Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process



Abstract
:1. Introduction
2. Cell Reprogramming Is a Process That Involves DNA Damage and Efficient Repair Mechanisms
3. Genetic Variations Identified in Human iPSCs
3.1. Chromosomal Instability
3.2. Copy Number Variations
3.3. Single Nucleotide Variants
4. Sources of Genetic Variations in iPSCs
4.1. Pre-Existing Genetic Variations in Parental Cells
4.2. Reprogramming-Induced Genetic Variations
4.3. Culture-Induced Genetic Variations
5. Phenotypic Consequences of Genetic Variations in iPSCs
6. Towards Production of Genetically Stable iPSCs
6.1. Culture Time Reduction
6.2. Reprogramming Methods
6.3. Reprogramming Factors
6.4. Oxidative Stress Reduction
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| iPSCs | Induced Pluripotent Stem Cells |
| ESCs | Embryonic Stem Cells |
| DDR | DNA Damage Response |
| DSB | Double Strand Break |
| ROS | Reactive Oxygen Species |
| HR | Homologous Recombination |
| LIG4 | DNA Ligase IV |
| NER | Nucleotide Excision Repair |
| NHEJ | Non-Homologous-End-Joining |
| CNVs | Copy Number Variations |
| SNVs | Single Nucleotide Variations |
| CGH | Comparative Genomic Hybridization |
| AML | Acute Myeloid Leukemia |
| MML | Mixed Lineage Leukemia |
| XP | Xeroderma Pigmentosum |
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Simara, P.; Motl, J.A.; Kaufman, D.S. Pluripotent stem cells and gene therapy. Transl. Res. 2013, 161, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Weissbein, U.; Benvenisty, N.; Ben-David, U. Quality control: Genome maintenance in pluripotent stem cells. J. Cell Biol. 2014, 204, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, O.; Knobloch, L.; Fornsaglio, J.; Varum, S.; Easley, C.; Schatten, G. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS ONE 2010, 5, e13410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A.; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folmes, C.D.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-MYC can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Pei, D.Q. Vitamin c improves the quality of somatic cell reprogramming. Nat. Genet. 2012, 44, 366–367. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Bester, A.C.; Kerem, B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012, 28, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Desmarais, J.A.; Hoffmann, M.J.; Bingham, G.; Gagou, M.E.; Meuth, M.; Andrews, P.W. Human embryonic stem cells fail to activate CHK1 and commit to apoptosis in response to DNA replication stress. Stem Cells 2012, 30, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Arad, G.; Weissbein, U.; Mandefro, B.; Maimon, A.; Golan-Lev, T.; Narwani, K.; Clark, A.T.; Andrews, P.W.; Benvenisty, N.; et al. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Lamm, N.; Ben-David, U.; Golan-Lev, T.; Storchova, Z.; Benvenisty, N.; Kerem, B. Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell 2016, 18, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A. DNA breakage and repair. Adv. Genet. 1998, 38, 185–218. [Google Scholar] [PubMed]
