Decision-Tree Based Meta-Strategy Improved Accuracy of Disorder Prediction and Identified Novel Disordered Residues Inside Binding Motifs


Abstract
:1. Introduction
2. Results
2.1. Prediction Performance of Component Predictors
2.2. Use Information Gain to Choose Threshold Values
2.3. Performance of the New Predictor
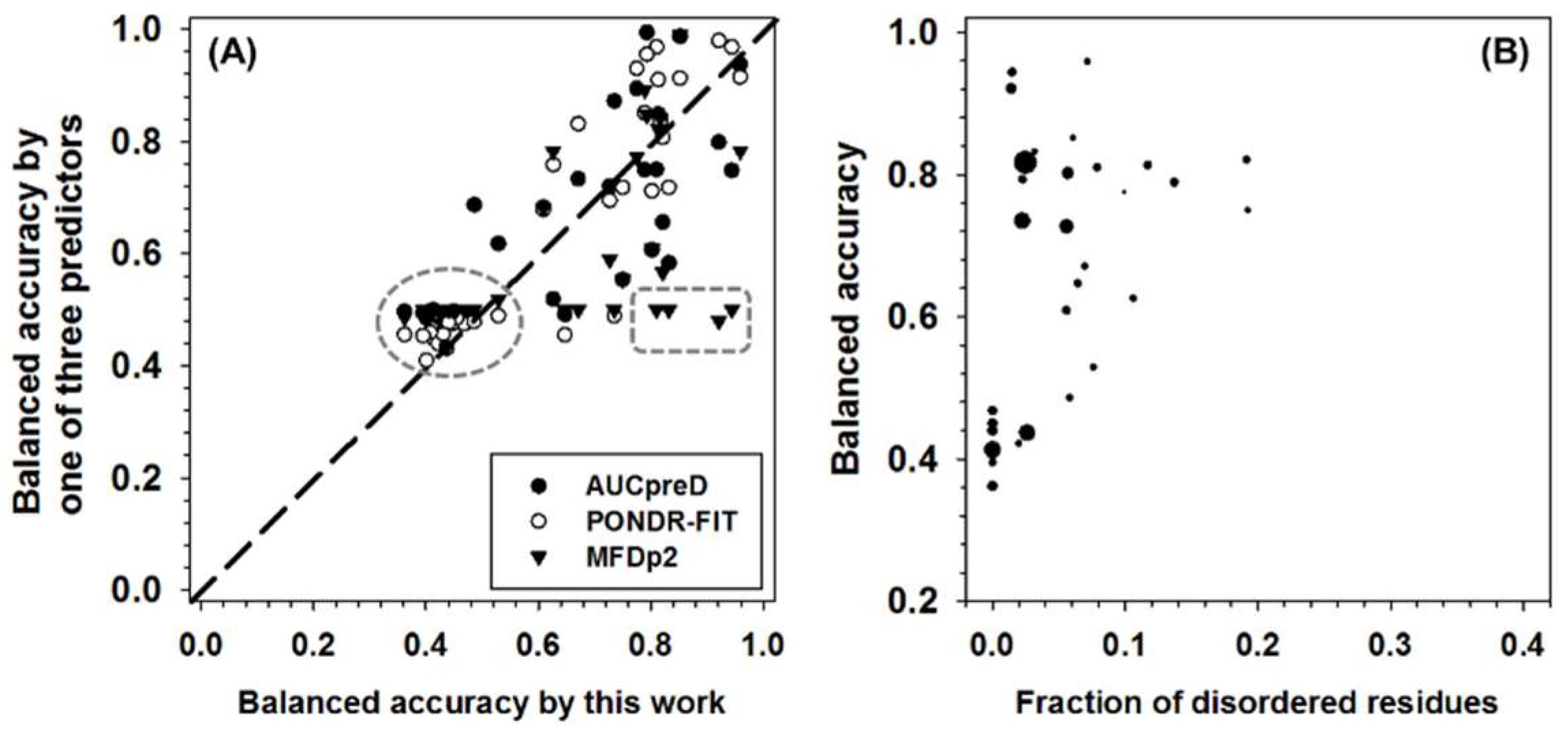
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| IDP | Intrinsically disordered protein |
| IDR | Intrinsically disordered region |
| IDAA | Intrinsically disordered amino acid |
| ANN | Artificial neural network |
| IG | Information gain |
| Sens | Sensitivity |
| Spec | Specificity |
| Acc | Accuracy |
| Acc-b | Balanced accuracy |
| MCC | Mathew’s correlation coefficient |
| AUC-ROC | Area under ROC curve |
| AUC-PR | Area under precision-recall curve |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sen | Spec | Acc | Acc-b | MCC | F1 | AUC_ROC | AUC_PR | |
|---|---|---|---|---|---|---|---|---|
| Disembl | 0.379 | 0.954 | 0.929 | 0.666 | 0.286 | 0.318 | 0.754 | 0.295 |
| IUPred | 0.168 | 0.958 | 0.924 | 0.563 | 0.122 | 0.161 | 0.618 | 0.175 |
| VSL2 | 0.612 | 0.811 | 0.803 | 0.712 | 0.214 | 0.213 | 0.774 | 0.275 |
| Espritz | 0.512 | 0.921 | 0.903 | 0.716 | 0.298 | 0.316 | 0.815 | 0.404 |
| DISOPRED3 | 0.362 | 0.993 | 0.966 | 0.678 | 0.495 | 0.481 | 0.860 | 0.495 |
| PONDRFIT | 0.586 | 0.929 | 0.914 | 0.758 | 0.362 | 0.374 | 0.830 | 0.358 |
| MFDp2 | 0.325 | 0.975 | 0.947 | 0.650 | 0.322 | 0.349 | 0.778 | 0.352 |
| IUPred2 | 0.164 | 0.959 | 0.924 | 0.561 | 0.119 | 0.158 | 0.616 | 0.170 |
| AUCpreD | 0.425 | 0.984 | 0.960 | 0.705 | 0.465 | 0.481 | 0.863 | 0.501 |
| This work | 0.629 | 0.840 | 0.831 | 0.734 | 0.249 | 0.245 | 0.793 | 0.305 |
References
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Multiparametric analysis of intrinsically disordered proteins: Looking at intrinsic disorder through compound eyes. Anal. Chem. 2012, 84, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Sandhu, K.S.; Hazai, E.; Hoksza, Z.; Kiss, H.J.; Miozzo, F.; Veres, D.V.; Piazza, F.; Nussinov, R. Disordered proteins and network disorder in network descriptions of protein structure, dynamics and function: Hypotheses and a comprehensive review. Curr. Protein Pept. Sci. 2012, 13, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. Intrinsically disordered proteins: A. 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dave, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Toth-Petroczy, A.; Kraut, D.A.; Matouschek, A.; Lim, R.Y.; Xue, B.; Kurgan, L.; Uversky, V.N. Disordered proteinaceous machines. Chem. Rev. 2014, 114, 6806–6843. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follis, A.V.; Hammoudeh, D.I.; Wang, H.; Prochownik, E.V.; Metallo, S.J. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 2008, 15, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulenburg, C.; Hilvert, D. Protein conformational disorder and enzyme catalysis. Top Curr. Chem. 2013, 337, 41–67. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef] [PubMed]
