Simple Sequence Repeat (SSR) Genetic Linkage Map of D Genome Diploid Cotton Derived from an Interspecific Cross between Gossypium davidsonii and Gossypium klotzschianum

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Parental Polymorphism
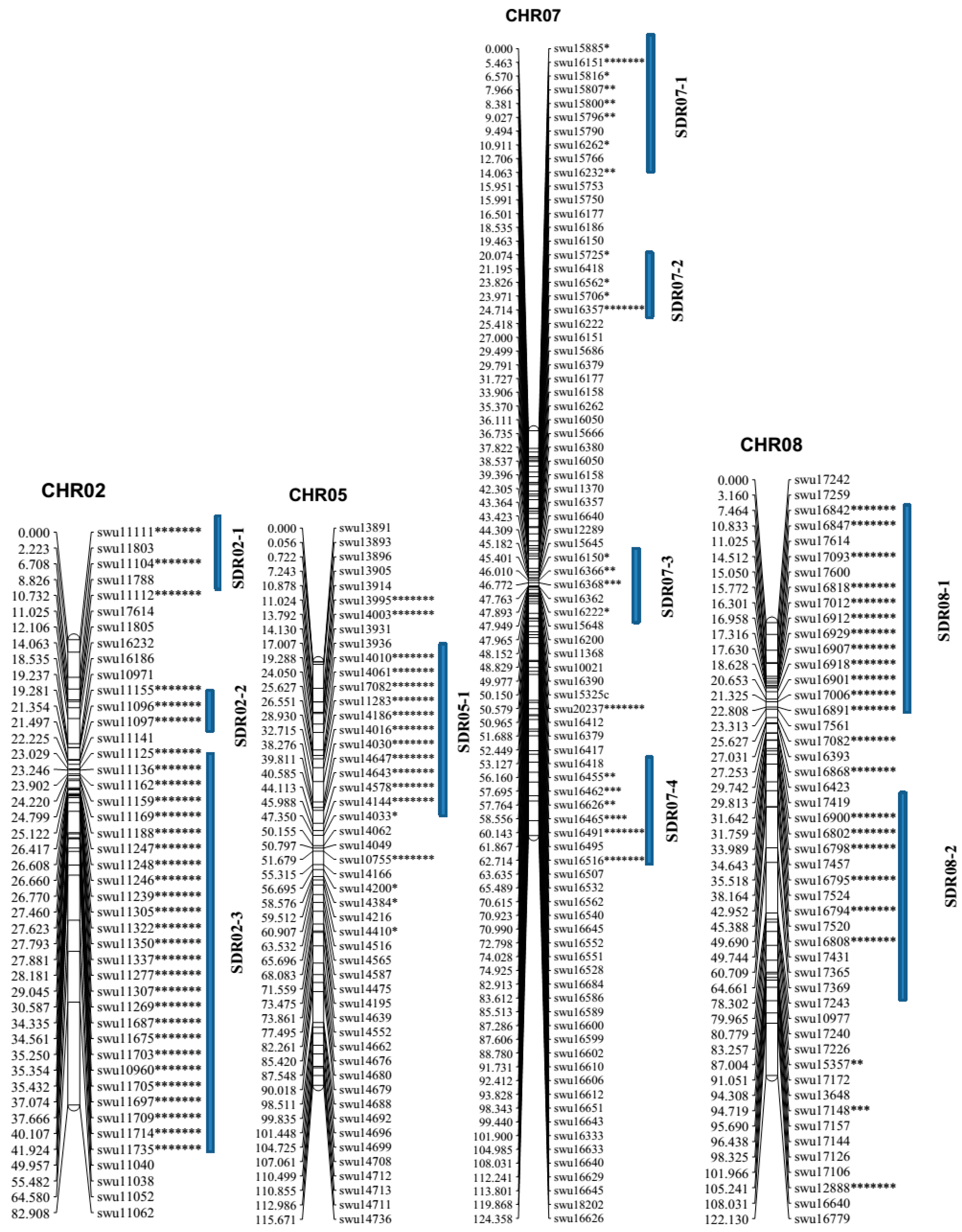
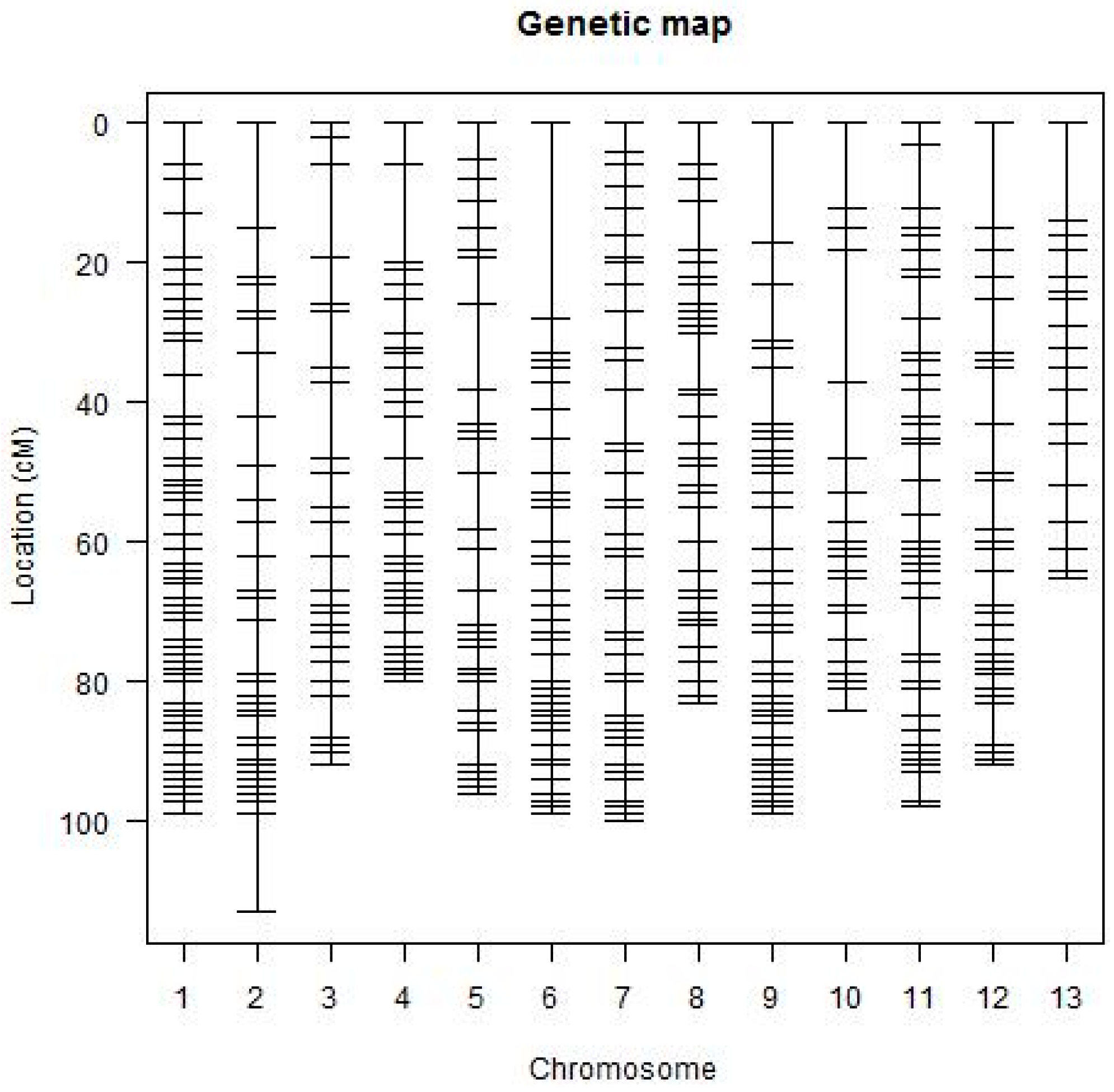
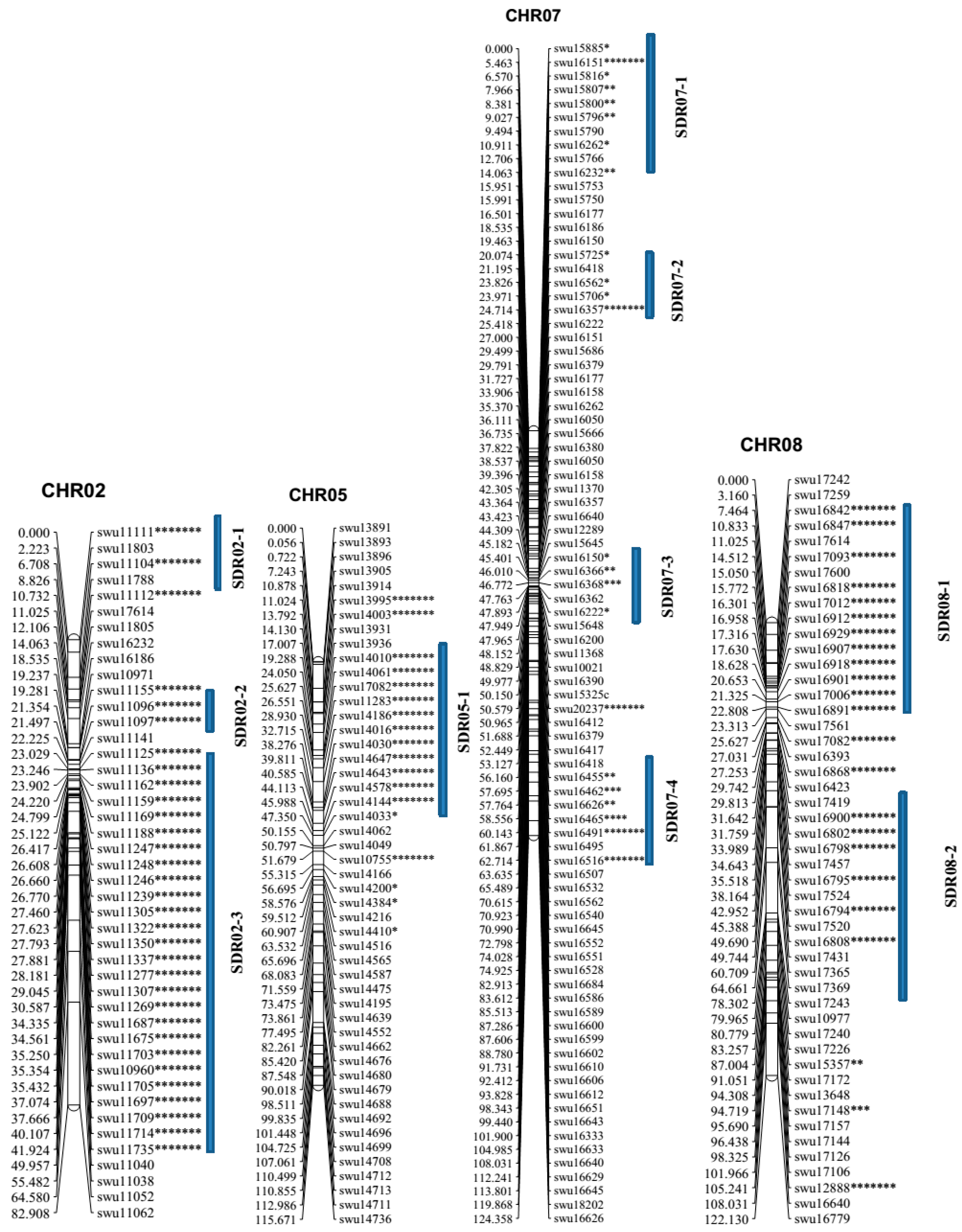
2.2. Linkage Analysis and Map Construction
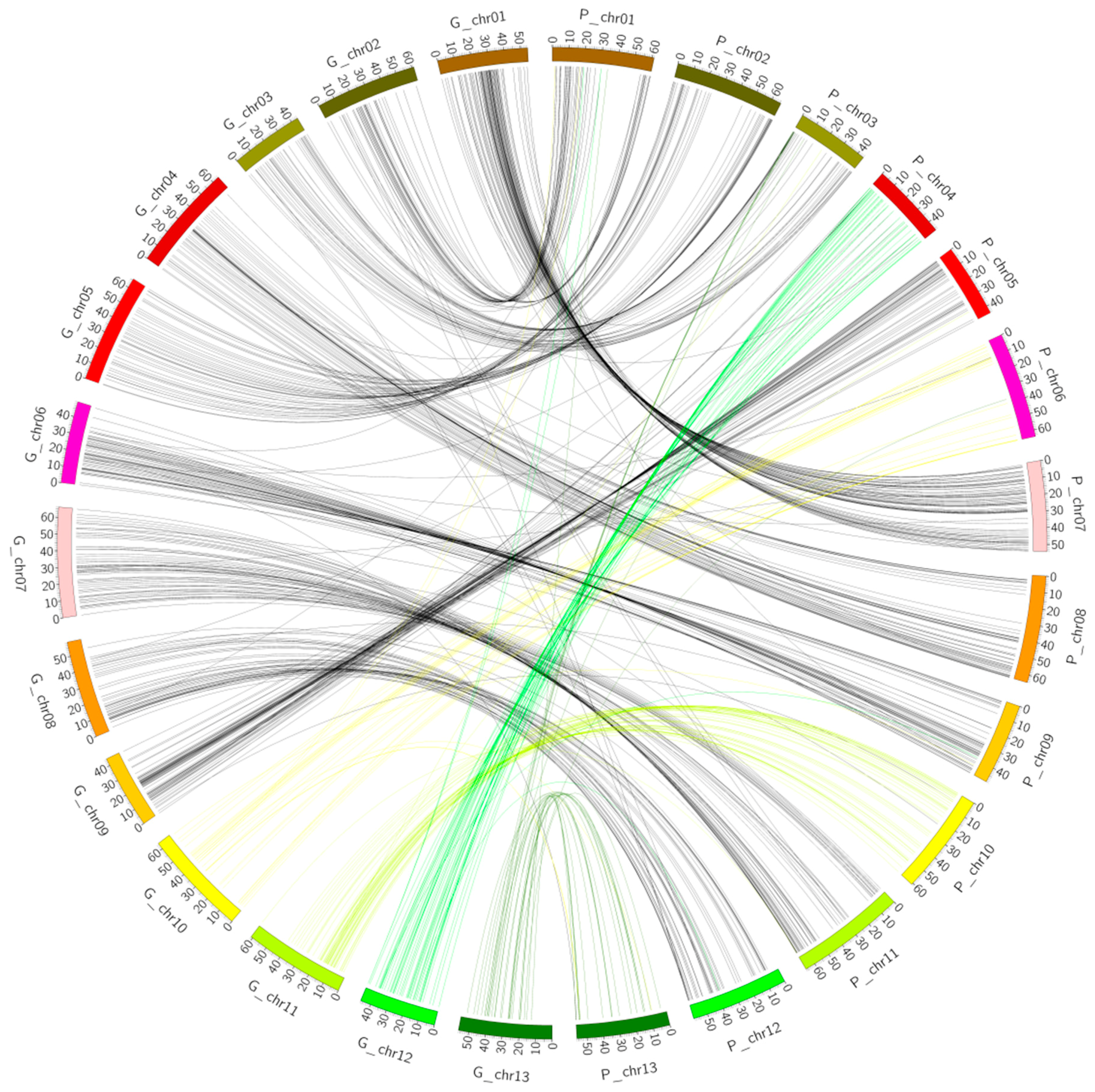
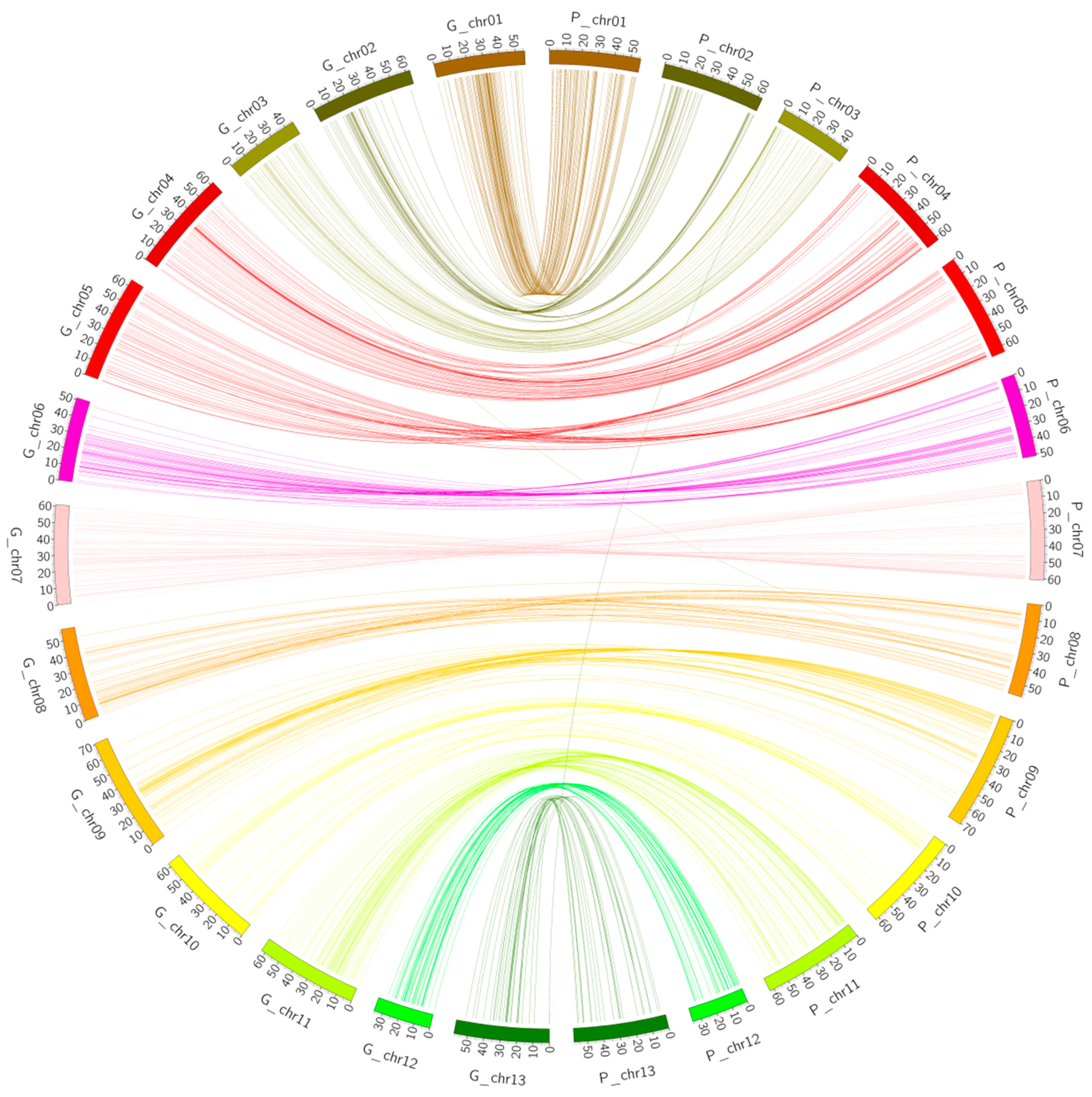
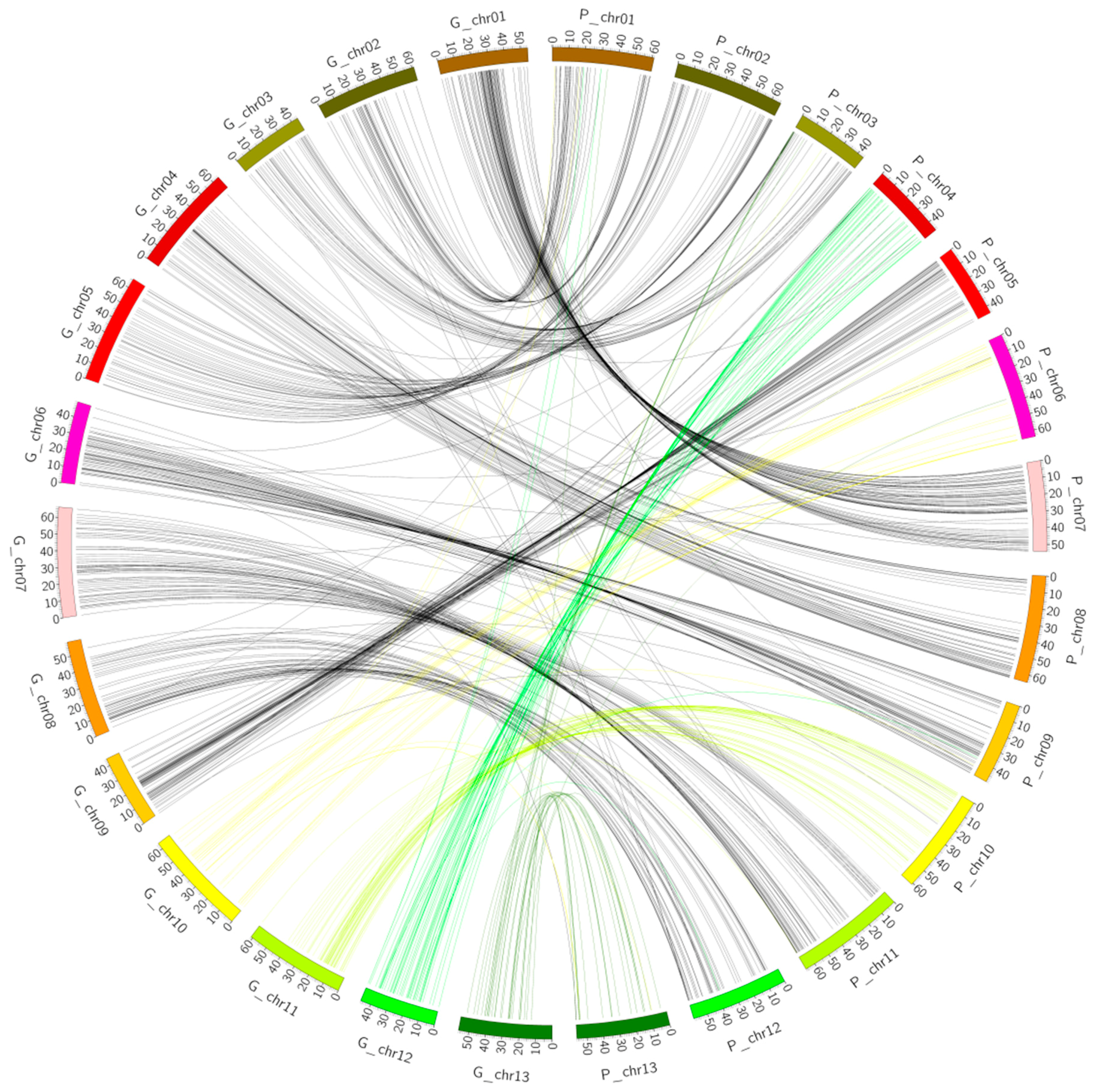
2.3. Collinearity Analysis
2.4. Collinearity Analysis of the Genetic Map and the Physical Map of the (Dt) Sub-Genome
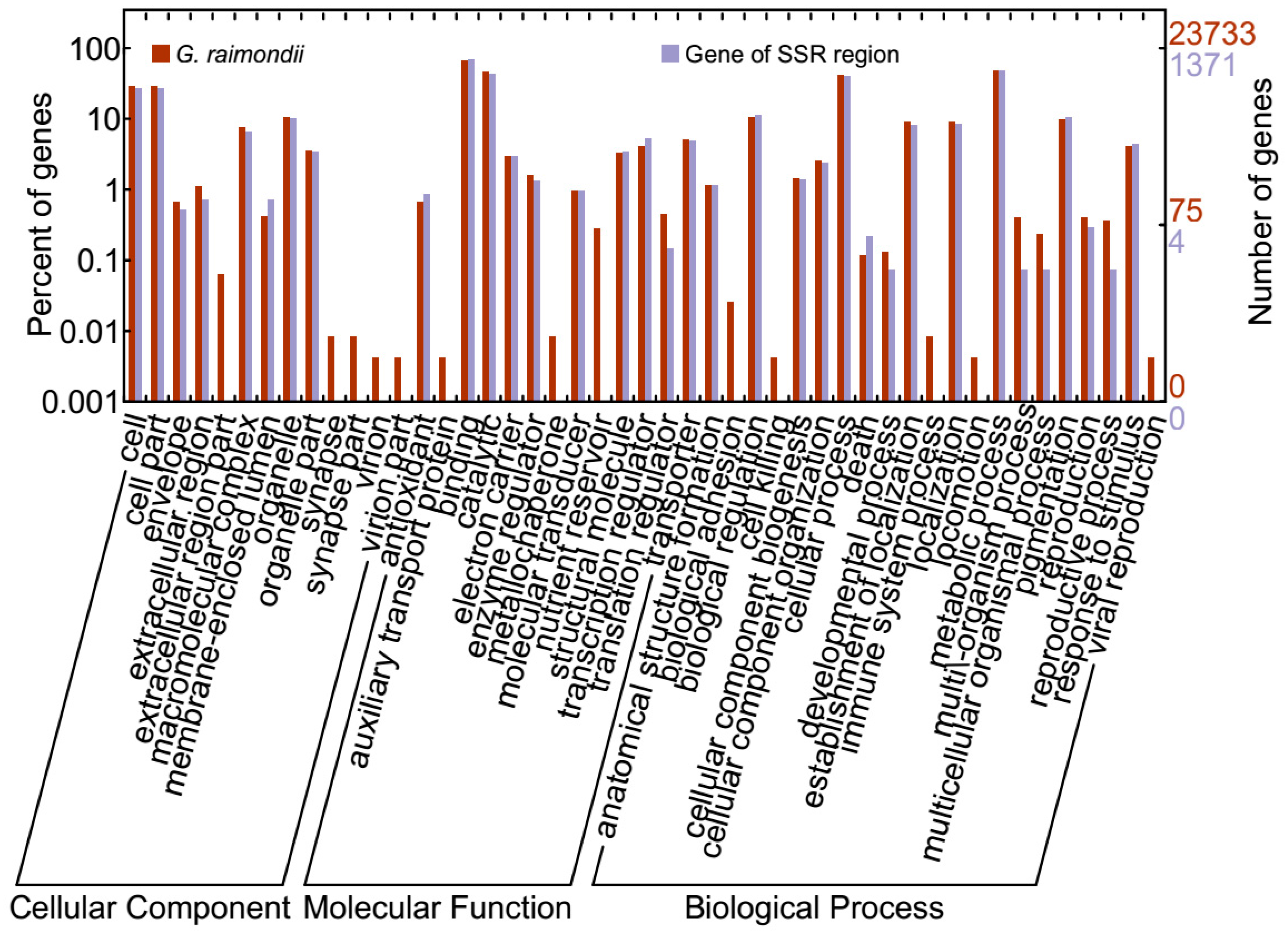
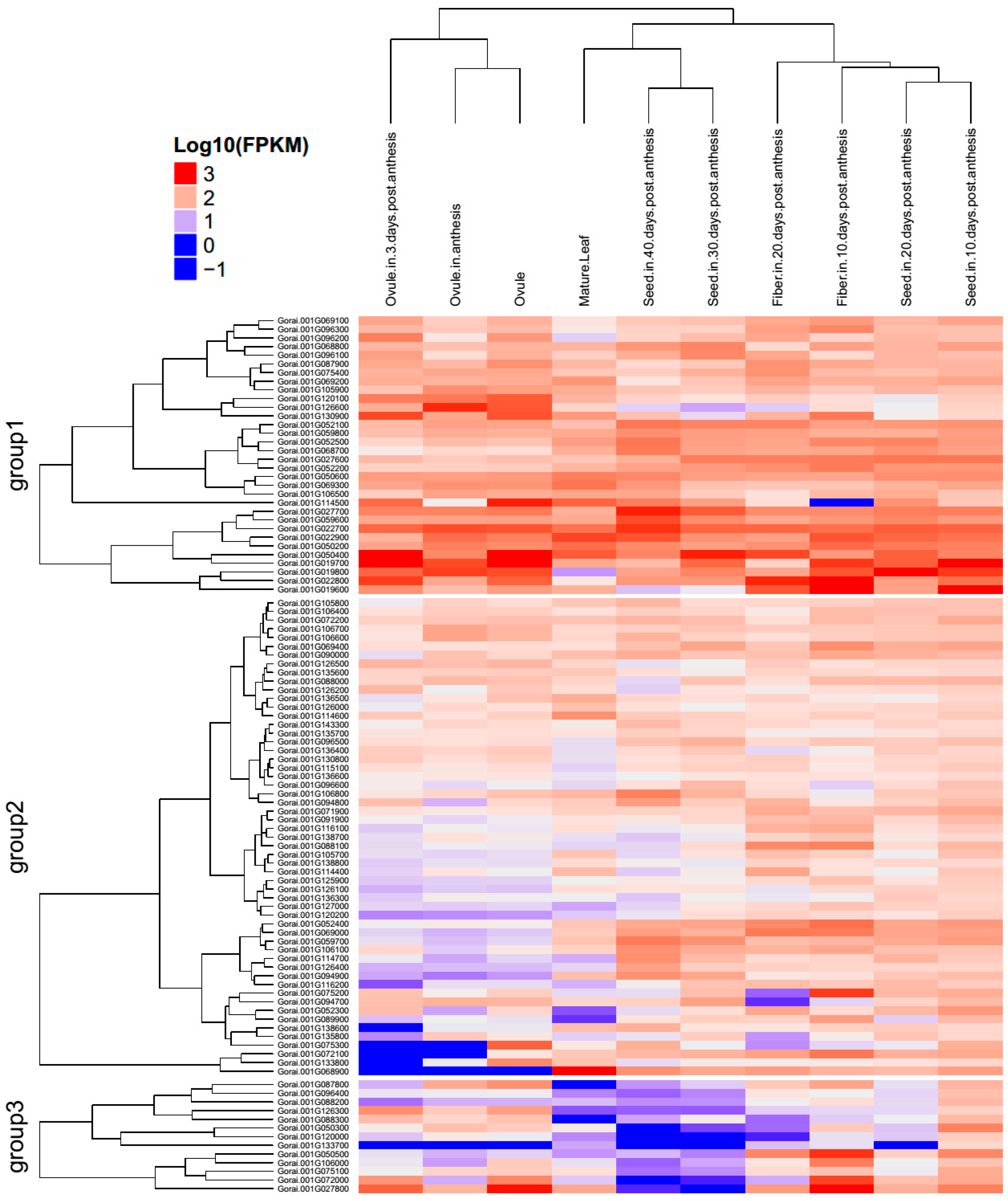
2.5. Gene Mining, GO Functional Annotation, and Expression
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction, Quantification, and Qualification
4.3. Screening of SWU Markers and Genotyping of F2:3 Populations
4.4. Construction of the Linkage Maps
4.5. Collinearity Analysis
4.6. Gene Mining, GO Functional Annotation, and Expression Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LG | linkage group |
| EST-SSR | Express sequence tag-simple sequence repeat |
| gSSR | Genomic simple sequence repeat |
| GO | Gene ontology |
| BP | Biological process |
| MF | Molecular function |
| CC | Cellular components |
| GRAVY | Grand average of hydropathy |
| SWU | South west university |
| MPK | Mitogen-activated protein kinase |
References
- Geber, M.A.; Griffen, L.R. Inheritance and Natural Selection on Functional Traits. Int. J. Plant Sci. 2003, 164, S21–S42. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, H.; Ren, X.; Chen, Y.; Xiao, Y.; Zhao, X.; Tang, M.; Huang, J.; Upadhyaya, H.D.; Liao, B. Abundant Microsatellite Diversity and Oil Content in Wild Arachis Species. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applequist, W.L.; Cronn, R.; Wendel, J.F. Comparative development of fiber in wild and cultivated cotton. Evol. Dev. 2001, 3, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Hulse-Kemp, A.M.; Ashrafi, H.; Zheng, X.; Wang, F.; Hoegenauer, K.A.; Maeda, A.B.V.; Yang, S.S.; Stoffel, K.; Matvienko, M.; Clemons, K.; et al. Development and bin mapping of gene-associated interspecific SNPs for cotton (Gossypium hirsutum L.) introgression breeding efforts. BMC Genom. 2014, 15, 945. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, E. Obtaining High Quality Textile Fibre from Industrial Hemp through Organic Cultivation by Elin Bengtsson; SLU: Alnarp, Sweden, 2009. [Google Scholar]
- Resta, B.; Gaiardelli, P.; Pinto, R.; Dotti, S. Enhancing environmental management in the textile sector: An Organisational-Life Cycle Assessment approach. J. Clean. Prod. 2016, 135, 620–632. [Google Scholar] [CrossRef]
