Inhibitory Effects of Trapping Agents of Sulfur Drug Reactive Intermediates against Major Human Cytochrome P450 Isoforms

 and
and
Abstract
:
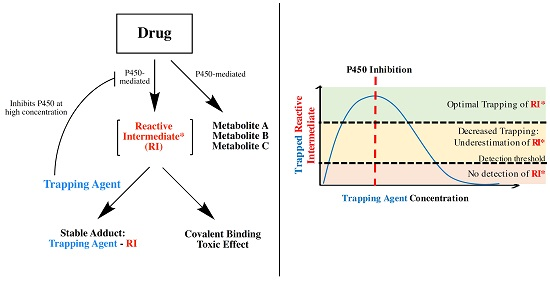
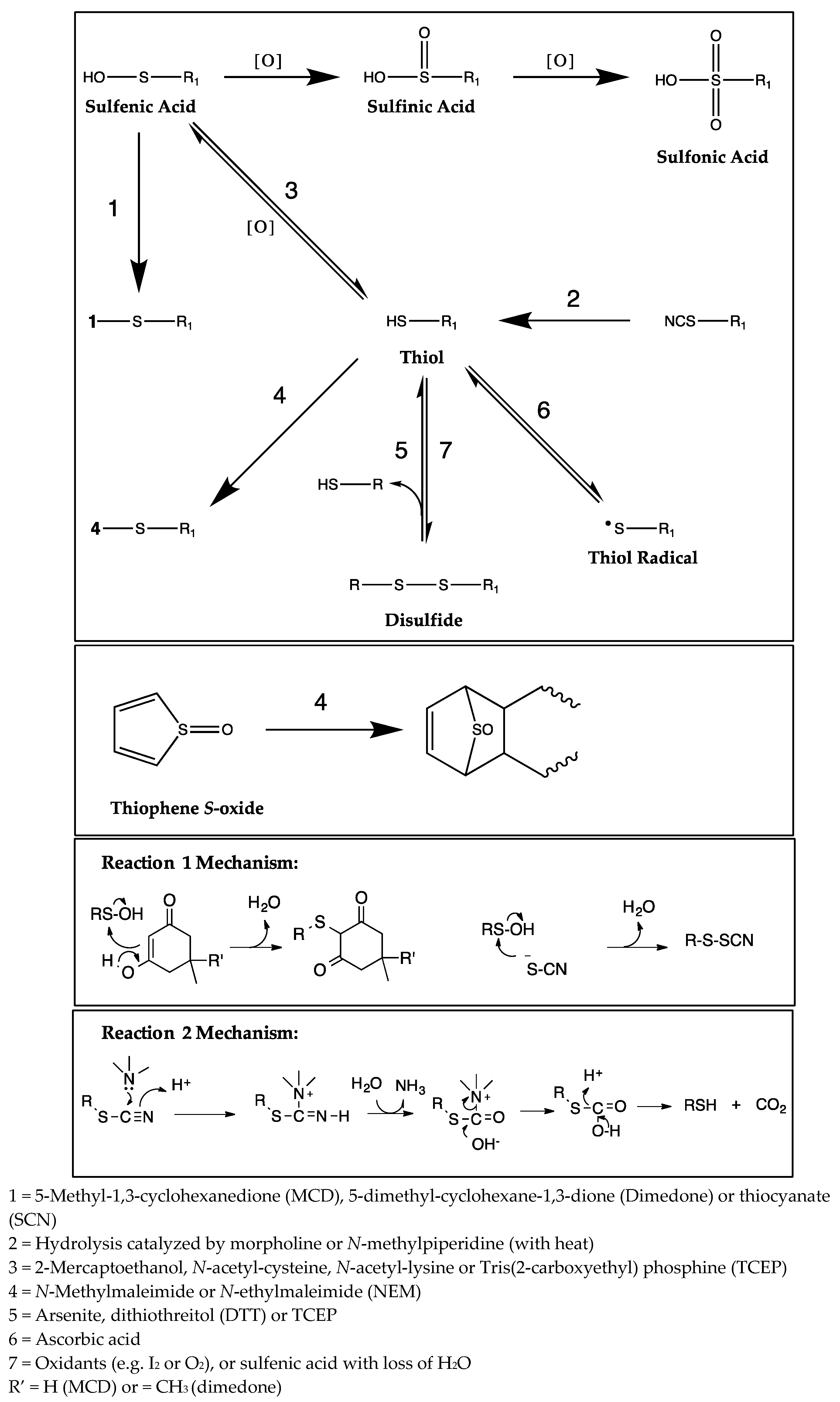
1. Introduction
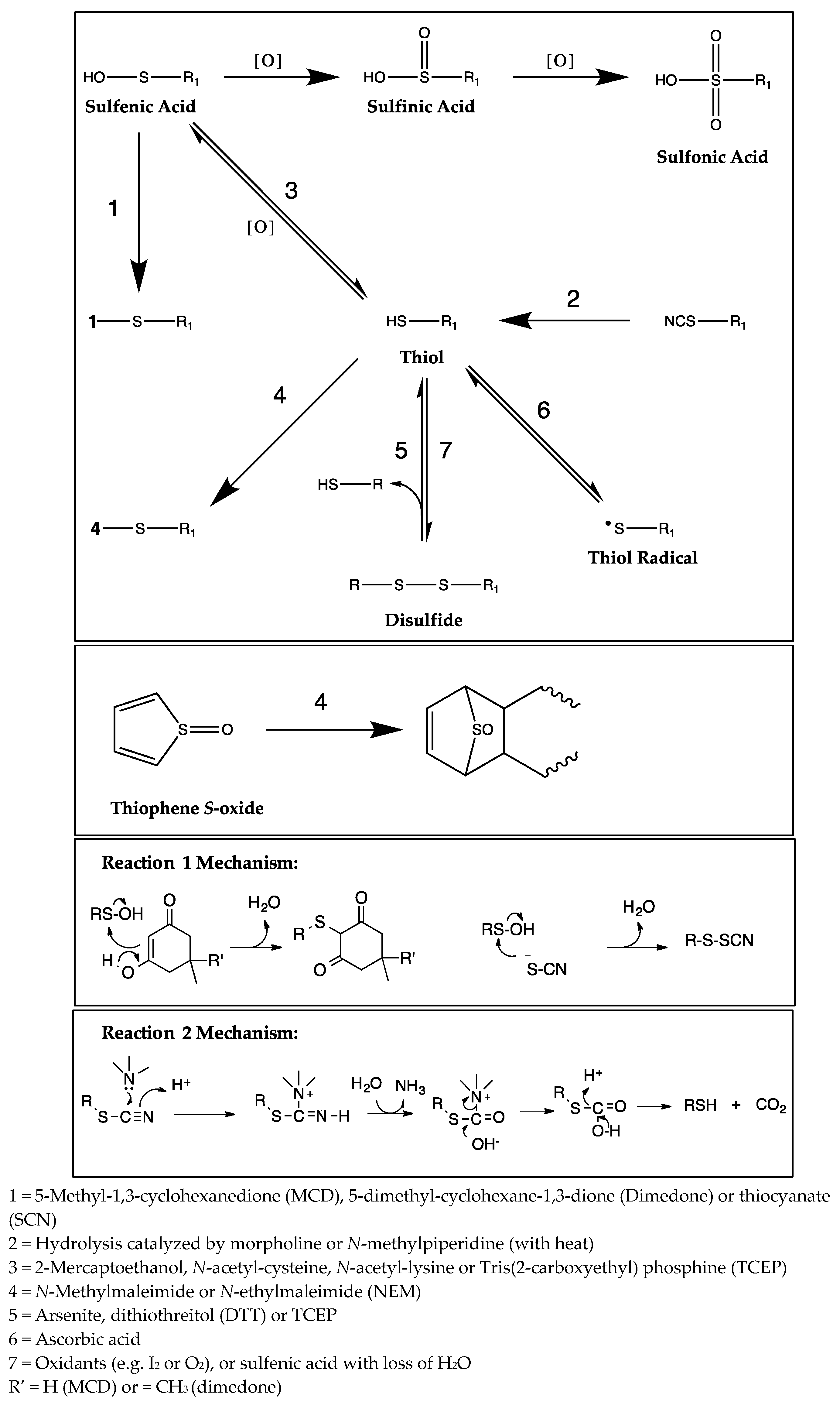
2. Results
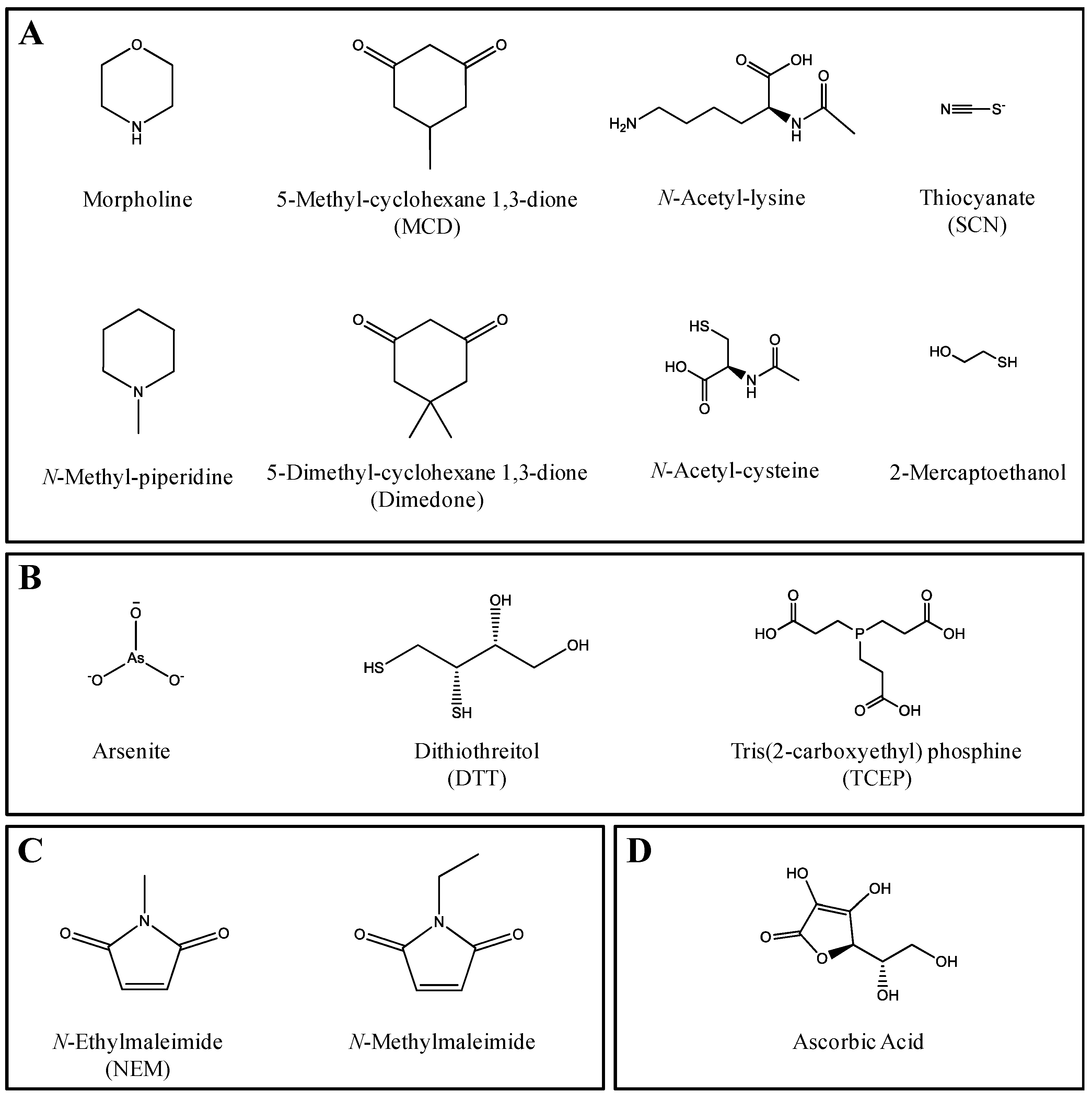
2.1. CYP Inhibition
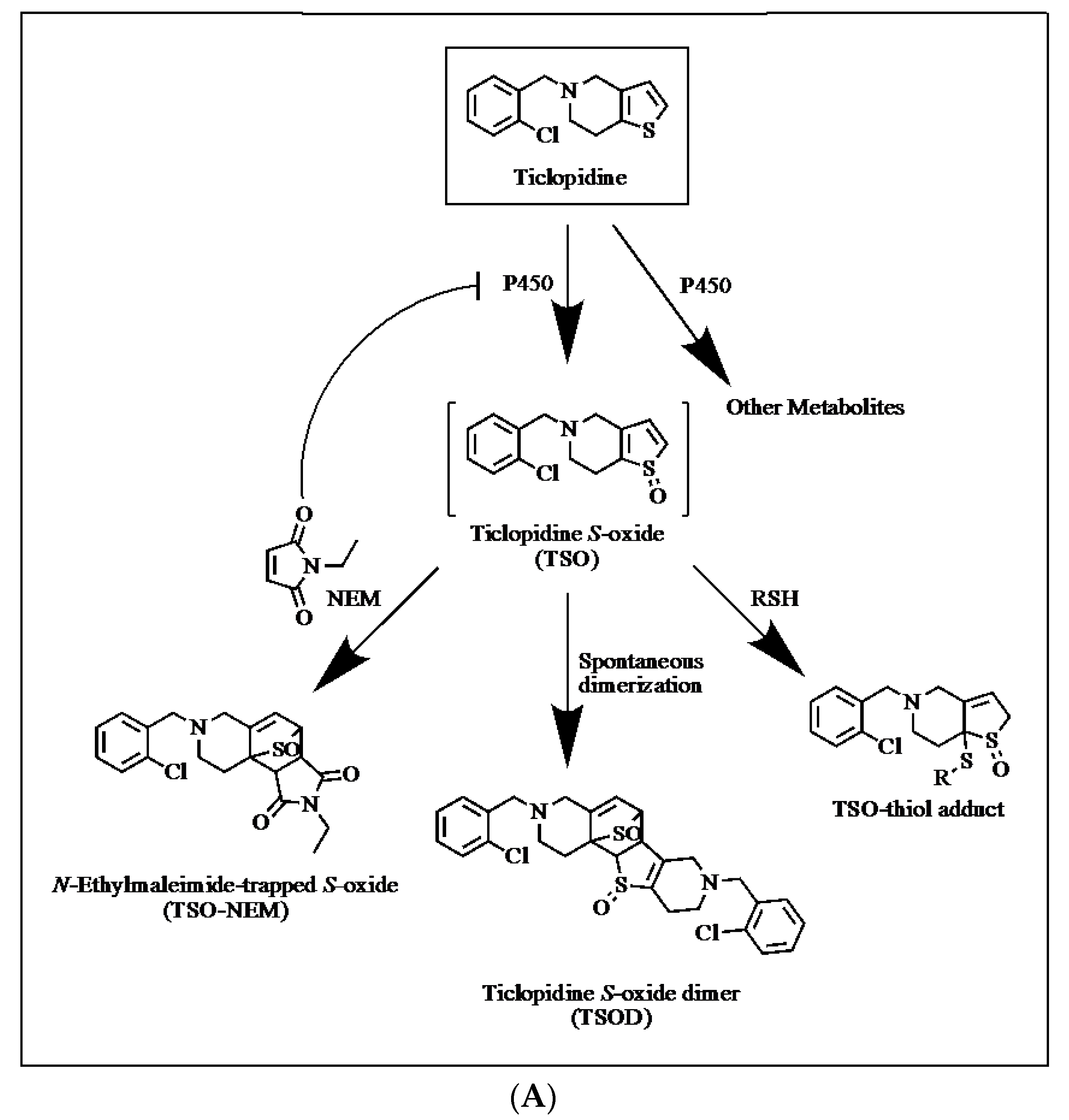
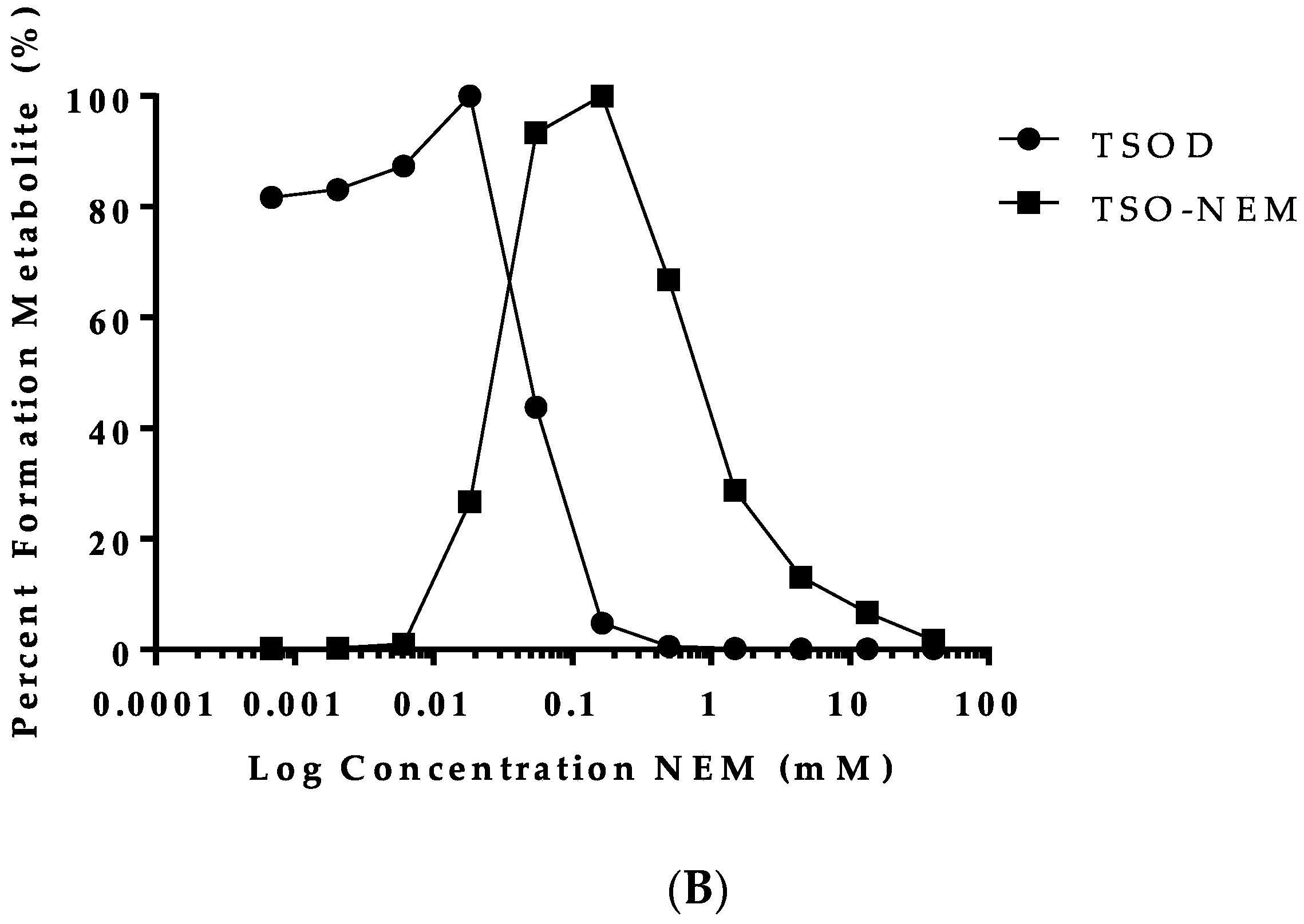
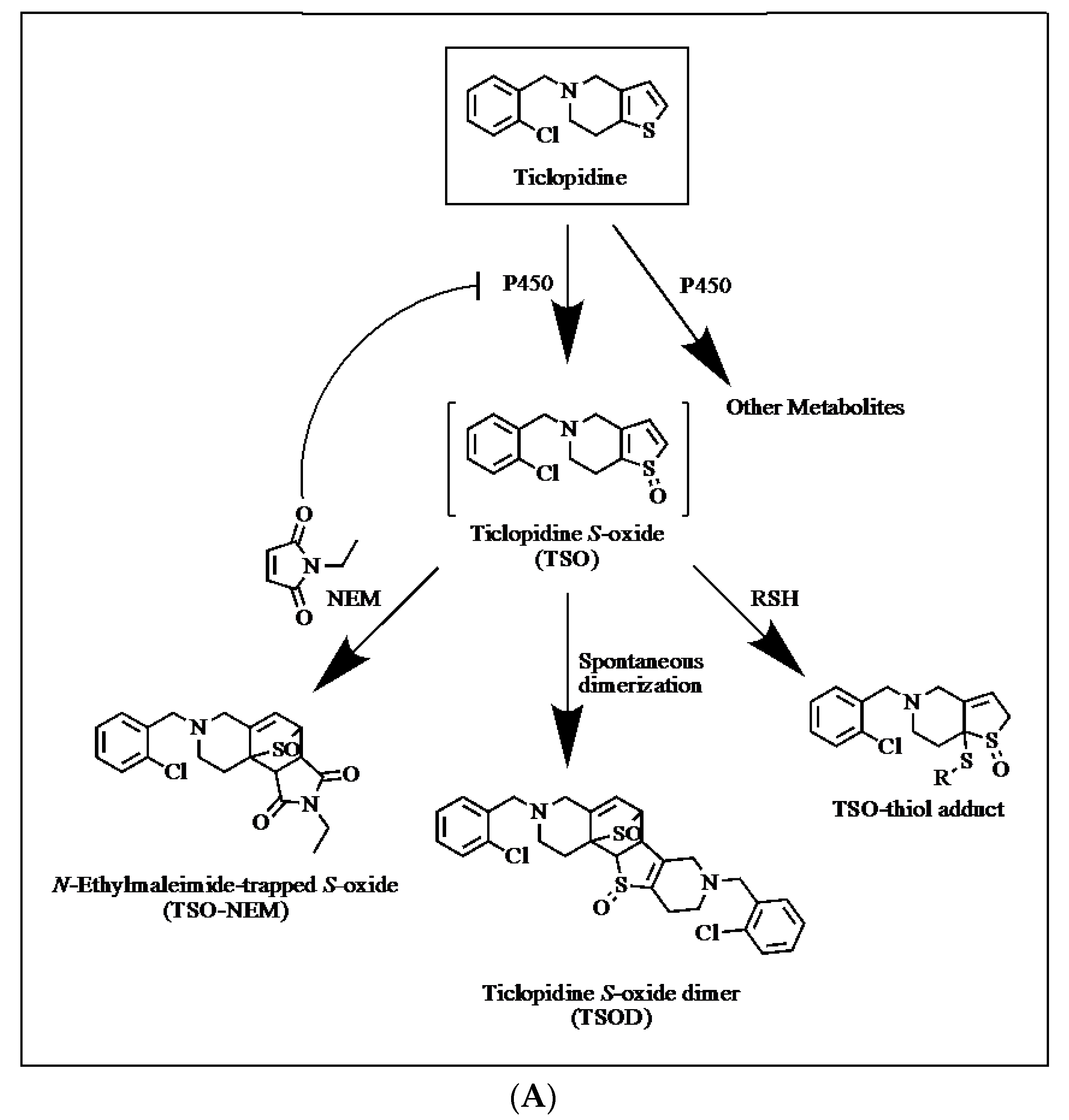
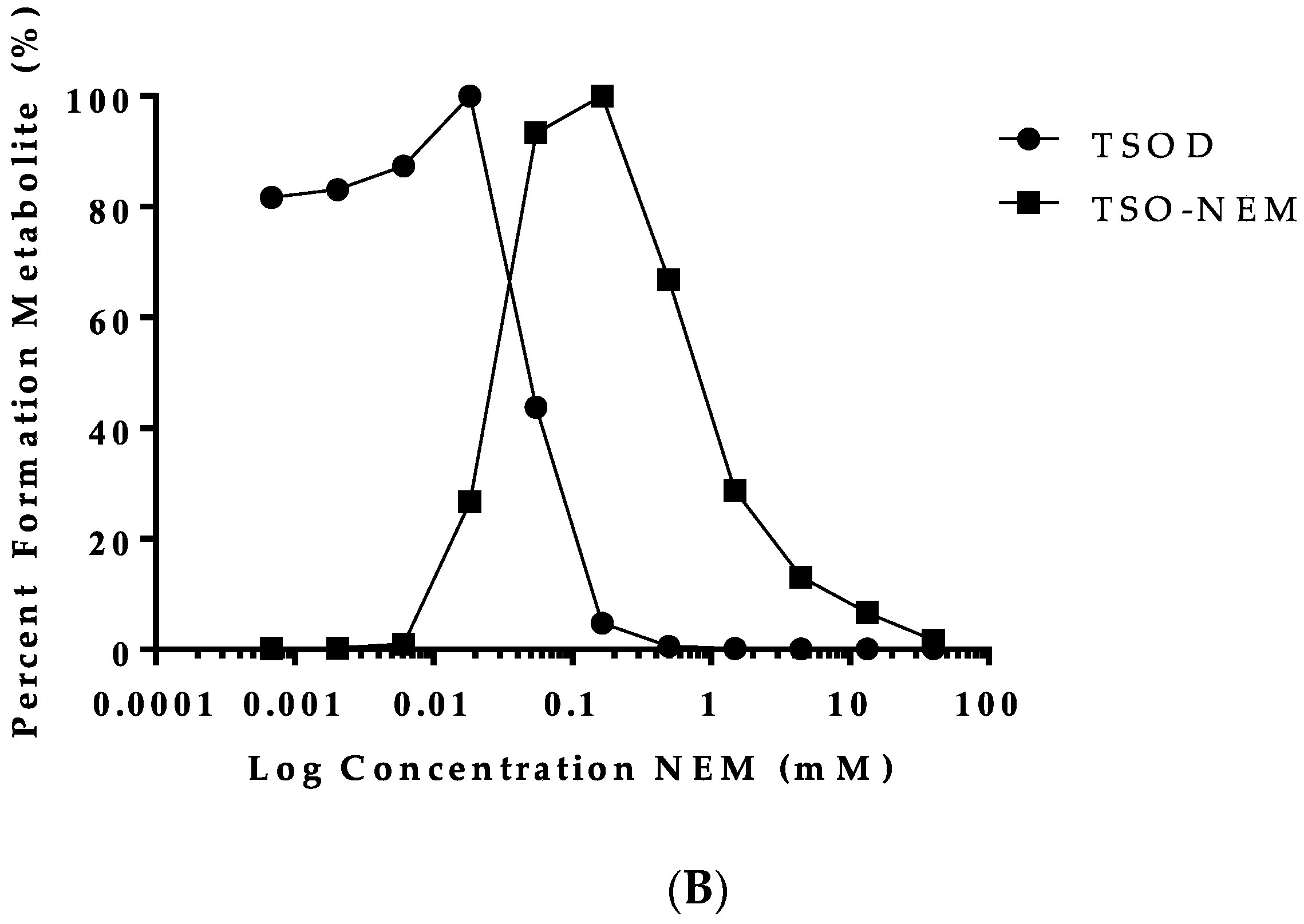
2.2. Trapping of Reactive Metabolites of Ticlopidine
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. CYP Inhibition
4.3. Trapping of Reactive Metabolites of Ticlopidine
4.4. LC-MS/MS Analysis
4.5. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CYP | Cytochrome P450 |
| Dimedone | 5,5-Dimethyl-1,3-cyclohexanedione |
| DTT | Dithiothreitol |
| GSH | Glutathione |
| HLM | Human liver microsomes |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| MCD | Methyl-cyclohexane 1,3-dione |
| NADPH | Reduced β-nicotinamide adenine dinucleotide phosphate |
| NEM | N-Ethylmaleimide |
| SCN | Thiocyanate |
| TCEP | Tris(2-carboxyethyl) phosphine |
| TSO | Ticlopidine S-oxide |
| TSOD | Ticlopidine S-oxide dimer |
| TSO-NEM | N-ethylmaleimide ticlopidine S-oxide adduct |
| TCEP | Tris(2-carboxyethyl) phosphine |
References
- Uetrecht, J. Immune-mediated adverse drug reactions. Chem. Res. Toxicol. 2008, 22, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.; Faulkner, L.; Wood, S.; Park, B.K.; Naisbitt, D.J. Identification of drug- and drug-metabolite immune responses originating from both naïve and memory T cells. J. Allergy Clin. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Didiuk, M.T. Structural alerts, reactive metabolites, and protein covalent binding: How reliable are these attributes as predictors of drug toxicity? Chem. Biodivers. 2009, 6, 2115–2137. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Boobis, A.; Clarke, S.; Goldring, C.E.; Jones, D.; Kenna, J.G.; Lambert, C.; Laverty, H.G.; Naisbitt, D.J.; Nelson, S.; et al. Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 2011, 10, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Soglia, J.R.; Contillo, L.G.; Kalgutkar, A.S.; Zhao, S.; Hop, C.E.; Boyd, J.G.; Cole, M.J. A semiquantitative method for the determination of reactive metabolite conjugate levels in vitro utilizing liquid chromatography-tandem mass spectrometry and novel quaternary ammonium glutathione analogues. Chem. Res. Toxicol. 2006, 19, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Maher, N.; Torres, R.; Huebert, N. Use of a trapping agent for simultaneous capturing and high-throughput screening of both “soft” and “hard” reactive metabolites. Anal. Chem. 2007, 79, 4206–4214. [Google Scholar] [CrossRef] [PubMed]
- Dalvie, D.; Kalgutkar, A.S.; Chen, W. Practical approaches to resolving reactive metabolite liabilities in early discovery. Drug Metab. Rev. 2015, 47, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.H.; Telfer, C.; McFadyen, M.C.E.; Melvin, W.T.; Murray, G.I. The role of cytochrome P450 in cytotoxic bioactivation: Future therapeutic directions. Curr. Cancer Drug Targets 2004, 4, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Gardner, I.; Obach, R.S.; Shaffer, C.L.; Callegari, E.; Henne, K.R.; Mutlib, A.E.; Dalvie, D.K.; Lee, L.S.; Nakai, Y.; et al. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metab. 2005, 6, 161–225. [Google Scholar] [CrossRef]
