Multiple Regulatory Networks Are Activated during Cold Stress in Medicago sativa L.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and discussion
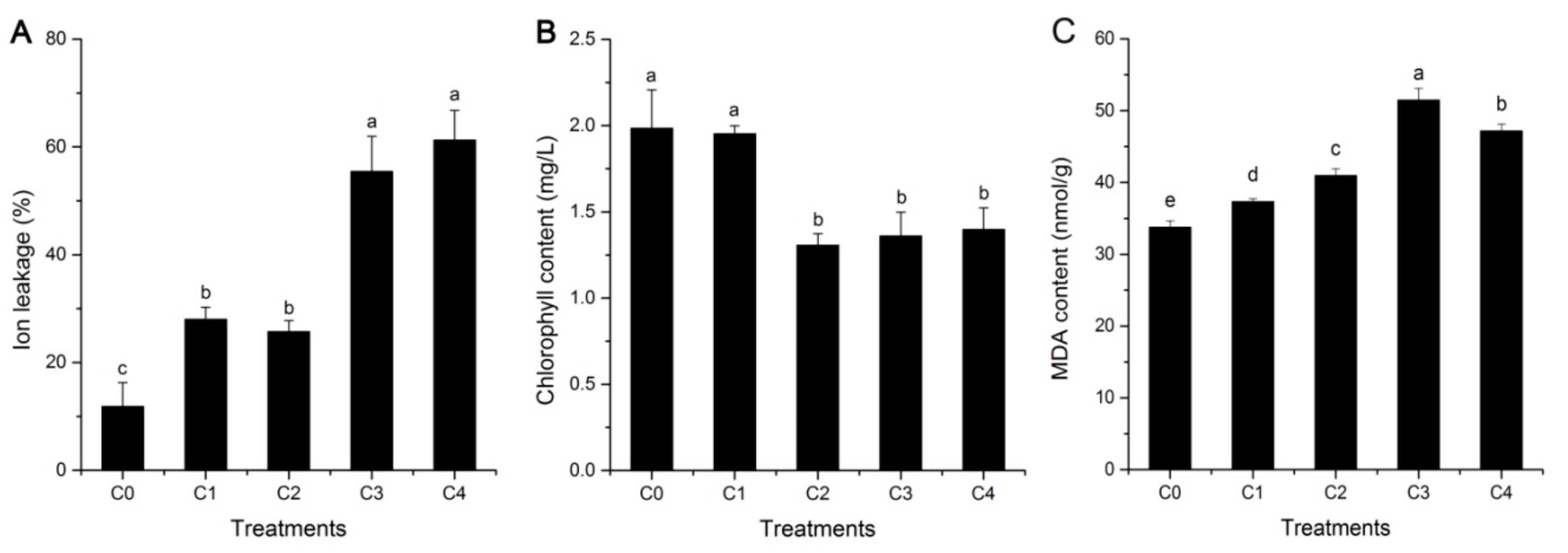
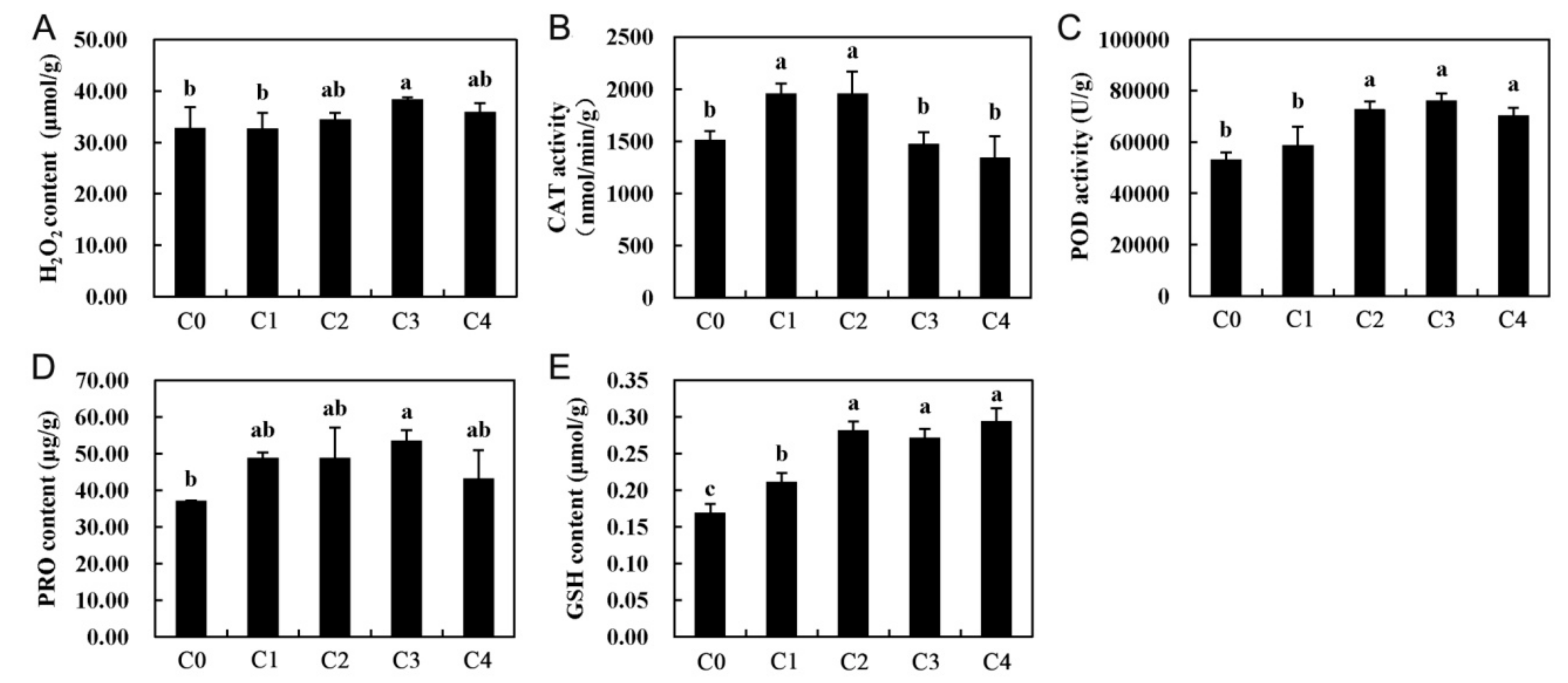
2.1. Cell Membrane Stability, Lipid Peroxidation, and Photosynthesis
2.2. De Novo Assembly of Transcriptome and the Functional Annotation of Unigenes
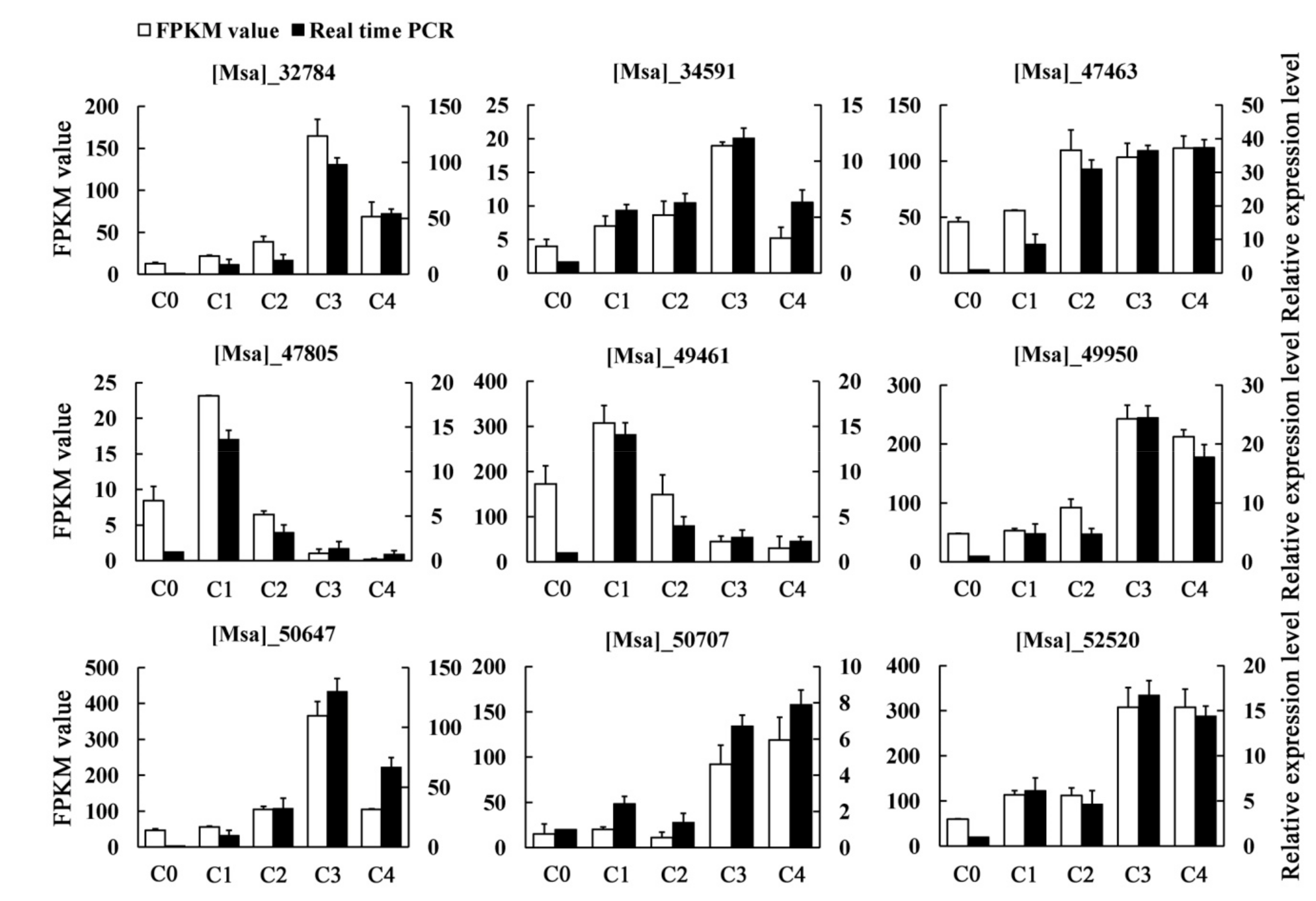
2.3. Quantitative Real-Time PCR Verification
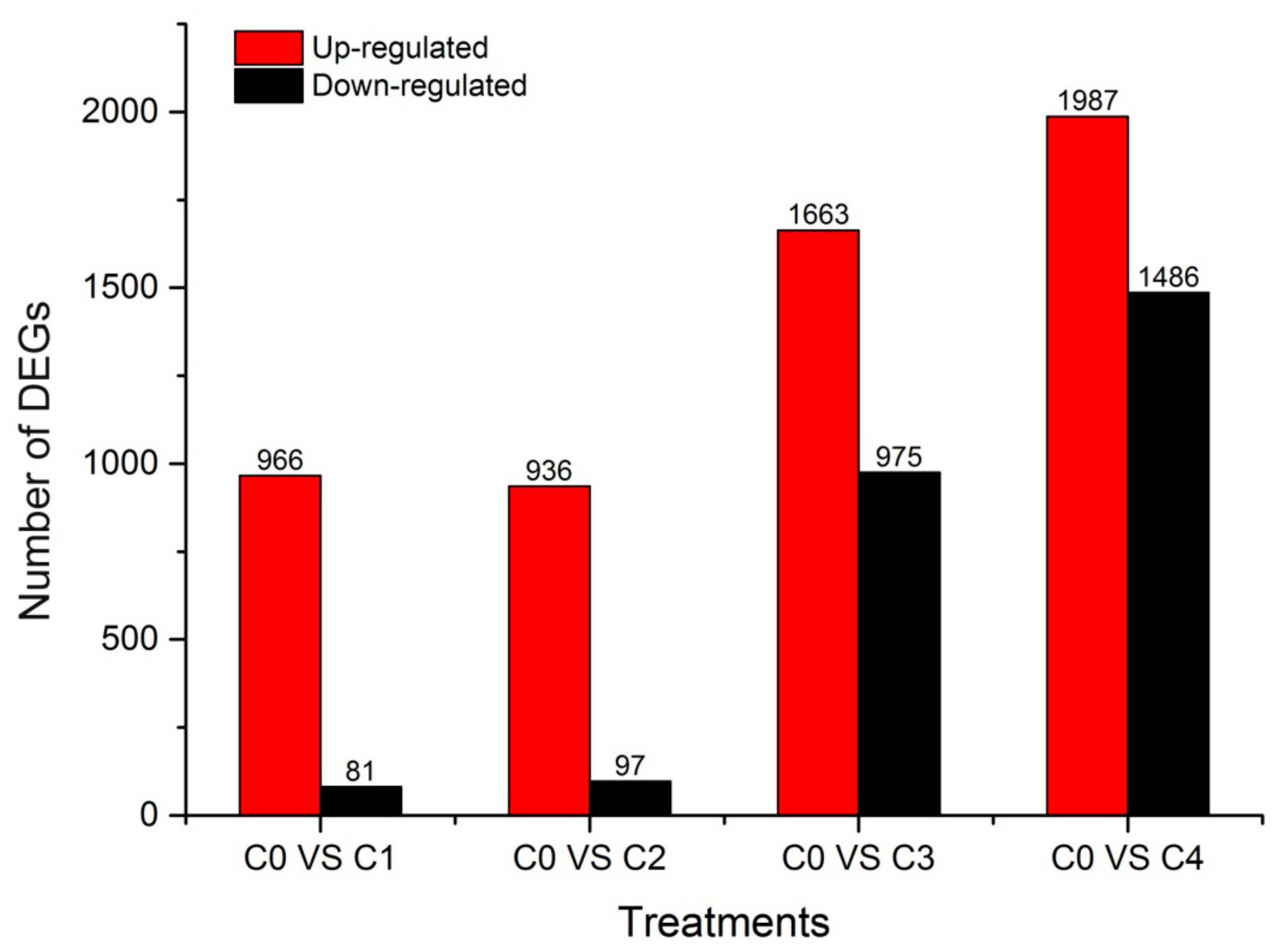
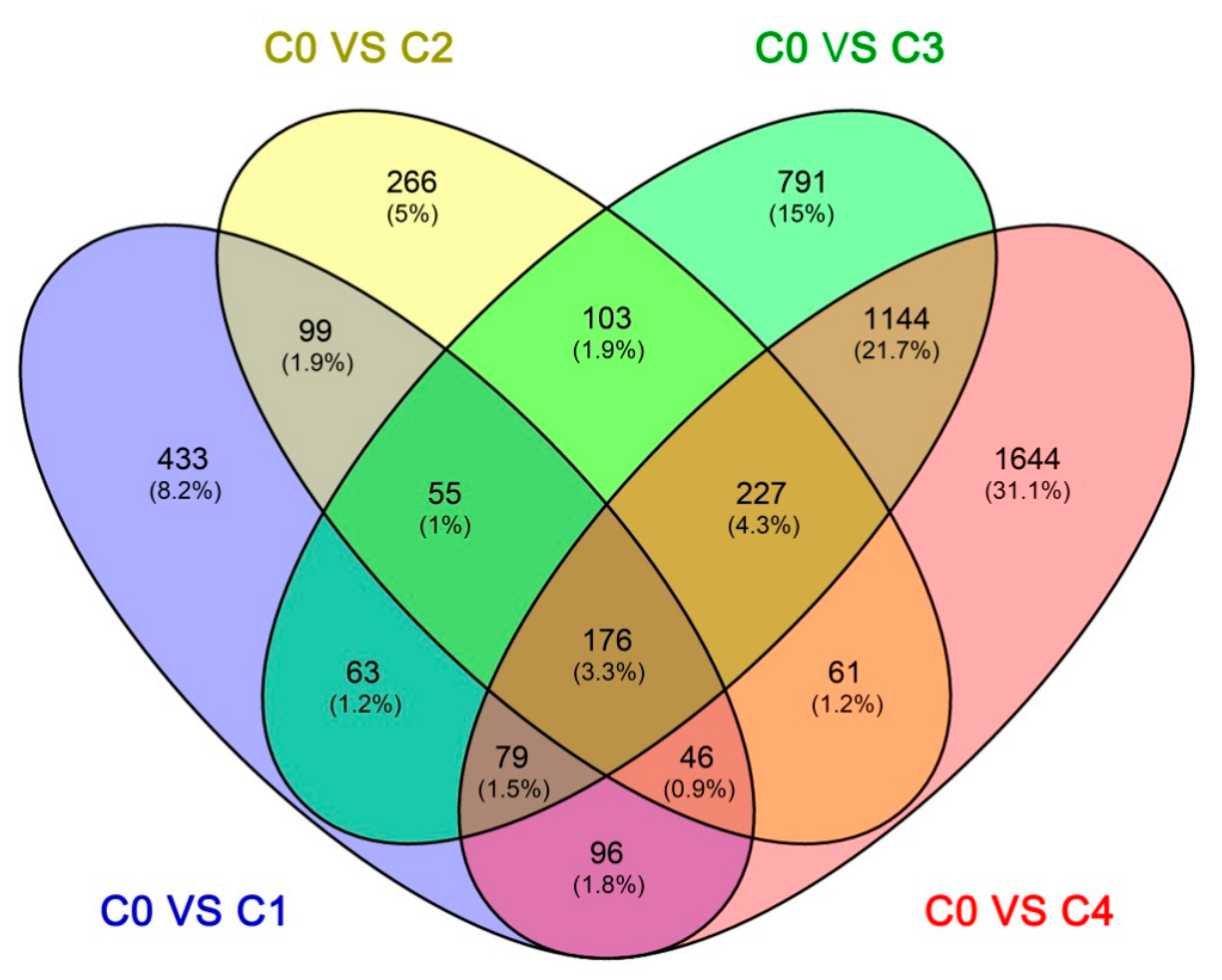
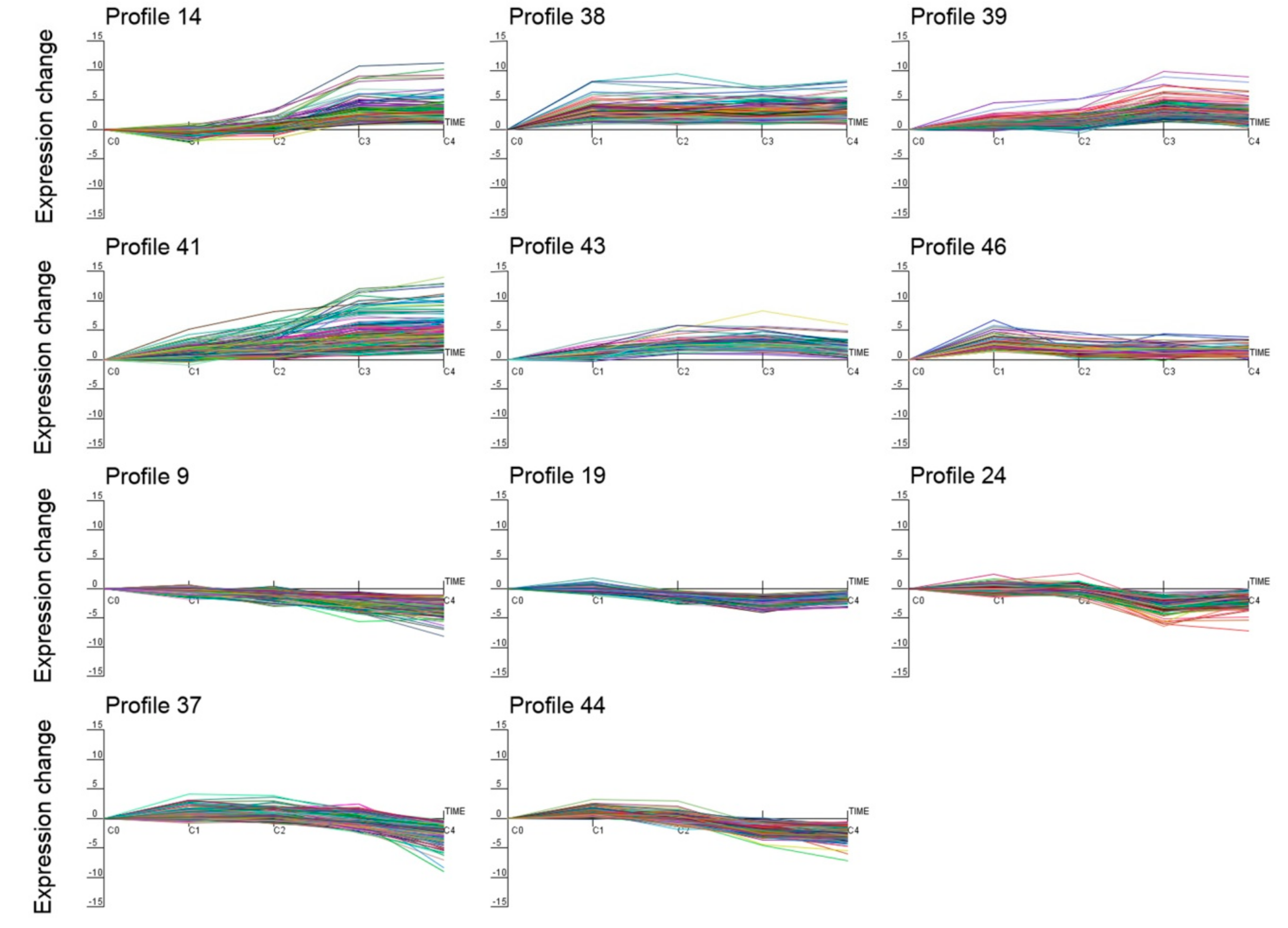
2.4. Identification and Clustering Analysis of Differentially Expressed Genes (DEGs)
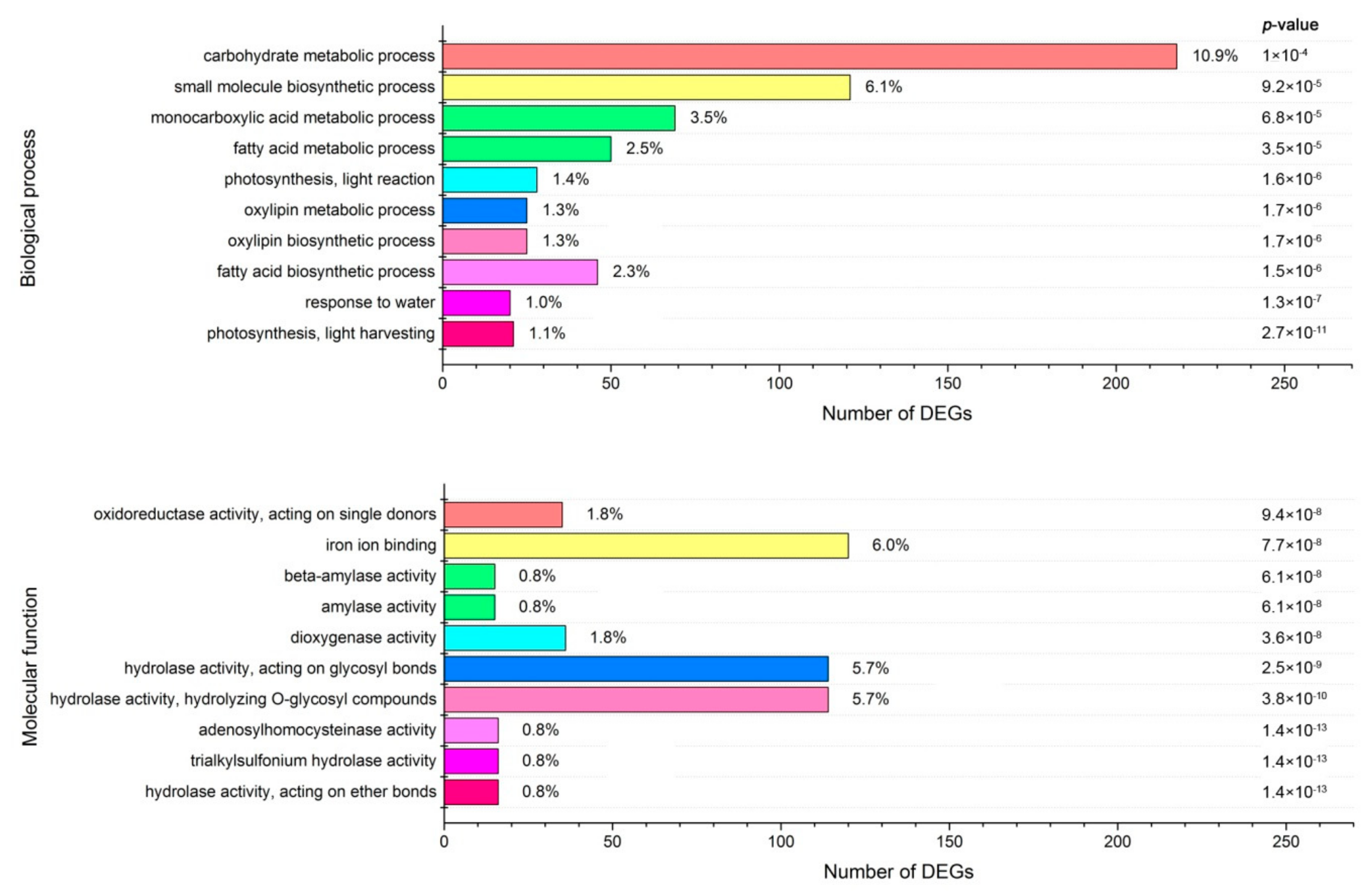
2.5. GO Functional Analysis of the DEGs
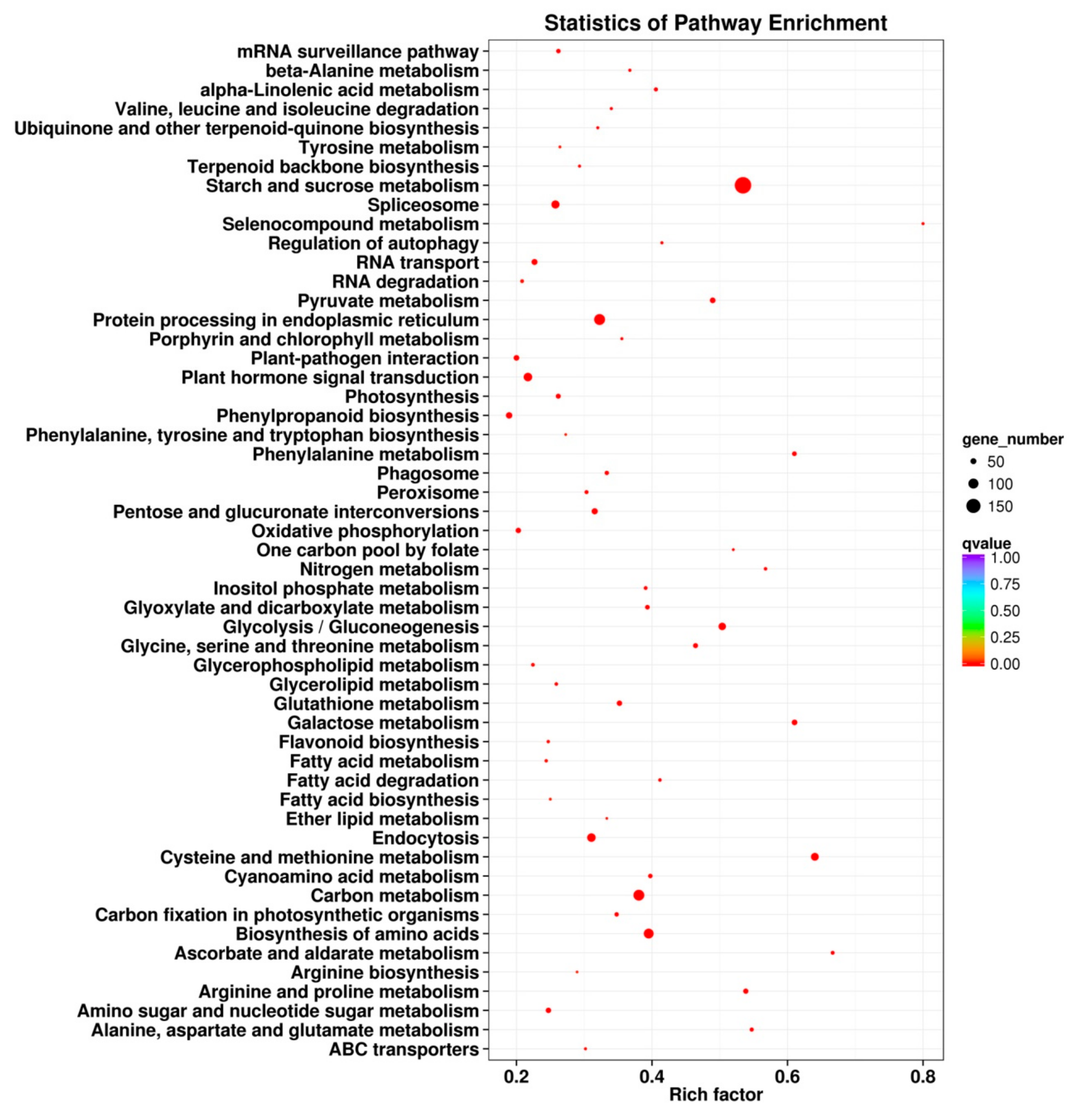
2.6. KEGG Pathway Enrichment Analysis of the DEGs
2.7. Antioxidant Defense System-Related Genes in Cold Stress
2.8. Transcriptional Regulatory Networks are Involved in the Adaptation of Alfalfa to Cold Stress
2.9. Plant Hormone-Related DEGs
2.10. Cold Stress-Related Genes Functionally Classified through MapMan Analysis
3. Materials and Methods
3.1. Plant Materials and Stress Treatments
3.2. Physiological Experiments and RNA Extraction
3.3. De Novo Assembly of Transcriptome
3.4. Quantitative Real-Time PCR Analysis
3.5. Differential Gene Expression, GO, and KEGG Enrichment Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FPKM | Fragments Per Kilobase Of Exon Per Million Fragments Mapped |
| Nr | NCBI non-redundant protein sequences |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| qRT-PCR | Quantitative real-time PCR |
| GSH | Glutathione |
| Pro | Proline |
| CAT | Catalase |
| POD | Peroxidase |
| NGS | Next-generation sequencing |
| DEGs | Differentially expressed genes |
| ROS | Reactive oxygen species |
| H2O2 | Hydrogen peroxide |
| MDA | Malondialdehyde |
| TFs | Transcription factors |
| ABA | Abscisic acid |