- Gonzalez, F.; Georgieva, D.; Vanoli, F.; Shi, Z.D.; Stadtfeld, M.; Ludwig, T.; Jasin, M.; Huangfu, D. Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep. 2013, 3, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, D.K.; Ko, J.J.; Kim, K.P.; Park, K.S. Rad51 regulates reprogramming efficiency through DNA repair pathway. Dev. Reprod. 2016, 20, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, K.; Neganova, I.; Moreno-Gimeno, I.; Al-Aama, J.Y.; Burks, D.; Yung, S.; Singhapol, C.; Saretzki, G.; Evans, J.; Gorbunova, V.; et al. A human ipsc model of ligase iv deficiency reveals an important role for nhej-mediated-dsb repair in the survival and genomic stability of induced pluripotent stem cells and emerging haematopoietic progenitors. Cell Death Differ. 2013, 20, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, K.; Neganova, I.; Singhapol, C.; Saretzki, G.; Al-Aama, J.Y.; Evans, J.; Gorbunova, V.; Gennery, A.; Przyborski, S.; Stojkovic, M.; et al. Brief report: A human induced pluripotent stem cell model of cernunnos deficiency reveals an important role for XLF in the survival of the primitive hematopoietic progenitors. Stem Cells 2013, 31, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Felgentreff, K.; Du, L.; Weinacht, K.G.; Dobbs, K.; Bartish, M.; Giliani, S.; Schlaeger, T.; DeVine, A.; Schambach, A.; Woodbine, L.J.; et al. Differential role of nonhomologous end joining factors in the generation, DNA damage response, and myeloid differentiation of human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 8889–8894. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Nagamatsu, G.; Kosaka, T.; Takubo, K.; Hotta, A.; Ellis, J.; Suda, T. Ataxia-telangiectasia mutated (ATM) deficiency decreases reprogramming efficiency and leads to genomic instability in iPS cells. Biochem. Biophys. Res. Commun 2011, 407, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.L.; D’Andrea, A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Rodriguez-Piza, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castella, M.; Rio, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Sakaguchi, H.; Sakamoto-Abutani, R.; Nakanishi, M.; Nishimura, K.; Yamazaki-Inoue, M.; Ohtaka, M.; Periasamy, V.S.; Alshatwi, A.A.; Higuchi, A.; et al. Distinctive features of single nucleotide alterations in induced pluripotent stem cells with different types of DNA repair deficiency disorders. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.M.; Strati, K.; Li, H.; Murga, M.; Blanco, R.; Ortega, S.; Fernandez-Capetillo, O.; Serrano, M.; Blasco, M.A. A p53-mediated DNA damage response limits reprogramming to ensure ips cell genomic integrity. Nature 2009. [Google Scholar] [CrossRef] [PubMed]
- Simara, P.; Tesarova, L.; Rehakova, D.; Matula, P.; Stejskal, S.; Hampl, A.; Koutna, I. DNA double-strand breaks in human induced pluripotent stem cell reprogramming and long-term in vitro culturing. Stem Cell Res. Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Liu, Y.; Wen, D.; Tseng, Z.; Tahmasian, M.; Zhong, M.; Rafii, S.; Stadtfeld, M.; Hochedlinger, K.; Xiao, A. Histone variant h2a.X deposition pattern serves as a functional epigenetic mark for distinguishing the developmental potentials of ipscs. Cell Stem Cell 2014, 15, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Orlando, L.; Sanchez-Ripoll, Y.; Kumpfmueller, B.; Storm, M.P.; Porcedda, P.; Minieri, V.; Saviozzi, S.; Accomasso, L.; Cibrario Rocchietti, E.; et al. High basal gammah2ax levels sustain self-renewal of mouse embryonic and induced pluripotent stem cells. Stem Cells 2012, 30, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Turinetto, V.; Giachino, C. Multiple facets of histone variant h2ax: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 2015, 43, 2489–2498. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Mayshar, Y.; Ben-David, U.; Lavon, N.; Biancotti, J.C.; Yakir, B.; Clark, A.T.; Plath, K.; Lowry, W.E.; Benvenisty, N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010, 7, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.Z.; Hansen, N.F.; Zhao, L.; Du, Y.T.; Zou, C.L.; Donovan, F.X.; Chou, B.K.; Zhou, G.Y.; Li, S.J.; Dowey, S.N.; et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell 2012, 10, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Hussein, S.M.; Batada, N.N.; Vuoristo, S.; Ching, R.W.; Autio, R.; Narva, E.; Ng, S.; Sourour, M.; Hamalainen, R.; Olsson, C.; et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011, 471, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Laurent, L.C.; Ulitsky, I.; Slavin, I.; Tran, H.; Schork, A.; Morey, R.; Lynch, C.; Harness, J.V.; Lee, S.; Barrero, M.J.; et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human escs and ipscs during reprogramming and time in culture. Cell Stem Cell 2011, 8, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; O’Malley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, M.; Hayashizaki, Y.; Murakawa, Y. Genomic instability of ipscs: Challenges towards their clinical applications. Stem Cell Rev. 2017, 13, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Nisler, B.S.; Taapken, S.M.; Compton, T.; Crandall, L.; Montgomery, K.D.; Lalande, M.; Xu, R.H. Recurrent copy number variations in human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Taapken, S.M.; Nisler, B.S.; Newton, M.A.; Sampsell-Barron, T.L.; Leonhard, K.A.; McIntire, E.M.; Montgomery, K.D. Karotypic abnormalities in human induced pluripotent stem cells and embryonic stem cells. Nat. Biotechnol. 2011, 29, 313–314. [Google Scholar] [CrossRef] [PubMed]
- Spits, C.; Mateizel, I.; Geens, M.; Mertzanidou, A.; Staessen, C.; Vandeskelde, Y.; Van der Elst, J.; Liebaers, I.; Sermon, K. Recurrent chromosomal abnormalities in human embryonic stem cells. Nat. Biotechnol. 2008, 26, 1361–1363. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.M.; Elliott, K.A.H.; Kammesheidt, A. High resolution array-cgh characterization of human stem cells using a stem cell focused microarray. Mol. Biotechnol. 2010, 46, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.F.; Ng, S.H.; Sharma, V.; Neculai, D.; Hussein, S.; Sam, M.; Trinh, Q.; Church, G.M.; Mcpherson, J.D.; Nagy, A.; et al. Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells 2012, 30, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Gore, A.; Li, Z.; Panopoulos, A.D.; Montserrat, N.; Fung, H.L.; Giorgetti, A.; Bilic, J.; Batchelder, E.M.; Zaehres, H.; et al. Analysis of protein-coding mutations in hipscs and their possible role during somatic cell reprogramming. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Summersgill, B.; Lu, Y.J.; Missiaglia, E.; Kitazawa, S.; Oosterhuis, J.W.; Looijenga, L.H.; Shipley, J. Genomic copy number and expression patterns in testicular germ cell tumours. Br. J. Cancer 2007, 97, 1707–1712. [Google Scholar] [CrossRef] [PubMed]