- Grillo, M.P. Detecting reactive drug metabolites for reducing the potential for drug toxicity. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Mansuy, D.; Dansette, P.M. Sulfenic acids as reactive intermediates in xenobiotic metabolism. Arch. Biochem. Biophys. 2011, 507, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 840, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Furdui, C.M.; Poole, L.B. Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom. Rev. 2014, 33, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.R.; Farmer, P.J. Trapping reactions of the sulfenyl and sulfinyl tautomers of sulfenic acids. ACS Chem. Biol. 2017, 12, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Benitez, L.V.; Allison, W.S. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J. Biol. Chem. 1974, 249, 6234–6243. [Google Scholar] [PubMed]
- Gupta, V.; Carroll, K.S. Profiling the reactivity of cyclic C-nucleophiles towards electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 2016, 7, 400–415. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Paritala, H.; Carroll, K.S. Reactivity, selectivity and stability in sulfenic acid detection: A comparative study of nucleophilic and electrophilic probes. Bioconjug. Chem. 2016, 27, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Shetty, V.; Spellman, D.S.; Neubert, T.A. Characterization by tandem mass spectrometry of stable cysteine sulfenic acid in a cysteine switch peptide of matrix metalloproteinases. J. Am. Soc. Mass Spectrom. 2007, 18, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Claiborne, A.; Mallett, T.C.; Yeh, J.I.; Luba, J.; Parsonage, D. Structural, redox, and mechanistic parameters for cysteine-sulfenic acid function in catalysis and regulation. Adv. Protein Chem. 2001, 58, 215–276. [Google Scholar] [PubMed]
- Saiki, T.; Goto, K.; Tokitoh, N.; Okazaki, R. Synthesis and structure of a bridged calix [6] arene with a sulfenic acid functionality in the cavity. J. Org. Chem. 1996, 61, 2924–2925. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Bertho, G.; Mansuy, D. First evidence that cytochrome P450 may catalyze both S-oxidation and epoxidation of thiophene derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Isin, E.M.; Guengerich, F.P. Complex reactions catalyzed by cytochrome P450 enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Thebault, S.; Durand-Gasselin, L.; Bertho, G.; Mansuy, D. Reactive metabolites of thiophenic compounds: a new trapping method for thiophene sulfoxides. Drug Metab. Rev. 2010, 41 S1, 233–234. [Google Scholar]
- Zhang, C.; Wong, S.; Delarosa, E.M.; Kenny, J.R.; Halladay, J.S.; Hop, C.E.; Khojasteh-Bakht, S.C. Inhibitory properties of trapping agents: glutathione, potassium cyanide and methoxyamine, againsts major cytochrome P450 isoforms. Drug Metab. Lett. 2009, 3, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Ha-Duong, N.T.; Dijols, S.; Macherey, A.C.; Goldstein, J.A.; Dansette, P.M.; Mansuy, D. Ticlopidine as a selective mechanism-based inhibitor of human cytochrome P450 2C19. Biochemistry 2001, 40, 12112–12122. [Google Scholar] [CrossRef] [PubMed]