| STEM | Short Timeseries Expression Miner |
| ANOVA | One-way analysis of variance |
References
- Liu, Z.P.; Chen, T.L.; Ma, L.C.; Zhao, Z.G.; Zhao, P.X.; Nan, Z.B.; Wang, Y.R. Global transcriptome sequencing using the Illumina platform and the development of EST-SSR markers in autotetraploid alfalfa. PLoS ONE 2013, 8, e83549. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, T.L.; Wang, Y.R.; Liu, Z.P. The development of 204 novel EST-SSRs and their use for genetic diversity analyses in cultivated alfalfa. Biochem. Syst. Ecol. 2014, 57, 227–230. [Google Scholar] [CrossRef]
- Yang, S.S.; Tu, Z.J.; Cheung, F.; Xu, W.W.; Lamb, J.F.; Jung, H.-J.G.; Vance, C.P.; Gronwald, J.W. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Samac, D.A.; Jung, H.-G.; Lamb, J.F.S. Development of alfalfa (Medicago sativa L.) as a feedstock for production of ethanol and other bioproducts. In Alcoholic Fuels; Minteer, S., Ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 79–98. [Google Scholar]
- Yang, S.S.; Xu, W.W.; Tesfaye, M.; Lamb, J.F.; Jung, H.-J.G.; VandenBosch, K.A.; Vance, C.P.; Gronwal, J.W. Transcript profiling of two alfalfa genotypes with contrasting cell wall composition in stems using a cross-species platform: Optimizing analysis by masking biased probes. BMC Genom. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Song, L.L.; Jiang, L.; Chen, Y.; Shu, Y.J.; Bai, Y.; Guo, C.H. Deep-sequencing transcriptome analysis of field-grown Medicago sativa L. crown buds acclimated to freezing stress. Funct. Integr. Genom. 2016, 16, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Alarcón-Zúñiga, B.; Kang, J.; Nadeem Tahir, M.H.; Jiang, Q.Z.; Wei, Y.L.; Reyno, R.; Robins, J.G.; Brummer, E.C. Mapping fall dormancy and winter injury in tetraploid alfalfa. Crop Sci. 2015, 55. [Google Scholar] [CrossRef]