- Zhou, M.; Sun, G.; Sun, Z.; Tang, Y.; Wu, Y. Cotton proteomics for deciphering the mechanism of environment stress response and fiber development. J. Proteom. 2014, 105, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Diouf, L.; Pan, Z.; He, S.-P.; Gong, W.-F.; Jia, Y.H.; Magwanga, R.O.; Romy, K.R.E.; Rashid, H.; Kirungu, J.N.; Du, X. High-Density Linkage Map Construction and Mapping of Salt-Tolerant QTLs at Seedling Stage in Upland Cotton Using Genotyping by Sequencing (GBS). Int. J. Mol. Sci. 2017, 18, 2622. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Thyssen, G.N.; Jenkins, J.N.; Fang, D.D. Detection, Validation, and Application of Genotyping-by-Sequencing Based Single Nucleotide Polymorphisms in Upland Cotton. Plant Genome 2015, 8. [Google Scholar] [CrossRef]
- Wegier, A.; Piñeyro-Nelson, A.; Alarcón, J.; Gálvez-Mariscal, A.; Álvarez-Buylla, E.R.; Piñero, D. Recent long-distance transgene flow into wild populations conforms to historical patterns of gene flow in cotton (Gossypium hirsutum) at its centre of origin. Mol. Ecol. 2011, 20, 4182–4194. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, V.S.K.; Reddy, T.P.; Reddy, V.D.; Rao, K.V. Current status of genetic engineering in cotton (Gossypium hirsutum L): An assessment. Crit. Rev. Biotechnol. 2012, 8551, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tuberosa, R.; Forster, B.P.; Ellis, R.P.; Thomas, W.T.; Newton, A.C.; This, D.; El-Enein, R.A.; Bahri, M.H.; Ben Salem, M. The development and application of molecular markers for abiotic stress tolerance in barley. J. Exp. Bot. 2000, 51, 19–27. [Google Scholar] [CrossRef]
- Khoury, C.K.; Castañeda-Alvarez, N.P.; Achicanoy, H.A.; Sosa, C.C.; Bernau, V.; Kassa, M.T.; Norton, S.L.; van der Maesen, L.J.G.; Upadhyaya, H.D.; Ramírez-Villegas, J.; et al. Crop wild relatives of pigeonpea [Cajanus cajan (L.) Millsp.]: Distributions, ex situ conservation status, and potential genetic resources for abiotic stress tolerance. Biol. Conserv. 2015, 184, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Becerra Lopez-Lavalle, L.A.; Matheson, B.; Brubaker, C.L.; Van Deynze, A. A genetic map of an Australian wild Gossypium C genome and assignment of homoeologies with tetraploid cultivated cotton. Genome 2011, 54, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Rungis, D.; Llewellyn, D.; Dennis, E.S.; Lyon, B.R. Simple sequence repeat (SSR) markers reveal low levels of polymorphism between cotton (Gossypium hirsutum L.) cultivars. Aust. J. Agric. Res. 2005, 56, 301–307. [Google Scholar] [CrossRef]
- Abdurakhmonov, I.Y.; Kohel, R.J.; Yu, J.Z.; Pepper, A.E.; Abdullaev, A.A.; Kushanov, F.N.; Salakhutdinov, I.B.; Buriev, Z.T.; Saha, S.; Scheffler, B.E.; et al. Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics 2008, 92, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Kuester, A.P.; Nason, J.D. Microsatellite loci for Gossypium davidsonii (Malvaceae) and other D-Genome, Sonoran Desert endemic cotton species. Am. J. Bot. 2012, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhu, G.; Du, L.; Shang, X.; Cheng, C.; Yang, B.; Hu, Y.; Cai, C.; Guo, W. Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci. Rep. 2016, 6, 20582. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xu, Y.; Lu, P.; Wang, X.; Li, Z.; Cai, X.; Zhou, Z.; Wang, Y.; Zhang, Z.; Lin, Z.; et al. Salt stress responsiveness of a wild cotton species (Gossypium klotzschianum) based on transcriptomic analysis. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.G.; Guo, W.Z.; Song, X.L.; Zhang, T.Z. Genetic mapping of EST-derived microsatellites from the diploid Gossypium arboreum in allotetraploid cotton. Mol. Genet. Genom. 2004, 272, 308–327. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Chee, P.W.; Draye, X.; Morrell, P.L.; Smith, C.W.; Paterson, A.H. Multilocus Interactions Restrict Gene Introgression in Interspecific Populations of Polyploid Gossypium (Cotton). Evolution (N. Y.) 2000, 54, 798–814. [Google Scholar] [CrossRef]
- Zhang, Z.S.; Hu, M.C.; Zhang, J.; Liu, D.J.; Zheng, J.; Zhang, K.; Wang, W.; Wan, Q. Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L.). Mol. Breed. 2009, 24, 49–61. [Google Scholar] [CrossRef]
- Jiang, C.; Wright, R.J.; El-Zik, K.M.; Paterson, A.H. Polyploid formation created unique avenues for response to selection in Gossypium (cotton). Proc. Natl. Acad. Sci. USA 1998, 95, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, A.J.; Dong, J.M.; Brubaker, C.L.; Stelly, D.M.; Wendel, J.F.; Paterson, A.H. A detailed RFLP map of cotton, Gossypium hirsutum x Gossypium barbadense: Chromosome organization and evolution in a disomic polyploid genome. Genetics 1994, 138, 829–847. [Google Scholar] [PubMed]
- Desai, A.; Chee, P.W.; Rong, J.; May, O.L.; Paterson, A.H. Chromosome structural changes in diploid and tetraploid A genomes of Gossypium. Genome 2006, 49, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Cota-Sánchez, J.H.; Remarchuk, K.; Ubayasena, K. Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol. Biol. Rep. 2006, 24, 161–167. [Google Scholar] [CrossRef]
- Rong, J.; Abbey, C.; Bowers, J.E.; Brubaker, C.L.; Chang, C.; Chee, P.W.; Delmonte, T.A.; Ding, X.; Garza, J.J.; Marler, B.S.; et al. A 3347-Locus Genetic Recombination Map of Sequence-Tagged Sites Reveals Features of Genome Organization, Transmission and Evolution of Cotton (Gossypium). Genetics 2004, 166, 389–417. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, C.L.; Brown, A.H. The use of multiple alien chromosome addition aneuploids facilitates genetic linkage mapping of the Gossypium G genome. Genome 2003, 46, 774–791. [Google Scholar] [CrossRef] [PubMed]
- Collard, B.C.Y.; Mackill, D.J. Marker-assisted selection : An approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B 2008, 363, 557–572. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xie, W.; Li, J.; Zhou, F.; Zhang, Q. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol. J. 2014, 12, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, X.; Yang, Y.; Xu, J.; Gu, J.; Fu, J.; Qian, X.; Zhang, S.; Wu, J.; Liu, K. Development and genetic mapping of microsatellite markers from whole genome shotgun sequences in Brassica oleracea. Mol. Breed. 2011, 28, 585–596. [Google Scholar] [CrossRef]