- Talakad, J.C.; Shah, M.B.; Walker, G.S.; Xiang, C.; Halpert, J.R.; Dalvie, D. Comparison of in vitro metabolism of ticlopidine by human cytochrome P450 2B6 and rabbit cytochrome 2B4. Drug Metab. Dispos. 2011, 39, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Valadon, P.; Dansette, P.M.; Girault, J.P.; Amar, C.; Mansuy, D. Thiophene sulfoxide as reactive metabolites: formation upon microsomal oxidation of a 3-aroylthiophene and fate in the presence of nucleophiles in vitro and in vivo. Chem. Res. Toxicol. 1996, 9, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Ménard, A.; Huang, Y.; Karam, P.; Cosa, G.; Auclair, K. Site-specific fluorescent labeling and oriented immobilization of a triple mutant of CYP3A4 via C64. Bioconjug. Chem. 2012, 23, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Librarie, J.; Bertho, G.; Mansy, D. Metabolic oxidative cleavage of thioesters: Evidence for the formation of sulfenic acid intermediates in the bioactivation of the antithrombotic prodrugs ticlopidine and clopidogrel. Chem. Res. Toxicol. 2009, 22, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Thebault, S.; Bertho, G.; Mansuy, D. Formation and fate of a sulfenic acid intermediate in the metabolic activation of the antithrombotic prodrug prasugrel. Chem. Res. Toxicol. 2010, 23, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Levent, D.; Hessani, A.; Bertho, G.; Mansuy, D. Thiolactone sulfoxides as new reactive metabolites acting as bis-electrophiles: Implication in clopidogrel and prasugrel bioactivation. Chem. Res. Toxicol. 