Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer

 ,
, 
 , and
, and
Abstract
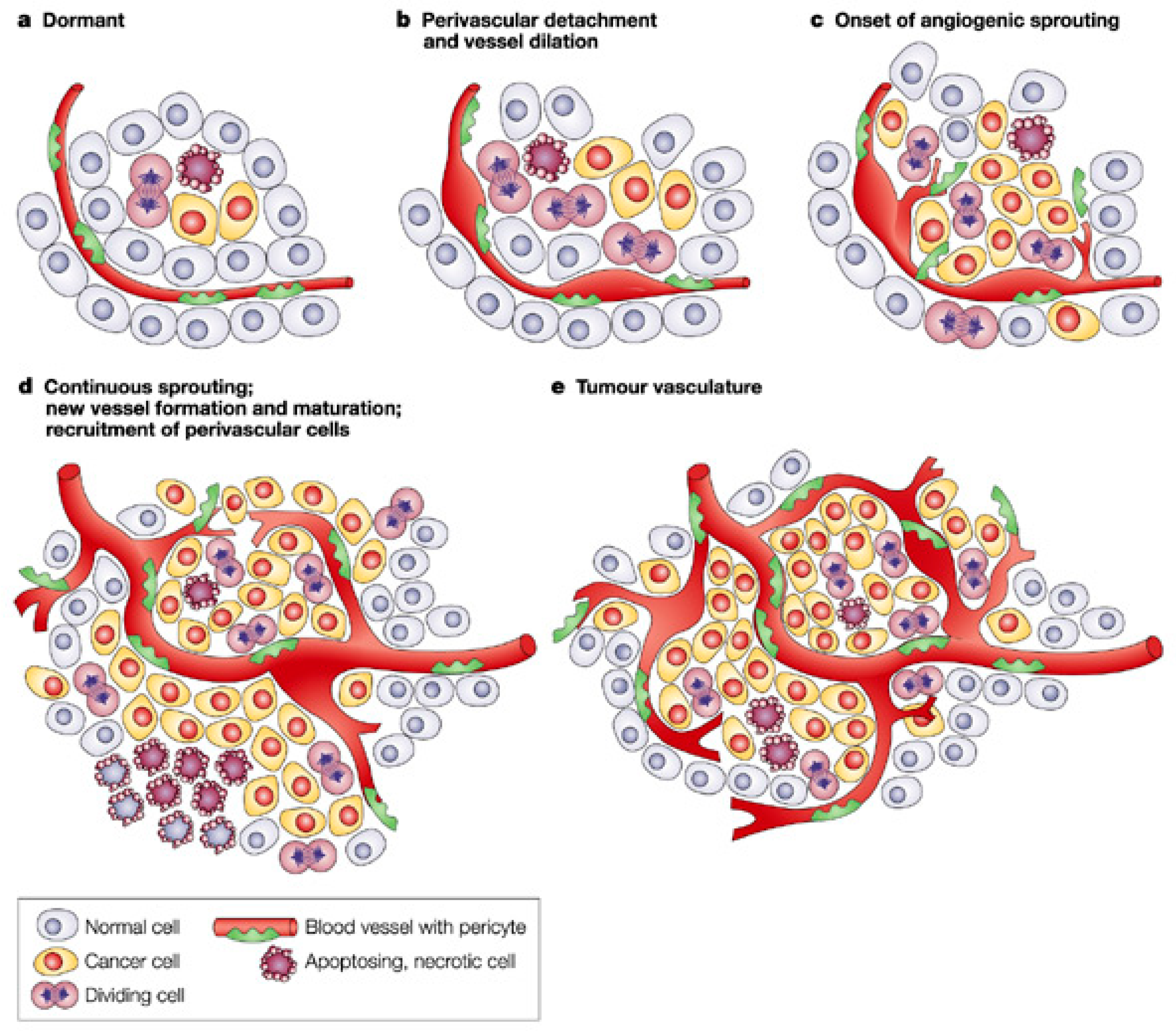
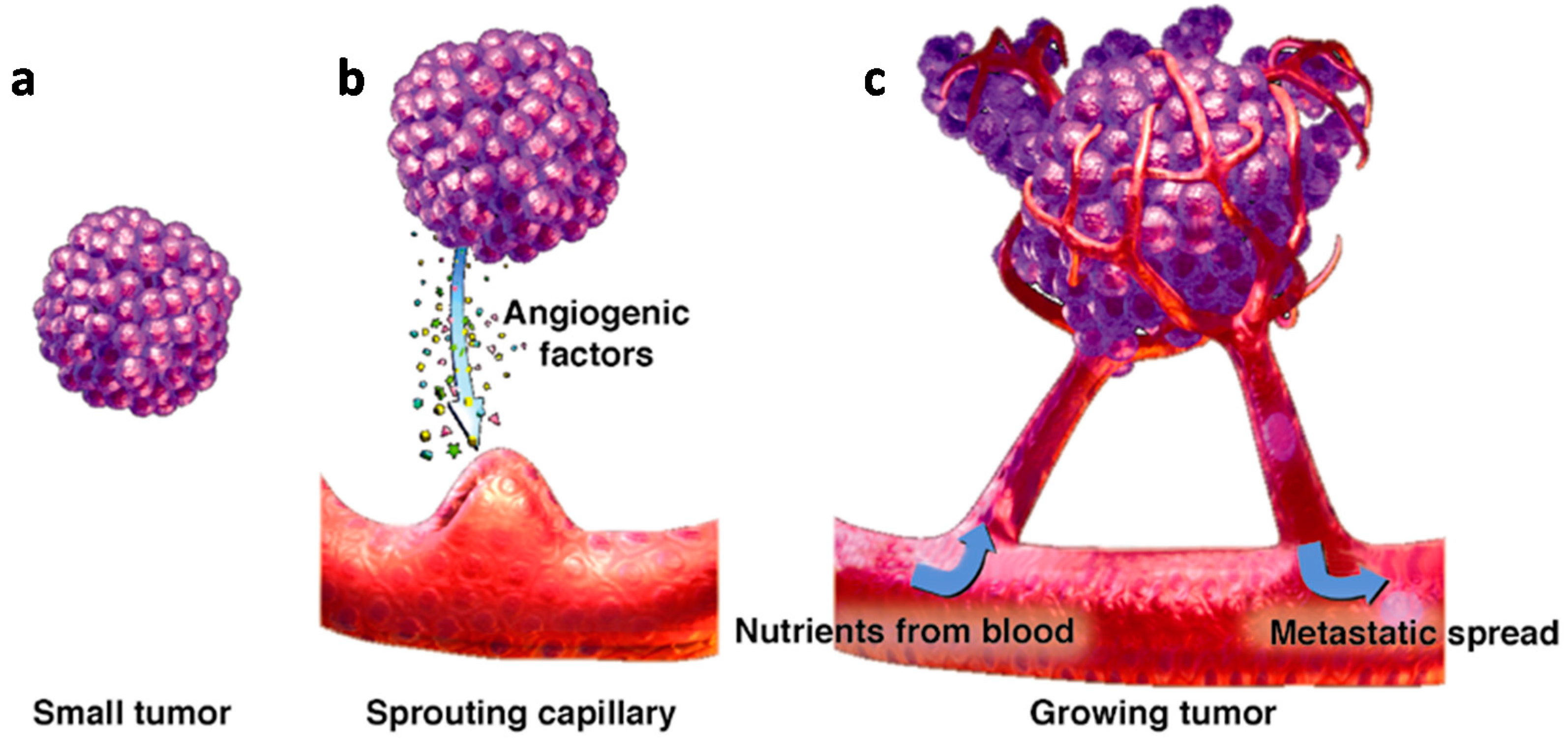
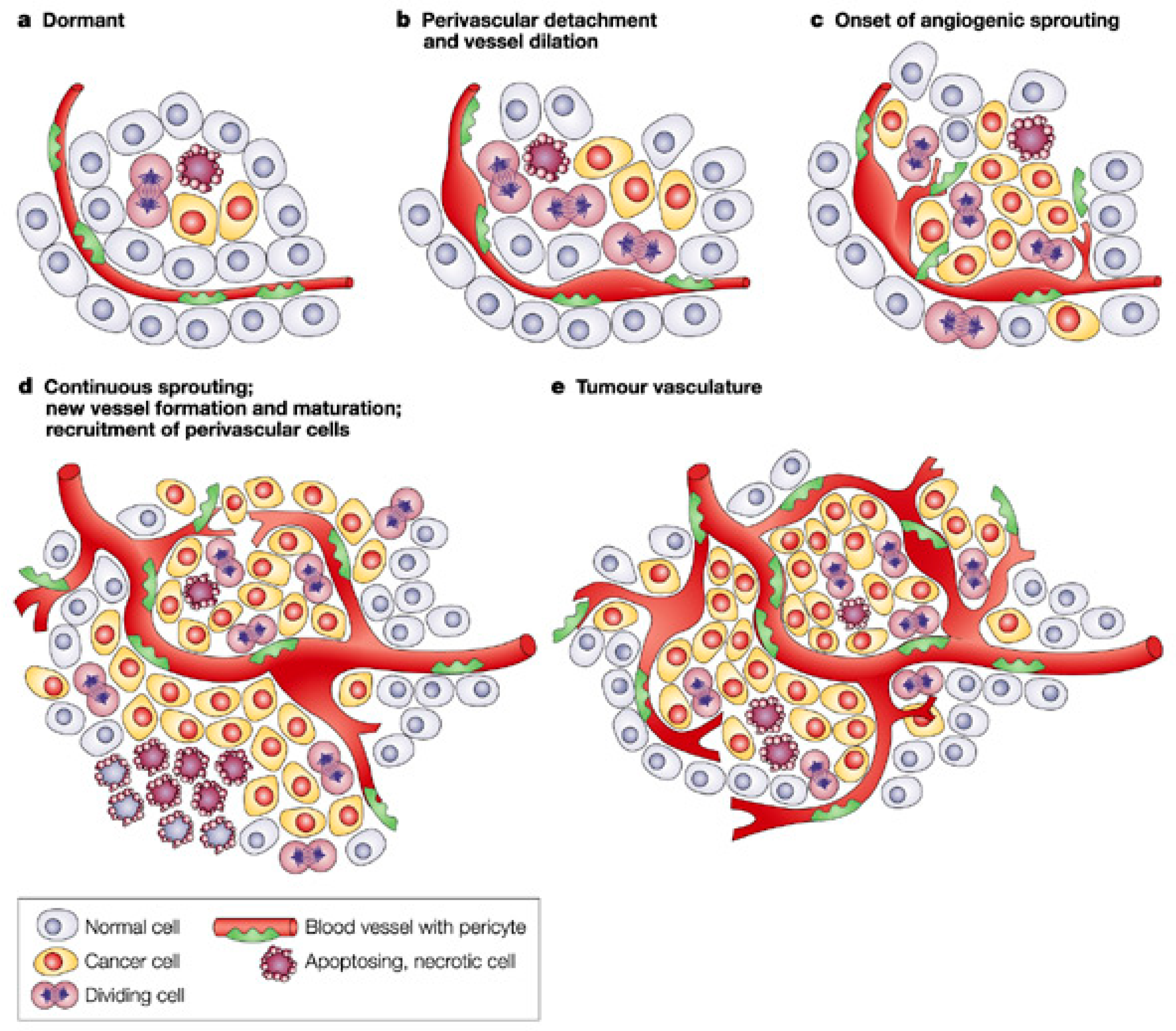
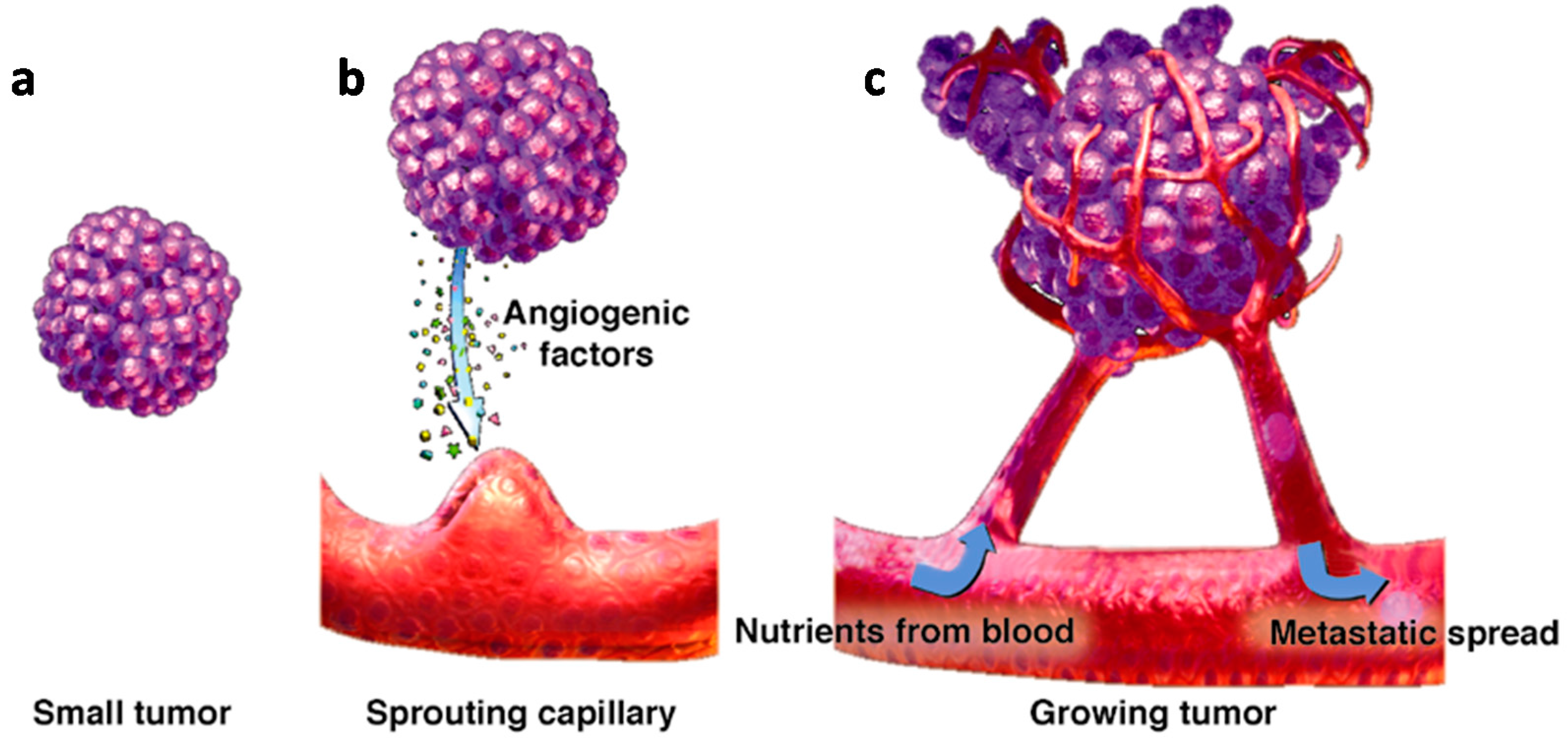
:1. Introduction
2. Main Pro-Angiogenic Factors
2.1. Vascular Endothelial Growth Factor (VEGF)
2.2. Platelet Derived Growth Factor (PDGF)
2.3. Fibroblast Growth Factor (FGF)
2.4. Epidermal Growth Factor (EGF)
2.5. Transforming Growth Factor (TGF)
2.6. Matrix Metalloproteinases (MMPs)
2.7. Tumor Necrosis Factor (TNF)
2.8. Angiopoietins
3. Bevacizumab: Structure Activity Data
4. Bevacizumab in Ovarian Cancer
4.1. Neoadjuvant Setting
4.2. First Line Setting
4.3. Recurrent EOC Setting
5. Conclusions
Conflicts of Interest
References
- Hansen, J.M.; Coleman, R.L.; Sood, A.K. Targeting the tumour microenvironment in ovarian cancer. Eur. J. Cancer 2016, 56, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Yadav, L.; Puri, N.; Rastogi, V.; Satpute, P.; Sharma, V. Tumour Angiogenesis and Angiogenic Inhibitors: A Review. J. Clin. Diagn. Res. 2015, 9, XE01–XE05. [Google Scholar] [CrossRef] [PubMed]
- Hoff, P.M.; Machado, K.K. Role of angiogenesis in the pathogenesis of cancer. Cancer Treat. Rev. 2012, 38, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Döme, B.; Hendrix, M.J.C.; Paku, S.; Tóvári, J.; Tímár, J. Alternative Vascularization Mechanisms in Cancer. Pathology and Therapeutic Implications. Am. J. Pathol. 2007, 170, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gavalas, N.G.; Liontos, M.; Trachana, S.-P.; Bagratuni, T.; Arapinis, C.; Liacos, C.; Dimopoulos, M.A.; Bamias, A. Angiogenesis-Related Pathways in the Pathogenesis of Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 15885–15909. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Kopf, B.; Burgio, S.L.; Bianchi, E.; Amadori, D.; De Giorgi, U. The emerging role of anti-angiogenic therapy in ovarian cancer (review). Int. J. Oncol. 2014, 44, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, J.-B.; Essaghir, A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014, 25, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.P.; Smyth, J.F. Growth factors and ovarian cancer. Endoc. Relat. Cancer 1998, 5, 283–291. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Hiroki, K.; Yamashita, Y. The Role of Epidermal Growth Factor Receptor in Cancer Metastasis and Microenvironment. BioMed Res. Int. 2013, 2013, 546318. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Zeineldin, R.; Silberberg, M.; Stack, M.S. Activated Epidermal Growth Factor Receptor in Ovarian Cancer. Cancer Treat. Res. 2009, 149, 203–226. [Google Scholar] [PubMed]
- Yeung, T.L.; Leung, C.S.; Wong, K.K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [PubMed]
- Le Page, C.; Puiffe, M.L.; Meunier, L.; Zietarska, M.; de Ladurantaye, M.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.M. BMP-2 signaling in ovarian cancer and its association with poor prognosis. J. Ovarian Res. 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Al-Alem, L.; Curry, T.E., Jr. Ovarian Cancer: Involvement of the matrix metalloproteinases. Reproduction 2015, 150, R55–R64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Q. Relationship between matrix metalloproteinases and the occurrence and development of ovarian cancer. Braz. J. Med. Biol. Res. 2017, 50, e6104. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. The Role of TNF in Cancer. Results Probl. Cell Differ. 2009, 49, 1–15. [Google Scholar] [PubMed]