- Dogan, J.; Gianni, S.; Jemth, P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Huang, Y. Advantages of proteins being disordered. Protein Sci. 2014, 23, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teilum, K.; Olsen, J.G.; Kragelund, B.B. Globular and disordered-the non-identical twins in protein-protein interactions. Front. Mol. Biosci. 2015, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Minde, D.P.; Dunker, A.K.; Lilley, K.S. Time, space, and disorder in the expanding proteome universe. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Tabaro, F.; Micetic, I.; Necci, M.; Quaglia, F.; Oldfield, C.J.; Aspromonte, M.C.; Davey, N.E.; Davidovic, R.; Dosztanyi, Z.; et al. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017, 45, D219–D227. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically Disordered proteins with Extensive Annotations and Literature. Nucleic Acids Res. 2012, 40, D507–D511. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.F.; Dou, X.H.; Sha, Y.J.; Wang, C.L.; Wang, H.B.; Chen, Y.T.; Zhang, F.; Zhou, Y.; Wang, J.H. DisBind: A database of classified functional binding sites in disordered and structured regions of intrinsically disordered proteins. BMC Bioinform. 2017, 18, 206. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Ficho, E.; Pancsa, R.; Simon, I.; Dosztanyi, Z.; Meszaros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2018, 34, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Ficho, E.; Remenyi, I.; Simon, I.; Meszaros, B. MFIB: A repository of protein complexes with mutual folding induced by binding. Bioinformatics 2017, 33, 3682–3684. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Oldfield, C.J.; Meng, J.; Romero, P.; Uversky, V.N.; Dunker, A.K. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar] [CrossRef] [PubMed]
- Malhis, N.; Gsponer, J. Computational identification of MoRFs in protein sequences. Bioinformatics 2015, 31, 1738–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Noguchi, T.; Tominaga, D.; Yamana, H. MFSPSSMpred: Identifying short disorder-to-order binding regions in disordered proteins based on contextual local evolutionary conservation. BMC Bioinform. 2013, 14, 300. [Google Scholar] [CrossRef] [PubMed]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, W488–W493. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bayarjargal, M.; Tsunoda, T.; Patil, A.; Sharma, A. MoRFPred-plus: Computational Identification of MoRFs in Protein Sequences using Physicochemical Properties and HMM profiles. J. Theor. Biol. 2018, 437, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Raicar, G.; Tsunoda, T.; Patil, A.; Sharma, A. OPAL: Prediction of MoRF regions in intrinsically disordered protein sequences. Bioinformatics 2018, 34, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Meszaros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef] [PubMed]
- Mooney, C.; Pollastri, G.; Shields, D.C.; Haslam, N.J. Prediction of short linear protein binding regions. J. Mol. Biol. 2012, 415, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.; Duffy, F.; Pollastri, G.; Shields, D.C.; Mooney, C. Predicting binding within disordered protein regions to structurally characterised peptide-binding domains. PLoS ONE 2013, 8, 72838. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Cai, Y.D.; Feng, K.Y.; Zhao, G.J. Prediction of protein cleavage site with feature selection by random forest. PLoS ONE 2012, 7, e45854. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dai, J.; Ning, Q.; Ma, Z.; Yin, M.; Sun, P. Position-specific analysis and prediction of protein pupylation sites based on multiple features. BioMed Res. Int. 2013, 2013, 109549. [Google Scholar] [CrossRef] [PubMed]
- Tretyachenko, V.; Vymetal, J.; Bednarova, L.; Kopecky, V., Jr.; Hofbauerova, K.; Jindrova, H.; Hubalek, M.; Soucek, R.; Konvalinka, J.; Vondrasek, J.; et al. Random protein sequences can form defined secondary structures and are well-tolerated in vivo. Sci. Rep. 2017, 7, 15449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, Y.; Zhang, Y.; Yu, D.J. ATPbind: Accurate Protein-ATP Binding Site Prediction by Combining Sequence-Profiling and Structure-Based Comparisons. J. Chem. Inf. Model. 2018, 58, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Soderquist, F.; Wallner, B. Proteus: A random forest classifier to predict disorder-to-order transitioning binding regions in intrinsically disordered proteins. J. Comput. Aided Mol. Des. 2017, 31, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Soenderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. BioRxiv 2018. [Google Scholar] [CrossRef]
- Xue, B.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. CDF it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, A.; Punta, M.; Yachdav, G.; Kajan, L.; Rost, B. Improved disorder prediction by combination of orthogonal approaches. PLoS ONE 2009, 4, e4433. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Shimizu, K.; Noguchi, T. POODLE-I: Disordered region prediction by integrating POODLE series and structural information predictors based on a workflow approach. In Silico Biol. 2010, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Acton, T.B.; Montelione, G.T. DisMeta: A meta server for construct design and optimization. Methods Mol. Biol. 2014, 1091, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Stach, W.; Chen, K.; Kedarisetti, K.D.; Disfani, F.M.; Kurgan, L. Improved sequence-based prediction of disordered regions with multilayer fusion of multiple information sources. Bioinformatics 2010, 26, i489–i496. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Lipps, D.; Devineni, S. Integrated Strategy Improves the Prediction Accuracy of miRNA in Large Dataset. PLoS ONE 2016, 11, e0168392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Xue, B. Improving prediction accuracy using decision-tree-based meta-strategy and multi-threshold sequential-voting exemplified by miRNA target prediction. Genomics 2017, 109, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dor, O.; Faraggi, E.; Zhou, Y. Real-value prediction of backbone torsion angles. Proteins 2008, 72, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Mann, M. Origins of mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 678. [Google Scholar] [CrossRef] [PubMed]
- Minde, D.P.; Ramakrishna, M.; Lilley, K.S. Biotinylation by proximity labelling favours unfolded proteins. BioRxiv 2018. [Google Scholar] [CrossRef]
- Wang, G.; Dunbrack, R.L., Jr. PISCES: Recent improvements to a PDB sequence culling server. Nucleic Acids Res. 2005, 33, W94–W98. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in CASP10. Proteins 2014, 82, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein disorder prediction: Implications for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Radivojac, P.; Vucetic, S.; Dunker, A.K.; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinform. 2006, 7, 208. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Mizianty, M.J.; Peng, Z.; Kurgan, L. MFDp2: Accurate predictor of disorder in proteins by fusion of disorder probabilities, content and profiles. Intrinsically Disord. Proteins 2013, 1, e24428. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ma, J.; Xu, J. AUCpreD: Proteome-level protein disorder prediction by AUC-maximized deep convolutional neural fields. Bioinformatics 2016, 32, i672–i679. [Google Scholar] [CrossRef] [PubMed]






| DisEMBL | IUPred | VSL2 | Espritz | PONDR-FIT | MFDp2 | IUPred2 | AUCpreD | This Work | |
|---|---|---|---|---|---|---|---|---|---|
| Sens | 0.440 ± 0.008 | 0.650 ± 0.003 | 0.817 ± 0.004 | 0.514 ± 0.009 | 0.713 ± 0.004 | 0.777 ± 0.004 | 0.640 ± 0.003 | 0.592 ± 0.006 | 0.807 ± 0.012 |
| Spec | 0.914± 0.002 | 0.874 ± 0.004 | 0.736 ± 0.003 | 0.939 ± 0.002 | 0.859 ± 0.004 | 0.859 ± 0.004 | 0.877 ± 0.004 | 0.909 ± 0.002 | 0.856 ± 0.007 |