2013, 26, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Dansette, P.M.; Levent, D.; Hessani, A.; Mansuy, D. Bioactivation of clopidogrel and prasugrel: Factors determining the stereochemistry of the thiol metabolite double bond. Chem. Res. Toxicol. 2015, 28, 1338–1345. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Desai, R.; Reynisson, J. Known drug space as a metric in exploring the boundaries of drug-like chemical space. Eur. J. Med. Chem. 2009, 44, 5006–5011. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, F.I.; Loi, D.; Ling, K.H.J.; Tang-Liu, D.D. Idiosyncratic reactions and metabolism of sulfur-containing drugs. Expert Opin. Drug Metab. Toxicol. 2012, 11, 467–485. [Google Scholar] [CrossRef] [PubMed]
- Savi, P.; Zachayus, J.L.; Delesque-Touchard, N.; Labouret, C.; Hervé, C.; Uzabiaga, M.F.; Pereillo, J.M.; Culouscou, J.M.; Bono, F.; Ferrara, P.; et al. The active metabolite of Clopidogrel disrupts P2Y12 receptor oligomers and partitions them out of lipid rafts. Proc. Natl. Acad. Sci. USA 2006, 103, 11069–11074. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Halladay, J.S.; Delarosa, E.M.; Tran, D.; Wang, L.; Wong, S.; Khojasteh, S.C. High-throughput, 384-well, LC-MS/MS CYP inhibition assay using automation, cassette-analysis technique and streamlined data analysis. Drug Metab. Lett. 2011, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
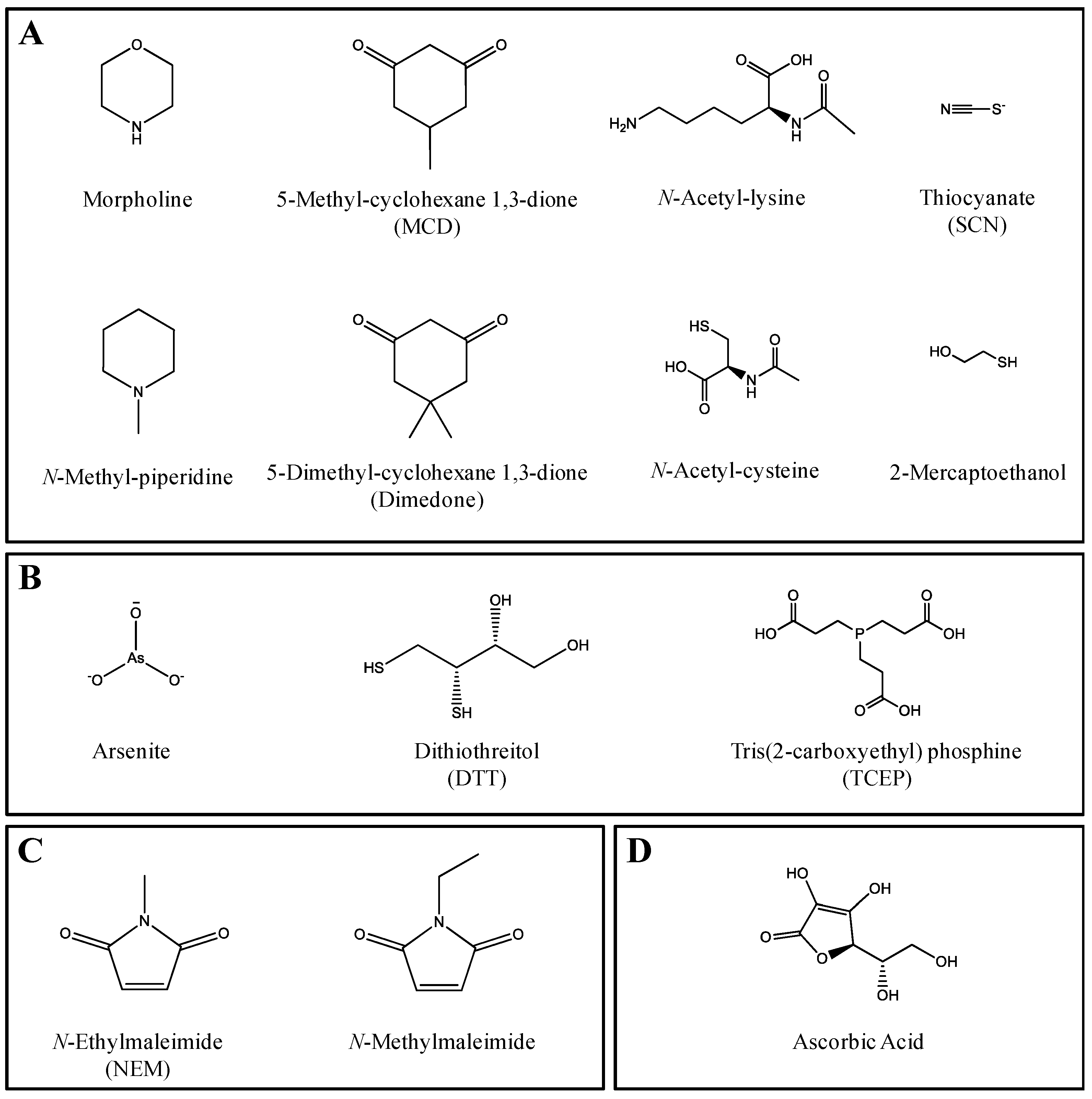
| Class | Trapping Agent | Typically Used In Vitro Concentration 1 (mM) | Highest Tested Concentration (mM) | IC50 (mM) (95% Confidence Interval) | |||||
|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 | CYP3A_T 2 | CYP3A_M 3 | ||||