- Kulbe, H.; Thompson, R.; Wilson, J.L.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill, F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 67, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Brunckhorst, M.K.; Xu, Y.; Lu, R.; Yu, Q. Angiopoietins Promote Ovarian Cancer Progression by Establishing a Procancer Microenvironment. Am. J. Pathol. 2014, 184, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Passantino, L.; Patruno, R.; Passantino, G.; Jirillo, F.; Catino, A.; Mattioli, V.; Gadaleta, C.; Ribatti, D. The dog mast cell tumour as a model to study the relationship between angiogenesis, mast cell density and tumour malignancy. Oncol. Rep. 2003, 10, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Patruno, R.; Lionetti, A.; Di Summa, A.; Mattioli, E.; Bufo, P.; Pellecchia, A.; Ribatti, D.; Zizzo, N. Endothelial area and microvascular density in a canine non-Hodgkin’s lymphoma: An interspecies model of tumor angiogenesis. Leuk. Lymph. 2005, 46, 1639–1643. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Vascular endothelial growth factor. Eur. J. Cancer 1996, 32A, 2413–2422. [Google Scholar] [CrossRef]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Gordon, M.; Margolin, K.; Talpaz, M.; Sledge, G.W., Jr.; Holmgren, E.; Benjamin, R.; Stalter, S.; Shak, S.; Adelman, D.J. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. Clin. Oncol. 2001, 19, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Ferrara, N. Pharmacology and pharmacodynamics of bevacizumab as monotherapy or in combination with cytotoxic therapy in preclinical studies. Cancer Res. 2005, 65, 671–680. [Google Scholar] [PubMed]