- Chen, J.H.; Tian, Q.Q.; Pang, T.; Jiang, L.B.; Wu, R.L.; Xia, X.L.; Yin, W.L. Deep-sequencing transcriptome analysis of low temperature perception in a desert tree, Populus euphratica. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.M.; Clarke, S.M.; Wood, J.E.; Mur, L.A. Salicylate accumulation inhibits growth at chilling temperature in Arabidopsis. Plant Physiol. 2004, 135, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sharma, N.; Deswal, R. The molecular biology of the low-temperature response in plants. BioEssays 2005, 27, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Solanke, A.U.; Sharma, A.K. Signal transduction during cold stress in plants. Physiol. Mol. Biol. Plants 2008, 14, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Korn, M.; Gartner, T.; Erban, A.; Kopka, J.; Selbig, J.; Hincha, D.K. Predicting Arabidopsis freezing tolerance and heterosis in freezing tolerance from metabolite composition. Mol. Plant 2010, 3, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.C.; Zhao, Q.Y.; Ma, C.L.; Zhang, Z.H.; Cao, H.L.; Kong, Y.M.; Yue, C.; Hao, X.Y.; Chen, L.; Ma, J.Q.; et al. Global transcriptome profiles of Camellia sinensis during cold acclimation. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Winfield, M.O.; Lu, C.; Wilson, I.D.; Coghill, J.A.; Edwards, K.J. Plant responses to cold: Transcriptome analysis of wheat. Plant Biotechnol. J. 2010, 8, 749–771. [Google Scholar] [CrossRef] [PubMed]
- Chinnusamy, V.; Schumaker, K.; Zhu, J.K. Molecular genetic perspectives on cross-talk and specificity in abiotic stress signaling in plants. J. Exp. Bot. 2004, 55, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ding, Y.L.; Shi, Y.T.; Zhang, X.Y.; Zhang, S.Q.; Gong, Z.Z.; Yang, S.H. MPK3- and MPK6-mediated ICE1 phosphorylation negatively regulates ICE1 stability and freezing tolerance in Arabidopsis. Dev. Cell 2017, 43, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Rihan, H.Z.; Al-Issawi, M.; Fuller, M.P. Upregulation of CBF/DREB1 and cold tolerance in artificial seeds of cauliflower (Brassica oleracea var. botrytis). Sci. Hortic. 2017, 225, 299–309. [Google Scholar] [CrossRef]
