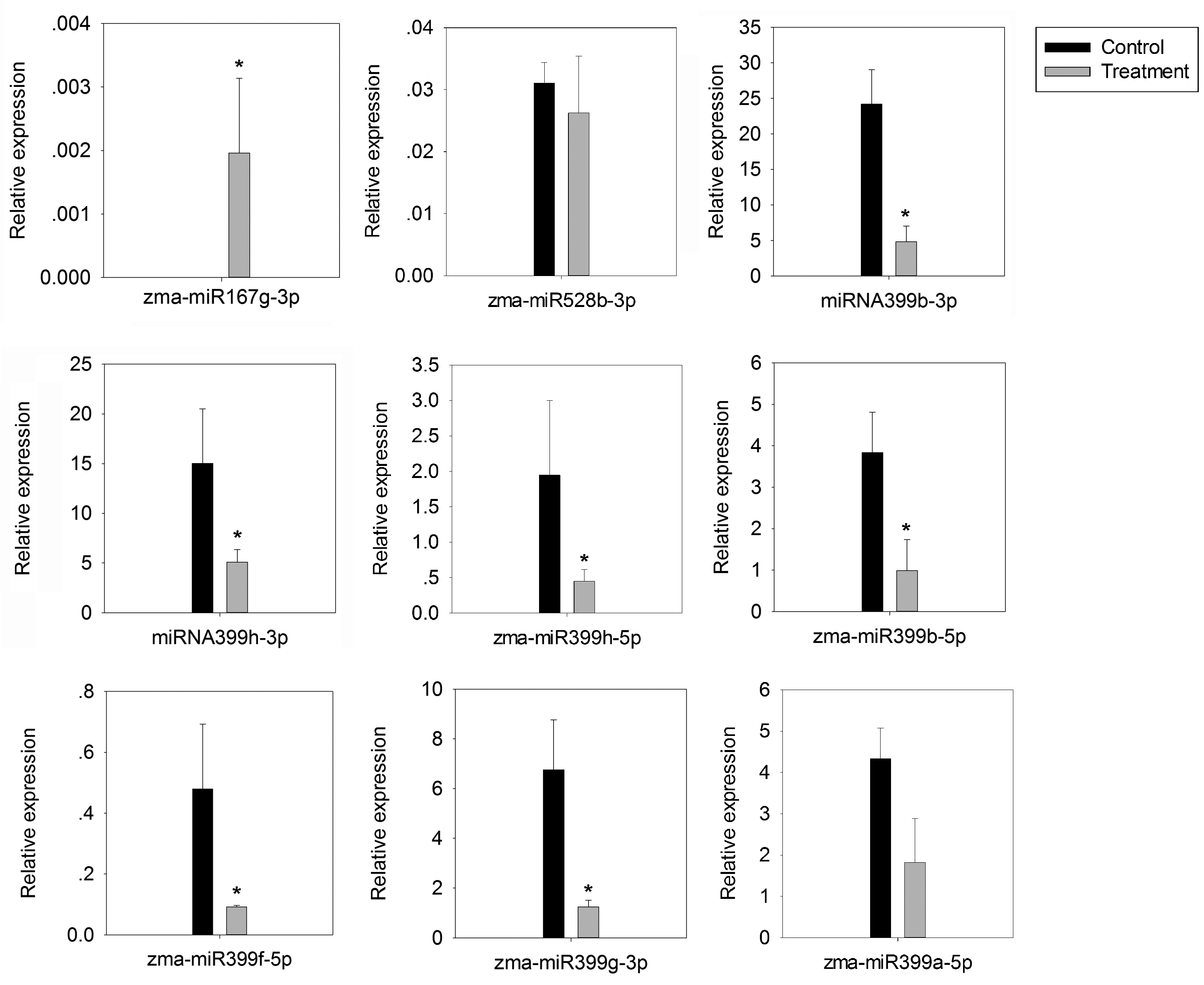
4.6. Stem-Loop Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
4.7. Identification of miRNA Targets
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, P.; Wu, Y.; Liu, C.C.; Liu, L.W.; Ma, F.F.; Wu, X.Y.; Wu, M.; Hang, Y.Y.; Chen, J.Q.; Shao, Z.Q. Identification of Arbuscular Mycorrhiza (AM)-responsive microRNAs in tomato. Front. Plant Sci. 2016, 7, 429. [Google Scholar] [CrossRef] [PubMed]
- Jonesrhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.; Chen, X. Biogenesis, turnover, and mode of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef] [PubMed]
- Pant, B.D.; Buhtz, A.; Kehr, J.; Scheible, W. MicroRNA399 is a long-distance signal for the regulation of plant phosphate homeostasis. Plant J. 2008, 53, 731–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Zhang, B. MicroRNAs in control of plant development. J. Cell Physiol. 2016, 231, 303. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Pant, B.D.; Stitt, M.; Scheible, W.R. PHO2, MicroRNA399, and PHR1 define a phosphate-signaling pathway in plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Kulcheski, F.R.; Oliveira, L.F.D.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C. Identification of novel soybean microRNAs involved in abiotic and biotic stresses. BMC Genom. 2011, 12, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, L.; Jones, J.D.G. A plant miRNA contributes to antibacterial resistance by repressing auxin signaling. Science 2006, 312, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.; Lii, Y.E.; Chellappan, P.; Lei, L.; Peralta, K.; Jiang, C.; Guo, J.; Coaker, G.; Jin, H. miRNA863-3p sequentially targets negative immune regulator ARLPKs and positive regulator SERRATE upon bacterial infection. Nat. Commun. 2016, 7, 11324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldroyd, G.E.D. Speak, friend, and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Xu, Y.J.; Jiang, H.H.; Jiang, C.S.; Du, Y.B.; Gong, C.; Wang, W.; Zhu, S.W.; Han, G.M.; Cheng, B.J. Systematic identification, evolution and expression analysis of the Zea mays PHT1 gene family reveals several new members involved in root colonization by Arbuscular Mycorrhizal fungi. Int. J. Mol. Sci. 2016, 17, 930. [Google Scholar] [CrossRef] [PubMed]
- Couzigou, J.M.; Lauressergues, D.; Andre, O.; Gutjahr, C.; Guillotin, B.; Becard, G.; Combier, J.P. Positive gene regulation by a natural protective miRNA enables Arbuscular Mycorrhizal symbiosis. Cell Host Microbe 2017, 21, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Liu, H.; Luo, D.; Nan, Y.; Dong, W.; Chao, W.; Zhang, X.; Dai, H.; Yang, J.; Wang, E. DELLA proteins are common components of symbiotic rhizobial and mycorrhizal signalling pathways. Nat. Commun. 2016, 7, 12433. [Google Scholar] [Green Version]
- Camille, F.F.; Tan, S.; Maël, B.; Mathias, B.; Wen, J.; Mysore, K.S.; Andreas, N.; Florian, F.; Anouck, D. DELLA-mediated gibberellin signalling regulates Nod factor signalling and rhizobial infection. Nat. Commun. 2016, 7, 12636. [Google Scholar] [Green Version]