- Ranieri, G.; Patruno, R.; Ruggieri, E.; Montemurro, S.; Valerio, P.; Ribatti, D. Vascular endothelial growth factor (VEGF) as a target of bevacizumab in cancer: From the biology to the clinic. Curr. Med. Chem. 2006, 13, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowle, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.; Witteveen, P.; Bamias, A.; et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J. Clin. Oncol. 2014, 32, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Piver, M.S.; Lele, S.B.; Marchetti, D.L.; Baker, T.R.; Tsukada, Y.; Emrich, L.J. The impact of aggressive debulking surgery and cisplatin-based chemotherapy on progression-free survival in stage III and IV ovarian carcinoma. J. Clin. Oncol. 1988, 6, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Van DerBurg, M.; Van Lent, M.; Buyse, M.; Kobierska, A.; Colombo, N.; Favalli, G.; Lacave, A.J.; Nardi, M.; Renard, J.; Pecorelli, S. The effect of debulking surgery after induction chemotherapy on the prognosis in advanced epithelial ovarian cancer. Gynecological Cancer Cooperative Group of the European Organization for Research and Treatment of Cancer. N. Engl. J. Med. 1995, 332, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.; Nerenstone, S.; Brady, M.F.; Clarke-Pearson, D.; Olt, G.; Rubin, S.C.; Moore, D.H.; Small, J.M.; Gynecologic Oncology Group. Secondary surgical cytoreduction for advanced ovarian carcinoma. N. Engl. J. Med. 2004, 351, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Chéreau, E.; Lambaudie, E.; Houvenaeghel, G. Morbidity of surgery after neoadjuvant chemotherapy including bevacizumab for advanced ovarian cancer. Int. J. Gynecol. Cancer 2013, 23, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- García, Y.; De Juan, A.; Mendiola, C.; Barretina-Ginesta, P.; Vidal, L.; Gil-Martin, M.; Manzano, A.; Rubio, M.J.; Romeo, M.; de Liaño, A.G.; et al. Phase II randomized trial of neoadjuvant (NA) chemotherapy(CT) with or without bevacizumab (Bev) in advanced epithelial ovarian cancer(EOC) (GEICO 1205/NOVA TRIAL) (abstract 5531). J. Clin. Oncol. 2015, 33, 5531. [Google Scholar]