- Zhang, B.B.; Guo, L.; Song, Z.Z.; Yu, M.L.; Ma, R.J. Effect of salicylic acid on freezing injury in peach floral organs and the expressions of CBF genes. Biol. Plant. 2017, 61, 622–630. [Google Scholar] [CrossRef]
- Kanchupati, P.; Wang, Y.F.; Anower, M.R.; Boe, A.; Hu, T.M.; Wu, Y.J. The CBF-Like gene family in alfalfa: Expression analyses and identification of potential functional homologs of Arabidopsis CBF3. Crop Sci. 2017, 57, 2051–2063. [Google Scholar] [CrossRef]
- Eremina, M.; Rozhon, W.; Poppenberger, B. Hormonal control of cold stress responses in plants. Cell. Mol. Life Sci. 2016, 73, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Seo, P.J. The MYB96-HHP module integrates cold and abscisic acid signaling to activate the CBF-COR pathway in Arabidopsis. Plant J. 2015, 82, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Mustilli, A.C.; Merlot, S.; Vavasseur, A.; Fenzi, F.; Giraudat, J. Arabidopsis OST1 protein kinase mediates the regulation of stomatal aperture by abscisic acid and acts upstream of reactive oxygen species production. Plant Cell 2002, 14, 3089–3099. [Google Scholar] [CrossRef] [PubMed]
- Calzadilla, P.I.; Maiale, S.J.; Ruiz, O.A.; Escaray, F.J. Transcriptome response mediated by cold stress in Lotus japonicus. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, S.X.; Yu, X.; Cheng, Z.H.; Yu, X.L.; Ruan, M.B.; Li, W.B.; Peng, M. Global gene expression analysis reveals crosstalk between response mechanisms to cold and drought stresses in cassava seedlings. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, S.F.; Xu, W.L.; Liu, X.X. Genome-wide transcriptome profiling of radish (Raphanus sativus L.) in response to vernalization. PLoS ONE 2017, 12, e0177594. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Xu, Y.Q.; Zhang, L.G.; Li, W.; Cai, Z.X.; Li, F.; Peng, M.; Li, F.L.; Hu, B.Z. De novo assembly and analysis of the transcriptome of Rumex patientia L. during cold stress. PLoS ONE 2017, 12, e0186470. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shen, Z.J.; Yan, J. Transcriptome analysis of peach [Prunus persica (L.) Batsch] stigma in response to low-temperature stress with digital gene expression profiling. J. Plant Biochem. Biotechnol. 2016, 26, 141–148. [Google Scholar] [CrossRef]
- Meng, D.L.; Yu, X.H.; Ma, L.Y.; Hu, J.; Liang, Y.L.; Liu, X.D.; Yin, H.Q.; Liu, H.W.; He, X.J.; Li, D.Q. Transcriptomic response of chinese yew (Taxus chinensis) to cold stress. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Shi, Y.H.; Cheng, N.N.; Du, H.Q.; Fan, W.N.; Wang, C.Z. De novo characterization of fall dormant and nondormant alfalfa (Medicago sativa L.) leaf transcriptome and identification of candidate genes related to fall dormancy. PLoS ONE 2015, 10, e0122170. [Google Scholar] [CrossRef] [PubMed]
- Beheshti Rooy, S.S.; Hosseini Salekdeh, G.; Ghabooli, M.; Gholami, M.; Karimi, R. Cold-induced physiological and biochemical responses of three grapevine cultivars differing in cold tolerance. Acta Physiol. Plant. 2017, 39. [Google Scholar] [CrossRef]
- Huang, X.B.; Shi, H.Y.; Hu, Z.R.; Liu, A.; Amombo, E.; Chen, L.; Fu, J.M. ABA is involved in regulation of cold stress response in bermudagrass. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Zhou, Q.; Ma, L.C.; Xie, W.G.; Wang, Y.R.; Hu, X.W.; Liu, Z.P. Novel polymorphic expressed-sequence tag-simple-sequence repeat markers in for genetic diversity analyses. Crop Sci. 2015, 55, 2712–2718. [Google Scholar] [CrossRef]
- Dong, R.; Dong, D.K.; Luo, D.; Zhou, Q.; Chai, X.T.; Zhang, J.Y.; Xie, W.G.; Liu, W.X.; Dong, Y.; Wang, Y.R.; et al. Transcriptome analyses reveal candidate pod shattering-associated genes involved in the pod ventral sutures of common vetch (Vicia sativa L.). Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Luo, D.; Ma, L.C.; Xie, W.G.; Wang, Y.; Wang, Y.R.; Liu, Z.P. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Zhang, Z.S.; Chen, S.Y.; Ma, L.C.; Wang, H.C.; Dong, R.; Wang, Y.R.; Liu, Z.P. Global transcriptome profiling analysis reveals insight into saliva-responsive genes in alfalfa. Plant Cell Rep. 2016, 35, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.X.; Xiong, C.H.; Yan, L.F.; Zhang, Z.S.; Ma, L.C.; Wang, Y.R.; Liu, Y.J.; Liu, Z.P. Transcriptome analyses reveal candidate genes potentially involved in Al stress response in alfalfa. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Huang, L.Y.; Wang, Y.X.; Wang, W.S.; Zhao, X.Q.; Zhang, S.L.; Zhang, J.; Hu, F.Y.; Fu, B.Y.; Li, Z.K. Differential transcriptome profiling of chilling stress response between shoots and rhizomes of Oryza longistaminata using RNA sequencing. PLoS ONE 2017, 12, e0188625. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Li, W.; Zhao, J.; Zhang, S.J.; Xu, H.Y.; Liu, Y.; Guo, C.H. Transcriptome sequencing analysis of alfalfa reveals CBF genes potentially playing important roles in response to freezing stress. Genet. Mol. Biol. 2017, 40, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Mittler, R. Reactive oxygen species and temperature stresses: A delicate balance between signaling and destruction. Physiol. Plant. 2005, 126, 45–51. [Google Scholar] [CrossRef]
- Li, G.W.; Santoni, V.; Maurel, C. Plant aquaporins: Roles in plant physiology. BBA Gen. Subjcts 2014, 1840, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Song, H.W.; Yuan, W.M.; Jin, P.; Wang, W.; Wang, X.F.; Yang, L.M.; Zhang, Y.F. Effects of chitosan/nano-silica on postharvest quality and antioxidant capacity of loquat fruit during cold storage. Postharvest Biol. Technol. 2016, 119, 41–48. [Google Scholar] [CrossRef]