- Ma, X.X.; Zhou, B.L.; Lü, Y.H.; Guo, W.Z.; Zhang, T.Z. Simple sequence repeat genetic linkage maps of A-genome diploid cotton (Gossypium arboreum). J. Integr. Plant Biol. 2008, 50, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Genome-Wide Analysis of the Cyclin Family in Arabidopsis and Comparative Phylogenetic Analysis of Plant Cyclin-Like Proteins. Plant Physiol. 2004, 135, 1084–1099. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S. Molecular markers from the transcribed/expressed region of the genome in higher plants. Funct. Integr. Genom. 2004, 4, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Luro, F.L.; Costantino, G.; Terol, J.; Argout, X.; Allario, T.; Wincker, P.; Talon, M.; Ollitrault, P.; Morillon, R. Transferability of the EST-SSRs developed on Nules clementine (Citrus clementina Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genom. 2008, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.G.; Ishii, T.; Temnykh, S.; Chen, X.; Lipovich, L.; McCouch, S.R.; Park, W.D.; Ayres, N.; Cartinhour, S. Diversity of microsatellites derived from genomic libraries and GenBank sequences in rice (Oryza sativa L.). Theor. Appl. Genet. 2000, 100, 713–722. [Google Scholar] [CrossRef]
- Eujayl, I.; Sorrells, M.; Baum, M.; Wolters, P.; Powell, W. Assessment of genotypic variation among cultivated durum wheat based on EST-SSRS and genomic SSRS. In Euphytica; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2001; Volume 119, pp. 39–43. [Google Scholar]
- Fraser, L.G.; Harvey, C.F.; Crowhurst, R.N.; De Silva, H.N. EST-derived microsatellites from Actinidia species and their potential for mapping. Theor. Appl. Genet. 2004, 108, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.C.; Mian, M.A.R.; Eujayl, I.; Zwonitzer, J.C.; Wang, L.; May, G.D. Tall fescue EST-SSR markers with transferability across several grass species. Theor. Appl. Genet. 2004, 109, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Giband, M.; Brottier, P.; Risterucci, A.M.; Lacape, J.M. Wide coverage of the tetraploid cotton genome using newly developed microsatellite markers. Theor. Appl. Genet. 2004, 109, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, J.N.; Brooks, T.; Bolek, Y.; El-zik, K.M. New Dinucleotide and Trinucleotide Microsatellite Marker Resources for Cotton Genome Research. J. Cotton Sci. 2001, 5, 103–113. [Google Scholar]
- Bowers, J.E.; Abbey, C.; Anderson, S.; Chang, C.; Draye, X.; Hoppe, A.H.; Jessup, R.; Lemke, C.; Lennington, J.; Li, Z.; et al. A high-density genetic recombination map of sequence-tagged sites for Sorghum, as a framework for comparative structural and evolutionary genomics of tropical grains and grasses. Genetics 2003, 165, 367–386. [Google Scholar] [PubMed]