- Han, E.; Monk, B. What is the risk of bowel perforation associated with bevacizumab therapy in ovarian cancer? Gynecol. Oncol. 2007, 105, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Morice, P.; Floquet, A.; Selle, F.; Lambaudie, E.; Fauvet, R.; Colombo, P.E.; Kalbacher, E.; Follana, P.; Martin, S.; et al. A randomized, open-label, phase II study assessing the efficacy and the safety of bevacizumab in neoadjuvant therapy in patients with FIGO stage IIIc/IV ovarian, tubal, or peritoneal adenocarcinoma, initially unresectable. J. Clin. Oncol. 2014, 32, TPS5614. [Google Scholar]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [PubMed]
- Bolis, G.; Scarfone, G.; Giardina, G.; Villa, A.; Mangili, G.; Melpignano, M.; Presti, M.; Tateo, S.; Franchi, M.; Parazzini, F.; et al. Carboplatin alone vs carboplatin plus epidoxorubicinas second-line therapy for cisplatin-orcarboplatin-sensitive ovarian cancer. Gynecol. Oncol. 2001, 81, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Ledermann, J.; Colombo, N.; du Bois, A.; Delaloye, J.F.; Kristensen, G.B.; Wheeler, S.; Swart, A.M.; Qian, W.; Torri, V.; et al. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: The ICON4/AGO-OVAR-2.2 trial. Lancet 2003, 361, 2099–2106. [Google Scholar] [PubMed]
- Pfisterer, J.; Plante, M.; Vergote, I.; du Bois, A.; Hirte, H.; Lacave, A.J.; Wagner, U.; Stähle, A.; Stuart, G.; Kimmig, R.; et al. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: An intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J. Clin. Oncol. 