| Acc | 0.779 ± 0.003 | 0.810 ± 0.003 | 0.759 ± 0.002 | 0.818 ± 0.004 | 0.817 ± 0.004 | 0.836 ± 0.003 | 0.810 ± 0.003 | 0.819 ± 0.003 | 0.842 ± 0.003 |
| Acc-b | 0.677 ± 0.006 | 0.762 ± 0.002 | 0.776 ± 0.002 | 0.726 ± 0.004 | 0.786 ± 0.003 | 0.818 ± 0.003 | 0.759 ± 0.002 | 0.751 ± 0.003 | 0.831 ± 0.004 |
| MCC | 0.410 ± 0.007 | 0.529 ± 0.006 | 0.504 ± 0.004 | 0.521 ± 0.007 | 0.561 ± 0.007 | 0.614 ± 0.006 | 0.526 ± 0.006 | 0.535 ± 0.006 | 0.635 ± 0.006 |
| F1 | 0.531 ± 0.006 | 0.660 ± 0.003 | 0.658 ± 0.003 | 0.616 ± 0.006 | 0.689 ± 0.004 | 0.729 ± 0.003 | 0.657 ± 0.004 | 0.651 ± 0.005 | 0.744 ± 0.004 |
| AUC_ROC | 0.776 ± 0.004 | 0.823 ± 0.001 | 0.841 ± 0.003 | 0.886 ± 0.003 | 0.857 ± 0.003 | 0.879 ± 0.002 | 0.822 ± 0.001 | 0.869 ± 0.003 | 0.899 ± 0.004 |
| AUC_PR | 0.607 ± 0.007 | 0.675 ± 0.007 | 0.656 ± 0.020 | 0.752 ± 0.006 | 0.696 ± 0.004 | 0.629 ± 0.006 | 0.657 ± 0.004 | 0.716 ± 0.007 | 0.788 ± 0.010 |
| DisEMBL | IUPred | VSL2 | Espritz | PONDR-FIT | MFDp2 | IUPred2 | AUCpreD | This Work | |
|---|---|---|---|---|---|---|---|---|---|
| Sens | 0.454 | 0.656 | 0.82 | 0.529 | 0.728 | 0.78 | 0.647 | 0.609 | 0.811 ± 0.007 |
| Spec | 0.915 | 0.872 | 0.735 | 0.932 | 0.856 | 0.857 | 0.87 | 0.908 | 0.856 ± 0.006 |
| Acc | 0.784 | 0.811 | 0.759 | 0.818 | 0.82 | 0.835 | 0.811 | 0.823 | 0.844 ± 0.003 |
| Acc-b | 0.684 | 0.764 | 0.777 | 0.731 | 0.792 | 0.819 | 0.761 | 0.759 | 0.834 ± 0.001 |
| MCC | 0.424 | 0.532 | 0.507 | 0.521 | 0.569 | 0.615 | 0.53 | 0.53 | 0.639 ± 0.003 |
| F1 | 0.544 | 0.663 | 0.659 | 0.622 | 0.696 | 0.729 | 0.66 | 0.66 | 0.747 ± 0.002 |
| AUC_ROC | 0.779 | 0.824 | 0.841 | 0.888 | 0.857 | 0.88 | 0.822 | 0.872 | 0.9 ± 0.002 |
| AUC_PR | 0.617 | 0.673 | 0.642 | 0.754 | 0.695 | 0.622 | 0.672 | 0.72 | 0.789 ± 0.005 |
| DisEMBL | IUPred | VSL2 | Espritz | PONDR-FIT | MFDp2 | IUPred2 | AUCpreD | This Work | ||
|---|---|---|---|---|---|---|---|---|---|---|
| N-ter | Sens | 0.553 ± 0.009 | 0.541 ± 0.017 | 0.782 ± 0.011 | 0.582 ± 0.010 | 0.837 ± 0.004 | 0.782 ± 0.009 | 0.539 ± 0.015 | 0.748 ± 0.011 | 0.829 ± 0.023 |
| Spec | 0.741 ± 0.016 | 0.841 ± 0.020 | 0.524 ± 0.024 | 0.789 ± 0.012 | 0.405 ± 0.020 | 0.582 ± 0.039 | 0.842 ± 0.020 | 0.590 ± 0.038 | 0.572 ± 0.049 | |
| Acc | 0.614 ± 0.003 | 0.639 ± 0.012 | 0.698 ± 0.005 | 0.650 ± 0.004 | 0.697 ± 0.006 | 0.718 ± 0.012 | 0.638 ± 0.014 | 0.697 ± 0.011 | 0.746 ± 0.014 | |
| Acc-b | 0.647 ± 0.004 | 0.691 ± 0.011 | 0.653 ± 0.010 | 0.686 ± 0.004 | 0.621 ± 0.011 | 0.682 ± 0.020 | 0.691 ± 0.014 | 0.669 ± 0.016 | 0.701 ± 0.020 | |
| MCC | 0.277 ± 0.009 | 0.364 ± 0.021 | 0.308 ± 0.019 | 0.349 ± 0.009 | 0.265 ± 0.024 | 0.361 ± 0.038 | 0.363 ± 0.027 | 0.330 ± 0.029 | 0.410 ± 0.035 | |
| F1 | 0.660 ± 0.007 | 0.669 ± 0.015 | 0.778 ± 0.006 | 0.692 ± 0.008 | 0.789 ± 0.007 | 0.789 ± 0.007 | 0.668 ± 0.014 | 0.770 ± 0.010 | 0.815 ± 0.013 | |