| Nucleophiles | Morpholine | 10 | 100 | >100 | 7.9 (6.0–10.1) | 14 (11–16) | 13 (11–16) | 16 (15–18) | 9.1 (7.3–11.5) |
| N-Methylpiperidine | 10 | 100 | ~50 4,a | 9.9 (8.0–12.1) | 19 (13–28) | 6.5 (5.7–7.4) | 14 (12–16) | 12 (11–14) | |
| 5-Methyl-1,3-cyclohexanedione (MCD) | 4 | 10 | 3.6 (2.9–4.3) | 6.7 (5.4–8.3) | 3.1 (2.1–4.7) | >10 | 5.5 (3.7–8.1) | >10 | |
| 5-Dimethyl-cyclohexane 1,3-dione (Dimedone) | 1–4 | 5 | >5 | >5 | >5 | >5 | >5 | >5 | |
| Thiocyanate (SCN) | 4 | 100 | >100 | 18 (14–22) | 20 (13–31) | 49 (42–59) | 43 (34–54) | 46 (35–61) | |
| 2-Mercaptoethanol | 20 | 100 | 42 (26–67) | 2.0 (1.8–2.3) | 0.33 (0.30–0.36) | 24 (22–27) | 7.8 (6.5–9.4) | 8.9 (6.3–12.7) | |
| N-Acetyl-cysteine1 | 5 | 50 | 9.3 (7.8–11.0) | >50 | 18 (15–22) | 23 (20–25) | 15 (13–18) | 20 (15–26) | |
| N-Acetyl-lysine1 | 5 | 50 | >50 | >50 | >50 | >50 | >50 | >50 | |
| N-Acetyl-lysine: N-Acetyl-cysteine | 5:5 | 50:50 | 4.3 (3.7–5.1) | 41 (18–96) | 14 (12–16) | 14 (11–17) | 13 (11–15) | 8.4 (7.0–10.1) | |
| Reducing Agents | Arsenite | 20 | 10 | >10 | >10 | >10 | >10 | >10 | >10 |
| Tris(2-carboxyethyl) phosphine (TCEP) | 4 | 100 | 3.4 (1.2–9.9) | 32 (13–79) | 1.3 (0.1–17.1) | >100 | 8.8 (8.0–9.7) | 20 (17–24) | |
| Dithiothreitol (DTT) | 4 | 100 | 16 (13–19) | 7.2 (6.2–8.3) | 5.6 (5.0–6.3) | 22 (19–25) | 3.3 (2.7–4.0) | 9.4 (8.9–9.9) | |
| Michaël Acceptors and Dienophiles | N-Ethylmaleimide (NEM) | 4 | 40 | 0.12 (0.01–0.14) | 3.2 4,b (0.7–14.7) | 2.9 4,c (1.2–7.1) | >40 | 0.14 (0.13–0.16) | 2.7 (2.4–3.1) |
| N-Methylmaleimide | 4 | 20 | 0.20 (0.16–0.26) | 6.2 4,d (0.8–51.0) | 1.5 4,d (0.6–3.5) | >20 | 3.3 (2.6–4.4) | 12 (9–15) | |