- Reuter, V.E. Origins and molecular biology of testicular germ cell tumors. Mod. Pathol. 2005, 18, S51–S60. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.H.; Mason, M.J.; Xie, W.; Volinia, S.; Singer, M.; Peterson, C.; Ambartsumyan, G.; Aimiuwu, O.; Richter, L.; Zhang, J.; et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell 2009, 5, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Guled, M.; Myllykangas, S.; Frierson, H.F.; Mills, S.E.; Knuutila, S.; Stelow, E.B. Array comparative genomic hybridization analysis of olfactory neuroblastoma. Mod. Pathol. 2008, 21, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Scotto, L.; Narayan, G.; Nandula, S.; Arias-Pulido, H.; Subramaniyam, S.; Schneider, A.; Kaufmann, A.M.; Wright, J.D.; Pothuri, B.; Mansukhani, M.; et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Gene Chromosome Cancer 2008, 47, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.A.F.; Holland, P.W.H. Eleven daughters of nanog. Genomics 2004, 84, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Johannesson, B.; Sagi, I.; Gore, A.; Paull, D.; Yamada, M.; Golan-Lev, T.; Li, Z.; LeDuc, C.; Shen, Y.F.; Stern, S.; et al. Comparable frequencies of coding mutations and loss of imprinting in human pluripotent cells derived by nuclear transfer and defined factors. Cell Stem Cell 2014, 15, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Young, M.A.; Larson, D.E.; Sun, C.W.; George, D.R.; Ding, L.; Miller, C.A.; Lin, L.; Pawlik, K.M.; Chen, K.; Fan, X.; et al. Background mutations in parental cells account for most of the genetic heterogeneity of induced pluripotent stem cells. Cell Stem Cell 2012, 10, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.M.; Connelly, J.P.; Hansen, N.F.; Donovan, F.X.; Winkler, T.; Davis, B.W.; Alkadi, H.; Chandrasekharappa, S.C.; Dunbar, C.E.; Mullikin, J.C.; et al. Ipscs and fibroblast subclones from the same fibroblast population contain comparable levels of sequence variations. Proc. Natl. Acad. Sci. USA 2017, 114, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lv, W.; Ye, X.; Wang, L.; Zhang, M.; Yang, H.; Okuka, M.; Zhou, C.; Zhang, X.; Liu, L.; et al. Zscan4 promotes genomic stability during reprogramming and dramatically improves the quality of ips cells as demonstrated by tetraploid complementation. Cell Res. 2013, 23, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Zaret, K.S. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. 2015, 34, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Pruszak, J.; Varadarajan, M.; Blomen, V.A.; Gokhale, S.; Camargo, F.D.; Wernig, M.; Jaenisch, R.; Brummelkamp, T.R. Generation of ipscs from cultured human malignant cells. Blood 2010, 115, 4039–4042. [Google Scholar] [CrossRef] [PubMed]