| Middle | Sens | 0.387 ± 0.009 | 0.682 ± 0.005 | 0.820 ± 0.004 | 0.481 ± 0.010 | 0.663 ± 0.004 | 0.777 ± 0.005 | 0.672 ± 0.005 | 0.539 ± 0.007 | 0.807 ± 0.013 |
| Spec | 0.927 ± 0.001 | 0.877 ± 0.004 | 0.751 ± 0.004 | 0.948 ± 0.002 | 0.888 ± 0.004 | 0.875 ± 0.005 | 0.880 ± 0.004 | 0.927 ± 0.001 | 0.877 ± 0.006 | |
| Acc | 0.801 ± 0.004 | 0.831 ± 0.004 | 0.767 ± 0.003 | 0.839 ± 0.004 | 0.835 ± 0.004 | 0.852 ± 0.005 | 0.831 ± 0.004 | 0.836 ± 0.003 | 0.861 ± 0.004 | |
| Acc-b | 0.657 ± 0.005 | 0.780 ± 0.003 | 0.786 ± 0.003 | 0.715 ± 0.005 | 0.776 ± 0.003 | 0.826 ± 0.004 | 0.776 ± 0.003 | 0.732 ± 0.003 | 0.842 ± 0.005 | |
| MCC | 0.376 ± 0.009 | 0.544 ± 0.005 | 0.497 ± 0.006 | 0.506 ± 0.008 | 0.546 ± 0.006 | 0.616 ± 0.008 | 0.540 ± 0.007 | 0.510 ± 0.006 | 0.643 ± 0.008 | |
| F1 | 0.477 ± 0.007 | 0.628 ± 0.003 | 0.622 ± 0.005 | 0.583 ± 0.008 | 0.653 ± 0.004 | 0.711 ± 0.004 | 0.650 ± 0.004 | 0.606 ± 0.005 | 0.731 ± 0.006 | |
| C-ter | Sens | 0.584 ± 0.014 | 0.615 ± 0.017 | 0.847 ± 0.016 | 0.609 ± 0.019 | 0.828 ± 0.017 | 0.771 ± 0.016 | 0.598 ± 0.016 | 0.681 ± 0.013 | 0.790 ± 0.018 |
| Spec | 0.787 ± 0.021 | 0.838 ± 0.023 | 0.586 ± 0.014 | 0.857 ± 0.015 | 0.615 ± 0.017 | 0.743 ± 0.015 | 0.845 ± 0.019 | 0.796 ± 0.019 | 0.769 ± 0.021 | |
| Acc | 0.686 ± 0.013 | 0.727 ± 0.005 | 0.715 ± 0.007 | 0.734 ± 0.009 | 0.720 ± 0.008 | 0.757 ± 0.007 | 0.723 ± 0.007 | 0.739 ± 0.009 | 0.780 ± 0.009 | |
| Acc-b | 0.685 ± 0.012 | 0.726 ± 0.006 | 0.716 ± 0.007 | 0.733 ± 0.007 | 0.721 ± 0.009 | 0.757 ± 0.007 | 0.722 ± 0.008 | 0.739 ± 0.008 | 0.780 ± 0.009 | |
| MCC | 0.379 ± 0.026 | 0.465 ± 0.014 | 0.448 ± 0.015 | 0.482 ± 0.015 | 0.453 ± 0.018 | 0.514 ± 0.014 | 0.459 ± 0.018 | 0.481 ± 0.018 | 0.560 ± 0.018 | |
| F1 | 0.649 ± 0.012 | 0.691 ± 0.008 | 0.747 ± 0.008 | 0.694 ± 0.009 | 0.746 ± 0.008 | 0.759 ± 0.006 | 0.682 ± 0.012 | 0.722 ± 0.007 | 0.781 ± 0.006 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B.; Xue, B. Decision-Tree Based Meta-Strategy Improved Accuracy of Disorder Prediction and Identified Novel Disordered Residues Inside Binding Motifs. Int. J. Mol. Sci. 2018, 19, 3052. https://doi.org/10.3390/ijms19103052
Zhao B, Xue B. Decision-Tree Based Meta-Strategy Improved Accuracy of Disorder Prediction and Identified Novel Disordered Residues Inside Binding Motifs. International Journal of Molecular Sciences. 2018; 19(10):3052. https://doi.org/10.3390/ijms19103052
Chicago/Turabian StyleZhao, Bi, and Bin Xue. 2018. "Decision-Tree Based Meta-Strategy Improved Accuracy of Disorder Prediction and Identified Novel Disordered Residues Inside Binding Motifs" International Journal of Molecular Sciences 19, no. 10: 3052. https://doi.org/10.3390/ijms19103052