- Li, M.; Zhu, L.; Zhou, C.Y.; Lin, L.; Fan, Y.J.; Zhuang, Z.M. Development and characterization of EST-SSR markers from Scapharca broughtonii and their transferability in Scapharca subcrenata and Tegillarca granosa. Molecules 2012, 17, 10716–10723. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.P.; Creel, S.; Kalinowski, S.T. Estimating relatedness and relationships using microsatellite loci with null alleles. Heredity 2006, 97, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Dakin, E.E.; Avise, J.C. Microsatellite null alleles in parentage analysis. Heredity 2004, 93, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Rustgi, S.; Sharma, S.; Singh, R.; Kumar, N.; Balyan, H.S. Transferable EST-SSR markers for the study of polymorphism and genetic diversity in bread wheat. Mol. Genet. Genom. 2003, 270, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Prasad, M.; Varshney, R.K.; Balyan, H.S.; Gupta, P.K.; Roy, J.K. The use of microsatellites for detecting DNA polymorphism, genotype identification and genetic diversity in wheat. TAG Theor. Appl. Genet. 2000, 100, 584–592. [Google Scholar] [CrossRef]
- Callen, D.F.; Thompson, A.D.; Shen, Y.; Phillips, H.A.; Richards, R.I.; Mulley, J.C.; Sutherland, G.R. Incidence and origin of “null” alleles in the (AC)n microsatellite markers. Am. J. Hum. Genet. 1993, 52, 922–927. [Google Scholar] [PubMed]
- La Rosa, R.; Angiolillo, A.; Guerrero, C.; Pellegrini, M.; Rallo, L.; Besnard, G.; Bervillé, A.; Martin, A.; Baldoni, L. A first linkage map of olive (Olea europaea L.) cultivars using RAPD, AFLP, RFLP and SSR markers. Theor. Appl. Genet. 2003, 106, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Lander, E.S.; Hewitt, J.D.; Peterson, S.; Lincoln, S.E.; Tanksley, S.D. Resolution of quantitative traits into Mendelian factors by using a complete linkage map of restriction fragment length polymorphisms. Nature 1988, 335, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, W.; Zhu, X.; Yuan, Y.; Yu, J.Z.; Kohel, R.J.; Zhang, T. Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breed. 2005, 15, 169–181. [Google Scholar] [CrossRef]
- Sibov, S.T.; Souza, C.L., Jr.; Garcia, A.A.F.; Silva, A.R.; Garcia, A.A.F.; Mangolin, C.A.; Benchimol, L.L.; Souza, A.P. Molecular mapping in tropical maize (Zea mays L.) using microsatellite markers. 1. Map construction and localization of loci showing distorted segregation. Hereditas 2003, 139, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.S. Patterns of chromosomal duplication in maize and their implications for comparative maps of the grasses. Genome Res. 2001, 11, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Helentjaris, T.; Weber, D.; Wright, S. Identification of the genomic locations of duplicate nucleotide sequences in maize by analysis of restriction fragment length polymorphisms. Genetics 1988, 118, 353–363. [Google Scholar] [PubMed]
- Brubaker, C.L.; Paterson, A.H.; Wendel, J.F. Comparative genetic mapping of allotetraploid cotton and its diploid progenitors. Genome 1999, 42, 184–203. [Google Scholar] [CrossRef]
- Juturu, V.N.; Mekala, G.K.; Kirti, P.B. Current status of tissue culture and genetic transformation research in cotton (Gossypium spp.). Plant Cell. Tissue Organ Cult. 2015, 120, 813–839. [Google Scholar] [CrossRef]