- Kumano, K.; Arai, S.; Hosoi, M.; Taoka, K.; Takayama, N.; Otsu, M.; Nagae, G.; Ueda, K.; Nakazaki, K.; Kamikubo, Y.; et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood 2012, 119, 6234–6242. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hoffman, J.P.; Alpaugh, R.K.; Rhim, A.D.; Reichert, M.; Stanger, B.Z.; Furth, E.E.; Sepulveda, A.R.; Yuan, C.X.; Won, K.J.; et al. An ipsc line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013, 3, 2088–2099. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Gentles, A.J.; Chatterjee, S.; Lan, F.; Reinisch, A.; Corces, M.R.; Xavy, S.; Shen, J.; Haag, D.; Chanda, S.; et al. Human aml-ipscs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell 2017, 20, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Kotini, A.G.; Chang, C.J.; Chow, A.; Yuan, H.; Ho, T.C.; Wang, T.; Vora, S.; Solovyov, A.; Husser, C.; Olszewska, M.; et al. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell 2017, 20, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Salci, K.R.; Reid, J.C.; Orlando, L.; Tanasijevic, B.; Shapovalova, Z.; Bhatia, M. Brief report: Human acute myeloid leukemia reprogramming to pluripotency is a rare event and selects for patient hematopoietic cells devoid of leukemic mutations. Stem Cells 2017, 35, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.E.; Harrison, N.J.; Maltby, E.; Smith, K.; Moore, H.D.; Shaw, P.J.; Heath, P.R.; Holden, H.; Andrews, P.W. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat. Biotechnol. 2007, 25, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.S.; Moore, H.D.; Ruban, L.N.; Gokhale, P.J.; Andrews, P.W. Culture and characterization of human embryonic stem cells. Stem Cells Dev. 2004, 13, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, C.; Liu, H.; Sun, Y. Induced pluripotent stem cells are sensitive to DNA damage. Genom. Proteom. Bioinform. 2013, 11, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Panopoulos, A.D.; Ruiz, S.; Izpisua Belmonte, J.C. Ipscs: Induced back to controversy. Cell Stem Cell 2011, 8, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, K.; Nazor, K.L.; Williams, R.; Tran, H.; Dai, H.; Dzakula, Z.; Cho, E.H.; Pang, A.W.C.; Rao, M.; Cao, H.; et al. Whole-genome mutational burden analysis of three pluripotency induction methods. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Li, H.; Jakubosky, D.; Benaglio, P.; Reyna, J.; Olson, K.M.; Huang, H.; Biggs, W.; Sandoval, E.; D’Antonio, M.; et al. Large-scale profiling reveals the influence of genetic variation on gene expression in human induced pluripotent stem cells. Cell Stem Cell 2017, 20, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.F.; Kaplan, A.; Yuan, B.; Hanna, J.H.; Lupski, J.R.; Reiner, O. Passage number is a major contributor to genomic structural variations in mouse ipscs. Stem Cells 2014, 32, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Liu, S.; Figueroa, M.E.; Kulalert, W.; Eminli, S.; Tan, K.Y.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. A more efficient method to generate integration-free human ips cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on sendai virus, an rna virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of mouse induced pluripotent stem cells without viral vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mrna. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Anokye-Danso, F.; Trivedi, C.M.; Juhr, D.; Gupta, M.; Cui, Z.; Tian, Y.; Zhang, Y.; Yang, W.; Gruber, P.J.; Epstein, J.A.; et al. Highly efficient mirna-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 2011, 8, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.P.; Li, Y.Q.; Zhang, X.; Liu, C.; Guan, J.Y.; Li, H.G.; Zhao, T.; Ye, J.Q.; Yang, W.F.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Yu, Q.; Huang, Y.; Song, B.; Chen, Y.; Gao, X.; He, W.; Sun, X.; Fan, Y. Effects of integrating and non-integrating reprogramming methods on copy number variation and genomic stability of human induced pluripotent stem cells. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Kim, D.; Kim, C.H.; Mills, R.E.; Chang, M.Y.; Iskow, R.C.; Ko, S.; Moon, J.I.; Choi, H.W.; Yoo, P.S.M.; et al. Increased genomic integrity of an improved protein-based mouse induced pluripotent stem cell method compared with current viral-induced strategies. Stem Cell Transl Med. 2014, 3, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Pasi, C.E.; Dereli-Oz, A.; Negrini, S.; Friedli, M.; Fragola, G.; Lombardo, A.; Van Houwe, G.; Naldini, L.; Casola, S.; Testa, G.; et al. Genomic instability in induced stem cells. Cell Death Differ. 2011, 18, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Meissner, A.; Cassady, J.P.; Jaenisch, R. C-myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2008, 2, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Jaenisch, R. Molecular control of induced pluripotency. Cell Stem Cell 2014, 14, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Lowe, S.W. Stem cells the promises and perils of p53. Nature 2009, 460, 1085–1086. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Belmonte, J.C.I. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Sarig, R.; Rivlin, N.; Brosh, R.; Bornstein, C.; Kamer, I.; Ezra, O.; Molchadsky, A.; Goldfinger, N.; Brenner, O.; Rotter, V. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J. Exp. Med. 2010, 207, 2127–2140. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ng, S.M.; Chang, C.; Zhang, Z.; Bourdon, J.C.; Lane, D.P.; Peng, J. P53 isoform delta113p53 is a p53 target gene that antagonizes p53 apoptotic activity via bclxl activation in zebrafish. Genes Dev. 2009, 23, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Gong, H.; Pan, X.; Chang, C.; Ou, Z.; Ye, S.; Yin, L.; Yang, L.; Tao, T.; Zhang, Z.; et al. P53 isoform delta113p53/delta133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 2015, 25, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Pan, X.; Chen, H.; Rao, L.; Zeng, Y.; Hang, H.; Peng, J.; Xiao, L.; Chen, J. P53 isoform delta133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S.; Lopez-Contreras, A.J.; Gabut, M.; Marion, R.M.; Gutierrez-Martinez, P.; Bua, S.; Ramirez, O.; Olalde, I.; Rodrigo-Perez, S.; Li, H.; et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Prakash Bangalore, M.; Adhikarla, S.; Mukherjee, O.; Panicker, M.M. Genotoxic effects of culture media on human pluripotent stem cells. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, J.; Zhou, W.Y.; Xing, Y.L.; Sperber, H.; Ferreccio, A.; Agoston, Z.; Kuppusamy, K.T.; Moon, R.T.; Ruohola-Baker, H. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell Stem Cell 2014, 14, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Wang, T.; Qin, B.M.; Yang, J.Y.; Qin, D.J.; Cai, J.L.; Li, W.; Weng, Z.H.; Chen, J.K.; Ni, S.; et al. Vitamin c enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sharma, V.; Qi, S.; Guarch, M.E.; Zhao, P.; Luo, Z.; Fan, W.; Wang, Y.; Mbabaali, F.; Neculai, D.; et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Gorgens, H.; Muller, A.; Kruger, S.; Kuhlisch, E.; Konig, I.R.; Ziegler, A.; Schackert, H.K.; Eckelt, U. Analysis of the base excision repair genes mth1, ogg1 and mutyh in patients with squamous oral carcinomas. Oral Oncol. 2007, 43, 791–795. [Google Scholar] [CrossRef] [PubMed]
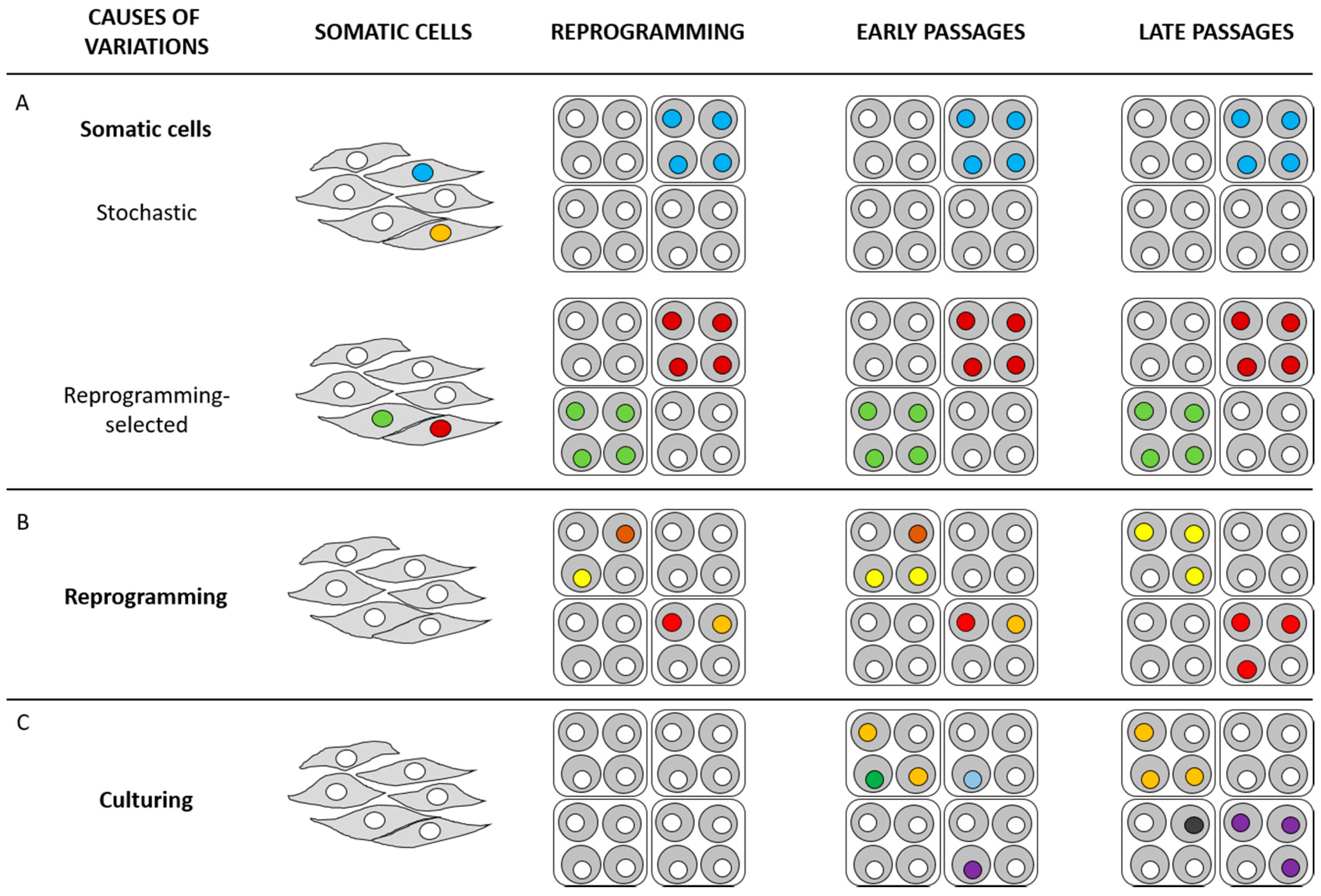

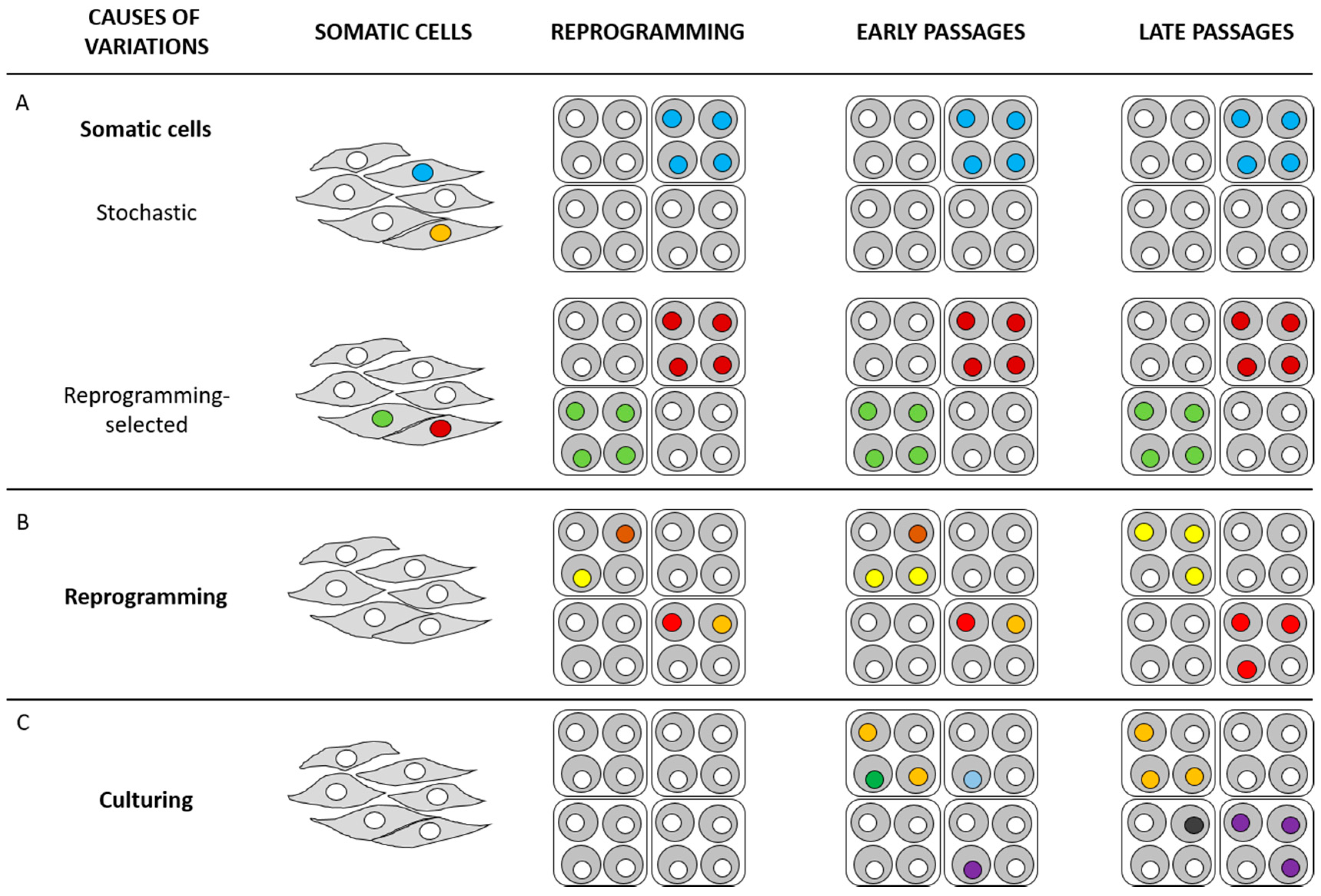{kind=link}
| Genetic Variants | Described Changes | References |
|---|---|---|
| Chromosomal Instability | Trisomy 12 | [34,37,40,41] |
| Trisomy 20q | [41] | |
| Trisomy X | ||
| Trisomy 8 | ||
| Copy Number Variations | Amplification of 20q11.21 | [37,40,42,43] |
| Amplification of 1q31.3 | [32,40] | |
| Deletion of 17q21.1 | ||
| Deletion of 8q24.3 | ||
| Duplication of 2p11.2 | [42] | |
| Single Nucleotide Variations | 22 hiPSC lines derived from 7 fibroblast cell lines: | [33] |
| 124 single nucleotide mutations (missense, nonsense, splice variants) identified. | ||
| 6 mutations per iPSC genome on average. | ||
| 8 mutations described in more than 1 cell line (OR52E8, SLC1A3, MYRIP, HK1, ANKRD12, SCN1A, NEK family genes, NTRK family genes). | ||
| 5 hiPSC lines derived from 1 fibroblast cell line: | [44] | |
| 59 single nucleotide mutations (missense, nonsense, splice variants) identified. | ||
| 12 mutations per iPSC genome on average. | ||
| 10 mutations described in more than 1 cell line (involved genes not specified). | ||
| 8 iPSC lines derived from 4 different somatic cell types (neural stem cells, astrocytes, umbilical vein endothelial cells, foreskin keratinocytes): | [45] | |
| 40 single nucleotide mutations (missense, nonsense, splice variants). | ||
| 5 mutations per iPSC genome on average. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turinetto, V.; Orlando, L.; Giachino, C. Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. Int. J. Mol. Sci. 2017, 18, 1952. https://doi.org/10.3390/ijms18091952
Turinetto V, Orlando L, Giachino C. Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. International Journal of Molecular Sciences. 2017; 18(9):1952. https://doi.org/10.3390/ijms18091952
Chicago/Turabian StyleTurinetto, Valentina, Luca Orlando, and Claudia Giachino. 2017. "Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process" International Journal of Molecular Sciences 18, no. 9: 1952. https://doi.org/10.3390/ijms18091952
APA StyleTurinetto, V., Orlando, L., & Giachino, C. (2017). Induced Pluripotent Stem Cells: Advances in the Quest for Genetic Stability during Reprogramming Process. International Journal of Molecular Sciences, 18(9), 1952. https://doi.org/10.3390/ijms18091952