- Zhang, W.Y.; Niu, C.J.; Liu, Y.K.; Chen, B.J. Glutathione redox balance in hibernating Chinese soft-shelled turtle Pelodiscus sinensis hatchlings. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2017, 207, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Ito, Y.; Yamaguchi-Shinozaki, K. Transcriptional regulatory networks in response to abiotic stresses in arabidopsis and grasses. Plant Physiol. 2009, 149, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wei, W.; Pang, X.Y.; Wang, X.F.; Zhang, H.L.; Dong, B.; Xing, Y.P.; Li, X.G.; Wang, M.Y. Comparative transcriptome profiling of a desert evergreen shrub, Ammopiptanthus mongolicus, in response to drought and cold stresses. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.M.; Jiang, M.; Li, P.; Chu, Z.Q. Genome-wide identification and evolutionary analyses of the PP2C gene family with their expression profiling in response to multiple stresses in Brachypodium distachyon. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, L.; Xiao, B.; Li, D.; Xing, X.; Kong, X.; Li, D. Enhanced tolerance to low temperature in tobacco by over-expression of a new maize protein phosphatase 2C, ZmPP2C2. J. Plant Physiol. 2010, 167, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wen, J.Q.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Yang, L.; Wu, K.C.; Gao, P.; Liu, X.J.; Li, G.P.; Wu, Z.J. GsLRPK, a novel cold-activated leucine-rich repeat receptor-like protein kinase from Glycine soja, is a positive regulator to cold stress tolerance. Plant Sci. 2014, 215–216, 19–28. [Google Scholar] [CrossRef]
- Yoo, Y.H.; Choi, H.K.; Jung, K.H. Genome-wide identification and analysis of genes associated with lysigenous aerenchyma formation in rice roots. J. Plant Biol. 2015, 58, 117–127. [Google Scholar] [CrossRef]
- Peng, X.J.; Teng, L.H.; Yan, X.Q.; Zhao, M.L.; Shen, S.H. The cold responsive mechanism of the paper mulberry: Decreased photosynthesis capacity and increased starch accumulation. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Hu, J.Y.; Salas Fernandez, M.G. Genetic architecture of photosynthesis in Sorghum bicolor under non-stress and cold stress conditions. J. Exp. Bot. 2017, 68, 4545–4557. [Google Scholar] [CrossRef] [PubMed]