- Lauressergues, D.; Delaux, P.M.; Formey, D.; Lelandais-Briere, C.; Fort, S.; Cottaz, S.; Becard, G.; Niebel, A.; Roux, C.; Combier, J.P. The microRNA miR171h modulates arbuscular mycorrhizal colonization of Medicago truncatula by targeting NSP2. Plant J. 2012, 72, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Bazin, J.; Khan, G.A.; Combier, J.P.; Bustossanmamed, P.; Debernardi, J.M.; Rodriguez, R.; Sorin, C.; Palatnik, J.; Hartmann, C.; Crespi, M. MiR396 affects mycorrhization and root meristem activity in the legume Medicago truncatula. Plant J. 2013, 74, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Branscheid, A.; Sieh, D.; Pant, B.D.; May, P.; Devers, E.A.; Elkrog, A.; Schauser, L.; Scheible, W.R.; Krajinski, F. Expression pattern suggests a role of miR399 in the regulation of the cellular response to local Pi increase during Arbuscular Mycorrhizal symbiosis. Mol. Plant Microbe Interact. 2010, 23, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, L.; Zou, Y.; Chen, L.; Cai, Z.; Zhang, S.; Zhao, F.; Tian, Y.; Jiang, Q.; Ferguson, B.J. Soybean miR172c targets the repressive AP2 transcription factor NNC1 to activate ENOD40 expression and regulate nodule initiation. Plant Cell 2014, 26, 4782–4801. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Ren, Z.; Wang, L.; Su, S.; Wei, X.; Zhang, X.; Wu, L.; Liu, D.; Tang, H.; Liu, H.; et al. Genome-wide identification of microRNAs responding to early stages of phosphate deficiency in maize. Physiol. Plantarum 2015, 157, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kumari, M.; Kumar, H.; Varadwaj, P.K. Genome-wide analysis of miRNAs and Tasi-RNAs in Zea mays in response to phosphate deficiency. Funct. Integr. Genomics 2017, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, Z.; Mo, Q.; Zou, C.; Li, W.; Xu, Y.; Xie, C. Combined small RNA and degradome sequencing reveals novel miRNAs and their targets in response to low nitrate availability in maize. Ann. Bot-Lond. 2013, 112, 633. [Google Scholar] [CrossRef] [PubMed]
- Aravind, J.; Rinku, S.; Pooja, B.; Shikha, M.; Kaliyugam, S.; Mallikarjuna, M.G.; Kumar, A.; Rao, A.R.; Nepolean, T. Identification, characterization, and functional validation of drought-responsive microRNAs in subtropical maize inbreds. Front. Plant Sci. 2017, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Zhang, M.; Zhao, Y.; He, X.; Ding, C.; Wang, S.; Feng, Y.; Song, X.; Li, P.; Wang, B. Identification of salt tolerance-related microRNAs and their targets in maize (Zea mays L.) using high-throughput sequencing and degradome analysis. Front. Plant Sci. 2017, 8, 864. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dong, J.; Bennetzen, J.L.; Zhong, M.; Yang, J.; Zhang, J.; Li, S.; Hao, X.; Zhang, Z.; Wang, X. Integrating transcriptome and microRNA analysis identifies genes and microRNAs for AHO-induced systemic acquired resistance in N. tabacum. Sci. Rep. 2017, 7, 12504. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhou, H.; Zhang, Q.; Zhang, J.; Ni, F.; Liu, C.; Qi, Y. DNA methylation mediated by a microRNA pathway. Mol. Cell 2010, 38, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Rubio, V.; Linhares, F.; Solano, R.; Martin, A.C.; Iglesias, J.; Leyva, A.; Paz-Ares, J. A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Gene Dev. 2001, 15, 2122–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, F.; Han, G.; Cheng, B. Genome-wide identification and comparative analysis of phosphate starvation-responsive transcription factors in maize and three other gramineous plants. Plant Cell Rep. 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Xu, K.; Chen, A.; Zhu, Y.; Tang, G.; Xu, G. Expression analysis suggests potential roles of microRNAs for phosphate and arbuscular mycorrhizal signaling in Solanum lycopersicum. Physiol. Plantarum 2010, 138, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Luginbuehl, L.H.; Menard, G.N.; Kurup, S.; Van, E.H.; Radhakrishnan, G.V.; Breakspear, A.; Ged, O.; Eastmond, P.J. Fatty acids in arbuscular mycorrhizal fungi are synthesized by the host plant. Science 2017, 356, 1175. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, W.; Xie, Q.; Liu, N.; Liu, L.; Wang, D.; Zhang, X.; Yang, C.; Chen, X.; Tang, D. Plants transfer lipids to sustain colonization by mutualistic mycorrhizal and parasitic fungi. Science 2017, 356, 1172. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; Mckay, R. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirerasalinas, D.; Rayner, K.; Suresh, U.; Pastorpareja, J.C. MiR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J. microRNA167-directed regulation of the auxin response factors, GmARF8a and GmARF8b, is required for soybean (Glycine max L.) nodulation and lateral root development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Wang, H.; Kasahara, H.; Liu, J.; Macpherson, C.; Machida, Y.; Kamiya, Y.; Hannah, M.A.; Chua, N.H. IAA-Ala Resistant3, an evolutionary conserved target of miR167, mediates Arabidopsis root architecture changes during high osmotic stress. Plant Cell 2012, 24, 3590–3602. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Meyers, B.C.; Green, P.J. Construction of small RNA cDNA libraries for deep sequencing. Methods 2007, 43, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.R.; Yeoh, J.M.; Coruh, C.; Axtell, M.J. Improved placement of multi-mapping small RNAs. G3-Genes Genom. Genet. 2016, 6, 2103–2111. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT-the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhu, S.; Liu, F.; Wang, W.; Wang, X.; Han, G.; Cheng, B. Identification of Arbuscular Mycorrhiza Fungi Responsive microRNAs and Their Regulatory Network in Maize. Int. J. Mol. Sci. 2018, 19, 3201. https://doi.org/10.3390/ijms19103201
Xu Y, Zhu S, Liu F, Wang W, Wang X, Han G, Cheng B. Identification of Arbuscular Mycorrhiza Fungi Responsive microRNAs and Their Regulatory Network in Maize. International Journal of Molecular Sciences. 2018; 19(10):3201. https://doi.org/10.3390/ijms19103201
Chicago/Turabian StyleXu, Yunjian, Suwen Zhu, Fang Liu, Wei Wang, Xuewen Wang, Guomin Han, and Beijiu Cheng. 2018. "Identification of Arbuscular Mycorrhiza Fungi Responsive microRNAs and Their Regulatory Network in Maize" International Journal of Molecular Sciences 19, no. 10: 3201. https://doi.org/10.3390/ijms19103201