2006, 24, 4699–4707. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Nycum, L.R.; Goff, B.; Nguyen, H.; Husain, A.; Blank, S.V. Updated overall survival analysis in OCEANS, a randomized phase 3 trial of gemcitabine (G), carboplatin (C) and bevacizumab (BV) or placebo(PL) followed by BV or PL in platinum sensitive recurrent epithelial ovarian (ROC), primary peritoneal (PPC) or fallopian tube cancer. Ann. Oncol. 2012, 23, 9319. [Google Scholar]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.G.; Fujiwara, K.; Tewari, K.S.; O'Malley, D.M.; et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef]
- Ten Bokkel Huinink, W.; Gore, M.; Carmichael, J.; Gordon, A.; Malfetano, J.; Hudson, I.; Broom, C.; Scarabelli, C.; Davidson, N.; Spanczynski, M.; et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J. Clin. Oncol. 1997, 15, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.N.; Tonda, M.; Sun, S.; Rackoff, W.; Doxil Study 30–49 investigators. Long-term survival advantage for women treated with pegylated liposomal doxorubicin compared with topotecan in a phase 3 randomized study of recurrent and refractory ovarian cancer. Gynecol. Oncol. 2004, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stockler, M.R.; Hilpert, F.; Friedlander, M.; King, M.T.; Wenzel, L.; Lee, C.K.; Joly, F.; de Gregorio, N.; Arranz, J.A.; Mirza, M.R.; et al. Patient-reported outcome results from the open-label phase III AURELIA trial evaluating bevacizumab-containing therapy for platinum-resistant ovarian cancer. J. Clin. Oncol. 2014, 32, 1309–1316. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Pro-Angiogenic Factors | Molecular Weight | Receptors |
|---|---|---|
| VEGF | 19 kDa | Tyrosine kinase receptors (VEGFR1, VEGFR2 and VEGFR3) |