- Gitelson, A.A.; Gritz, Y.; Merzlyak, M.N. Relationships between leaf chlorophyll content and spectral reflectance and algorithms for non-destructive chlorophyll ssessment in higher plant leaves. J. Plant Physiol. 2003, 160, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Su, L.Y.; Dai, Z.W.; Li, S.H.; Xin, H.P. A novel system for evaluating drought-cold tolerance of grapevines using chlorophyll fluorescence. BMC Plant Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, A.J. Java Treeview-extensible visualization of microarray data. Bioinformatics 2004, 20, 3246–3248. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinf. 2006, 7. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Q.; Luo, D.; Chai, X.; Wu, Y.; Wang, Y.; Nan, Z.; Yang, Q.; Liu, W.; Liu, Z. Multiple Regulatory Networks Are Activated during Cold Stress in Medicago sativa L. Int. J. Mol. Sci. 2018, 19, 3169. https://doi.org/10.3390/ijms19103169
Zhou Q, Luo D, Chai X, Wu Y, Wang Y, Nan Z, Yang Q, Liu W, Liu Z. Multiple Regulatory Networks Are Activated during Cold Stress in Medicago sativa L. International Journal of Molecular Sciences. 2018; 19(10):3169. https://doi.org/10.3390/ijms19103169
Chicago/Turabian StyleZhou, Qiang, Dong Luo, Xutian Chai, Yuguo Wu, Yanrong Wang, Zhibiao Nan, Qingchuan Yang, Wenxian Liu, and Zhipeng Liu. 2018. "Multiple Regulatory Networks Are Activated during Cold Stress in Medicago sativa L." International Journal of Molecular Sciences 19, no. 10: 3169. https://doi.org/10.3390/ijms19103169
APA StyleZhou, Q., Luo, D., Chai, X., Wu, Y., Wang, Y., Nan, Z., Yang, Q., Liu, W., & Liu, Z. (2018). Multiple Regulatory Networks Are Activated during Cold Stress in Medicago sativa L. International Journal of Molecular Sciences, 19(10), 3169. https://doi.org/10.3390/ijms19103169