- Zhao, D.; Derkx, A.P.; Liu, D.C.; Buchner, P.; Hawkesford, M.J. Overexpression of a NAC transcription factor delays leaf senescence and increases grain nitrogen concentration in wheat. Plant Biol. 2015, 17, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lin, R.; Feng, J.; Chen, W.; Qiu, D.; Xu, S. TaNAC1 acts as a negative regulator of stripe rust resistance in wheat, enhances susceptibility to Pseudomonas syringae, and promotes lateral root development in transgenic Arabidopsis thaliana. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gongbuzhaxi; Wang, C.; Xue, F.; Zhang, H.; Ji, W. Wheat NAC transcription factor TaNAC29 is involved in response to salt stress. Plant Physiol. Biochem. 2015, 96, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Wang, Y.; Li, B.; Chang, J.; Chen, M.; Li, K.; Yang, G.; He, G. TaNAC29, a NAC transcription factor from wheat, enhances salt and drought tolerance in transgenic Arabidopsis. BMC Plant Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signaling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Furuya, T.; Matsuoka, D.; Nanmori, T. Membrane rigidification functions upstream of the MEKK1-MKK2-MPK4 cascade during cold acclimation in Arabidopsis thaliana. FEBS Lett. 2014, 588, 2025–2030. [Google Scholar] [CrossRef] [PubMed]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. Map kinase signalling cascade in Arabidopsis innate immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Stanko, V.; Giuliani, C.; Retzer, K.; Djamei, A.; Wahl, V.; Wurzinger, B.; Wilson, C.; Heberle-Bors, E.; Teige, M.; Kragler, F. Timing is everything: Highly specific and transient expression of a map kinase determines auxin-induced leaf venation patterns in arabidopsis. Mol. Plant 2014, 7, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Rudrabhatla, P.; Reddy, M.M.; Rajasekharan, R. Genome-wide analysis and experimentation of plant serine/ threonine/tyrosine-specific protein kinases. Plant Mol. Biol. 2006, 60, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ren, F.; Zhong, H.; Jiang, W.; Li, X. Identification and expression analysis of genes in response to high-salinity and drought stresses in Brassica napus. 2010, 42, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Menard, G.; Martin Moreno, J.; Bryant, F.; Munoz-Azcarate, O.; Kelly, A.A.; Hassani-Pak, K.; Kurup, S.; Eastmond, P.J. Genome wide analysis of fatty acid desaturation and its response to temperature. Plant Physiol. 2017, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant Receptor-Like Serine Threonine Kinases: Roles in Signaling and Plant Defense. Mol. Plant-Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Marivet, J.; Frendo, P.; Burkard, G. Effects of abiotic stresses on cyclophilin gene expression in maize and bean and sequence analysis of bean cyclophilin cDNA. Plant Sci. 1992, 84, 171–178. [Google Scholar] [CrossRef]
- Godoy, A.V.; Lazzaro, A.S.; Casalongué, C.A.; San Segundo, B. Expression of a Solanum tuberosum cyclophilin gene is regulated by fungal infection and abiotic stress conditions. Plant Sci. 2000, 152, 123–134. [Google Scholar] [CrossRef]