| PDFG | 30 kDa | Tyrosine kinase receptors (PDGFRα and β) |
| FGF | 18 kDa | Tyrosine kinase receptors (FGFR1, FGFR2, FGFR3, and FGFR4) |
| EGF | 6.4 kDa | Tyrosine kinase receptors: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4) |
| TGF | 25 kDa | Serine/threonine kinase receptors (type I and type II) |
| MMP’S | 125 kDa | Low-density lipoprotein receptor-related protein (LRP) |
| TNF | 51 kDa | Tyrosine kinase receptors (TNFRI and TNFRII) |
| ANGIOPOIETIN | 57 kDa | Tyrosine kinase receptors (Tie-1 and Tie-2) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loizzi, V.; Del Vecchio, V.; Gargano, G.; De Liso, M.; Kardashi, A.; Naglieri, E.; Resta, L.; Cicinelli, E.; Cormio, G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. Int. J. Mol. Sci. 2017, 18, 1967. https://doi.org/10.3390/ijms18091967
Loizzi V, Del Vecchio V, Gargano G, De Liso M, Kardashi A, Naglieri E, Resta L, Cicinelli E, Cormio G. Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. International Journal of Molecular Sciences. 2017; 18(9):1967. https://doi.org/10.3390/ijms18091967
Chicago/Turabian StyleLoizzi, Vera, Vittoria Del Vecchio, Giulio Gargano, Maria De Liso, Anila Kardashi, Emanuele Naglieri, Leonardo Resta, Ettore Cicinelli, and Gennaro Cormio. 2017. "Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer" International Journal of Molecular Sciences 18, no. 9: 1967. https://doi.org/10.3390/ijms18091967
APA StyleLoizzi, V., Del Vecchio, V., Gargano, G., De Liso, M., Kardashi, A., Naglieri, E., Resta, L., Cicinelli, E., & Cormio, G. (2017). Biological Pathways Involved in Tumor Angiogenesis and Bevacizumab Based Anti-Angiogenic Therapy with Special References to Ovarian Cancer. International Journal of Molecular Sciences, 18(9), 1967. https://doi.org/10.3390/ijms18091967