| Radical Scavenger | Ascorbic Acid | 3 | 50 | 16 (13–20) | >50 | 16 (7–37) | 12 (8–18) | 2.7 (1.6–4.9) | 11 (8–16) |
| CYP Isoform | Probe Substrate (Concentration in µM) | Metabolite Monitored | Incubation Time (min) | Human Liver Microsomal Concentration (mg/mL) |
|---|---|---|---|---|
| 1A2 | Tacrine (1) | 1′-Hydroxytacrine | 10 | 0.2 |
| 2C9 | (S)-Warfarin (2) | 7-Hydroxywarfarin | 30 | 0.2 |
| 2C19 | (S)-Mephenytoin (60) | 4′-Hydroxymephenytoin | 40 | 0.2 |
| 2D6 | Dextromethorphan (5) | Dextrorphan | 10 | 0.03 |
| 3A | Midazolam (2) | 1′-Hydroxymidazolam | 10 | 0.03 |
| 3A | Testosterone (50) | 6β-Hydroxytestosterone | 10 | 0.07 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sodhi, J.K.; Delarosa, E.M.; Halladay, J.S.; Driscoll, J.P.; Mulder, T.; Dansette, P.M.; Khojasteh, S.C. Inhibitory Effects of Trapping Agents of Sulfur Drug Reactive Intermediates against Major Human Cytochrome P450 Isoforms. Int. J. Mol. Sci. 2017, 18, 1553. https://doi.org/10.3390/ijms18071553
Sodhi JK, Delarosa EM, Halladay JS, Driscoll JP, Mulder T, Dansette PM, Khojasteh SC. Inhibitory Effects of Trapping Agents of Sulfur Drug Reactive Intermediates against Major Human Cytochrome P450 Isoforms. International Journal of Molecular Sciences. 2017; 18(7):1553. https://doi.org/10.3390/ijms18071553
Chicago/Turabian StyleSodhi, Jasleen K., Erlie Marie Delarosa, Jason S. Halladay, James P. Driscoll, Teresa Mulder, Patrick M. Dansette, and S. Cyrus Khojasteh. 2017. "Inhibitory Effects of Trapping Agents of Sulfur Drug Reactive Intermediates against Major Human Cytochrome P450 Isoforms" International Journal of Molecular Sciences 18, no. 7: 1553. https://doi.org/10.3390/ijms18071553