- Cheng, C.; Xu, X.; Gao, M.; Li, J.; Guo, C.; Song, J.; Wang, X. Genome-wide analysis of respiratory burst oxidase homologs in grape (Vitis vinifera L.). Int. J. Mol. Sci. 2013, 14, 24169–24186. [Google Scholar] [CrossRef] [PubMed]
- Krizman, M.; Jakse, J.; Baricevic, D.; Javornik, B.; Prosekm, M. Robust CTAB-activated charcoal protocol for plant DNA extraction. Acta Agric. Slov. 2006, 87, 427–433. [Google Scholar]
- Wilfinger, W.W.; Mackey, K.; Chomczynski, P. 260/280 and 260/230 Ratios NanoDrop® ND-1000 and ND-8000 8-Sample Spectrophotometers. BioTechniques 1997, 22, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ThermoFisher Scientific Qubit dsDNA HS Assay Kit. Bioinformatics 2016, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Wang, Y.; Cai, S.; Wang, X.; Li, Y.; Abduweli, A.; Hua, J. Partial Dominance, Overdominance, Epistasis and QTL by Environment Interactions Contribute to Heterosis in Two Upland Cotton Hybrids. Genes Genomes Genet. 2016, 6, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sun, J.; Jia, Y.; Ma, Z.; Du, X. Genetic Diversity of Colored Cotton Analyzed by Simple Sequence Repeat Markers. Int. J. Plant Sci. 2009, 170, 76–82. [Google Scholar] [CrossRef]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, P.; Venables, W.N. An Introduction to R: Software for Statistical Modelling & Computing; Commonwealth Scientific and Industrial Research Organisation (CSIRO): Canberra, Australia, 2005; pp. 1–364. [Google Scholar]
- You, M.; Boersma, J.G.; Buirchell, B.J.; Sweetingham, M.W.; Siddique, K.H.; Yang, H. A PCR-based molecular marker applicable for marker-assisted selection for anthracnose disease resistance in lupin breeding. Cell. Mol. Biol. Lett. 2005, 10, 123–134. [Google Scholar] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | No. of Markers | No. of Polymorphic Primers | Dominant Markers | Non-Dominant Markers | No. of Linked Markers | Polymorphic Rate |
|---|---|---|---|---|---|---|
| SWU | 12,650 | 1000 | 200 (27.17%) | 528 (72.83%) | 728 | 8.00% |
| Chr. | Markers per Chromosome | SD | Average Distance (cM) | Average % SD | Map Size (cM) | Marker Distance Gaps (cM) per Chromosome | Ratio | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Smallest Gap (cM) | Largest Gap in (cM) | <5 cM | 5–10 cM | 10–20 cM | >20 cM | |||||||
| Chr01 | 89 | 3 | 1.304 | 3.371 | 116.045 | 0.005 | 8.756 | 83 | 5 | 0 | 0 | 0.9326 |
| Chr02 | 44 | 35 | 1.884 | 76.087 | 82.908 | 0.044 | 18.328 | 39 | 3 | 1 | 0 | 0.8864 |
| Chr03 | 45 | 8 | 2.59 | 17.778 | 116.528 | 0.14 | 10.009 | 39 | 1 | 1 | 0 | 0.8667 |
| Chr04 | 56 | 2 | 1.997 | 3.571 | 111.846 | 0.008 | 17.538 | 53 | 1 | 1 | 0 | 0.9464 |
| Chr05 | 49 | 17 | 2.361 | 34.694 | 115.671 | 0.056 | 8.493 | 45 | 5 | 0 | 0 | 0.9184 |
| Chr06 | 58 | 5 | 2.001 | 8.621 | 116.045 | 0.002 | 14.926 | 52 | 3 | 2 | 0 | 0.8966 |
| Chr07 | 86 | 35 | 1.446 | 40.698 | 124.358 | 0.016 | 7.988 | 81 | 4 | 0 | 0 | 0.9419 |
| Chr08 | 49 | 25 | 2.492 | 51.02 | 122.13 | 0.054 | 14.099 | 45 | 0 | 3 | 0 | 0.9184 |
| Chr09 | 69 | 12 | 1.697 | 16.216 | 117.06 | 0.046 | 11.167 | 63 | 3 | 2 | 0 | 0.9130 |
| chr10 | 34 | 2 | 2.998 | 5.882 | 101.93 | 0.117 | 12.472 | 25 | 7 | 1 | 0 | 0.7353 |
| Chr11 | 63 | 5 | 1.806 | 7.937 | 113.801 | 0.042 | 36.597 | 60 | 1 | 0 | 1 | 0.9524 |
| Chr12 | 49 | 6 | 2.301 | 12.245 | 112.739 | 0.065 | 9.588 | 43 | 5 | 0 | 0 | 0.8776 |
| Chr13 | 37 | 4 | 3.491 | 10.526 | 129.164 | 0.137 | 25.3 | 28 | 7 | 0 | 1 | 0.7568 |
| Genetic map | 728 | 159 | 2.182 | 22.20 | 1480.23 | 0.0563 | 15.020 | 656 | 45 | 11 | 2 | 0.9011 |
| Chromosome | Chi Square (χ2) Values | Loci Number | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| p < 0.05 | p < 0.01 | p < 0.005 | p < 0.001 | p < 0.0005 | p < 0.0001 | p < 0.00005 | G. davidsonii | G. klotzschianum | Heterozygote’s | Totals | |
| Chr01 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 1 | 3 |
| Chr02 | 1 | 0 | 0 | 0 | 0 | 0 | 34 | 0 | 34 | 1 | 35 |
| Chr03 | 2 | 2 | 1 | 0 | 0 | 0 | 3 | 1 | 0 | 7 | 8 |
| Chr04 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 2 |
| Chr05 | 3 | 1 | 0 | 0 | 0 | 0 | 13 | 0 | 11 | 6 | 17 |
| Chr06 | 0 | 1 | 0 | 0 | 0 | 0 | 4 | 4 | 0 | 1 | 5 |
| Chr07 | 11 | 10 | 5 | 1 | 1 | 0 | 7 | 1 | 5 | 29 | 35 |
| Chr08 | 2 | 0 | 1 | 0 | 0 | 0 | 22 | 0 | 22 | 3 | 25 |
| Chr09 | 5 | 5 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 12 | 12 |
| Chr10 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 1 | 2 |
| Chr11 | 3 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 5 | 5 |
| Chr12 | 3 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 1 | 3 | 6 |
| Chr13 | 2 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 | 3 | 4 |
| Total | 32 | 22 | 8 | 2 | 3 | 0 | 92 | 12 | 74 | 73 | 159 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirungu, J.N.; Deng, Y.; Cai, X.; Magwanga, R.O.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Wang, K.; Liu, F. Simple Sequence Repeat (SSR) Genetic Linkage Map of D Genome Diploid Cotton Derived from an Interspecific Cross between Gossypium davidsonii and Gossypium klotzschianum. Int. J. Mol. Sci. 2018, 19, 204. https://doi.org/10.3390/ijms19010204
Kirungu JN, Deng Y, Cai X, Magwanga RO, Zhou Z, Wang X, Wang Y, Zhang Z, Wang K, Liu F. Simple Sequence Repeat (SSR) Genetic Linkage Map of D Genome Diploid Cotton Derived from an Interspecific Cross between Gossypium davidsonii and Gossypium klotzschianum. International Journal of Molecular Sciences. 2018; 19(1):204. https://doi.org/10.3390/ijms19010204
Chicago/Turabian StyleKirungu, Joy Nyangasi, Yanfeng Deng, Xiaoyan Cai, Richard Odongo Magwanga, Zhongli Zhou, Xingxing Wang, Yuhong Wang, Zhenmei Zhang, Kunbo Wang, and Fang Liu. 2018. "Simple Sequence Repeat (SSR) Genetic Linkage Map of D Genome Diploid Cotton Derived from an Interspecific Cross between Gossypium davidsonii and Gossypium klotzschianum" International Journal of Molecular Sciences 19, no. 1: 204. https://doi.org/10.3390/ijms19010204