
Transposable Elements in Human Cancer: Causes and Consequences of Deregulation


1
Division of Surgical Oncology, Department of Surgery Faculty of Medicine, Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
2
Institute of Pathology, Medizinische Hochschule Hannover, Hannover 30625, Germany
3
PILAR (Philippine and Indonesian Scholar) Research and Education, 20 Station Road, Cambridge CB1 2JD, UK
4
MRC (Medical Research Council) Unit for Lifelong Health and Ageing, University College London, London WC1B 5JU, UK
5
Division of Haematology/Oncology, Faculty of Medicine Universitas Gadjah Mada, Yogyakarta 55281, Indonesia
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(5), 974; https://doi.org/10.3390/ijms18050974
Submission received: 21 March 2017
/
Revised: 26 April 2017
/
Accepted: 29 April 2017
/
Published: 4 May 2017
(This article belongs to the Special Issue Cancer Epigenetics)
Abstract
:Transposable elements (TEs) comprise nearly half of the human genome and play an essential role in the maintenance of genomic stability, chromosomal architecture, and transcriptional regulation. TEs are repetitive sequences consisting of RNA transposons, DNA transposons, and endogenous retroviruses that can invade the human genome with a substantial contribution in human evolution and genomic diversity. TEs are therefore firmly regulated from early embryonic development and during the entire course of human life by epigenetic mechanisms, in particular DNA methylation and histone modifications. The deregulation of TEs has been reported in some developmental diseases, as well as for different types of human cancers. To date, the role of TEs, the mechanisms underlying TE reactivation, and the interplay with DNA methylation in human cancers remain largely unexplained. We reviewed the loss of epigenetic regulation and subsequent genomic instability, chromosomal aberrations, transcriptional deregulation, oncogenic activation, and aberrations of non-coding RNAs as the potential mechanisms underlying TE deregulation in human cancers.
1. Introduction
Over two-thirds of the human genome is composed of repetitive sequences [1]. Transposable elements (TEs), which make up the majority of repetitive sequences (up to 50% of the human genome), can jump within the genome and play an important role as an engine for the dynamics of human evolution and the pathogenesis of human cancers [2]. TEs are classified according to their transposition mechanisms into retrotransposons (Class I) and DNA transposons (Class II) [3,4]. Class I retrotransposons are further sub-classified into long terminal repeats (LTRs) or endogenous retrovirus (ERV) elements that make up 8% of the human genome, and non-LTRs, which include the two most important TEs in cancers, long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) [3,4]. LTRs are identical repeated DNA sequences originating from the integration of ancient retroviruses into the human genome that have lost their ability as mobile elements. On the other hand, non-LTRs with autonomous and nonautonomous retrotranspositions (LINEs and SINEs, respectively) retain their ability as active mobile elements in the human genome. LINE1 sequences represent up to 17% of the human genome [3,4]. Although LINE1 is considered the most active mobile element in a human, around 500,000 copies no longer have the ability to mediate retrotransposition due to significant LINE1 truncations [5]. However, new full length LINE1 would be able to encode proteins that are very efficient to “copy and paste” into a new genetic location and hamper the associated gene expression and/or drive oncogenic process through the transcription of chimeric proteins [5].
Active TEs are considered highly mutagenic and are associated with the multiple steps of cancer development and progression [2,6]. TEs have been demonstrated to play an active role in regulating the human genome by governing endogenous gene expression, as well as generating novel genetic loci [7]. However, TE activity might have different impacts on the human genome ranging from positive to negative consequences, including the maintenance of centromere and telomere integrity, recombinant genome remodeling, and deleterious gene expression [3,7]. In addition, the accumulation of TEs throughout human evolution has been adapted into novel functions through several mechanisms, known as domestication. The co-opting of TEs can be delivered through several ways such as the formation of a new gene entity, integration into an existing gene generating a chimeric protein, and insertion into a regulatory region upstream of a gene to further regulate gene expression by forming alternative promoters or altering transcriptional binding sites [8,9]. In addition, the integration of TEs into introns might interfere with transcription, alternative-splicing, polyadenylation, and messenger RNA stability [3,10,11].
A growing body of evidence has documented the essential role of TEs in human carcinogenesis. The insertion of TEs into genes that are responsible for DNA repair including BRCA2 [12], APC [13], and RB1 [14] can cause the disruption of gene expression and further affect genome instability [15]. Methylation loss of a specific LINE1 promoter is able to activate an alternative transcript that encodes a truncated and constitutively active MET protein in bladder cancer [16]. De novo insertions of LTR and LINE sequences cause an alternative transcription of a new isoform in an ALK (anaplastic lymphoma kinase) gene [17]. The new ALK isoform is specifically expressed in around 11% of melanomas that further show a specific response to the ALK inhibitor [17]. A comprehensive approach in diffuse large B-cell lymphomas (DLBCLs) has detected multiple LTR transcripts in several genes, including fatty-acid binding protein 7 (FABP7); that are known to promote lymphomagenesis [18]. The reactivation of ancient LTR has also been associated with the novel oncogenic transcription of ERBB4 in ALK-negative anaplastic large cell lymphoma (ALCL) [19]. Two aberrant ERBB4 transcripts are found in almost a quarter of ALK-negative ALCL patients [19]. Despite their important roles in genome regulation, the detailed mechanisms of TE reactivation in tumor development remain largely unexplained.
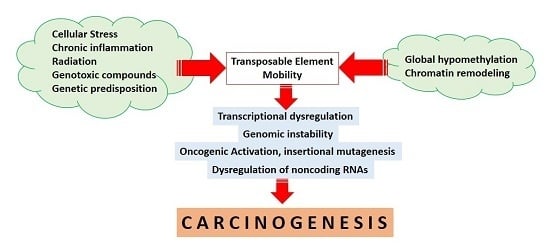
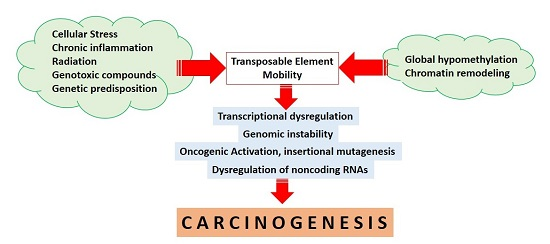
TEs are tightly regulated from early embryonic development and during the entire human life [20]. Epigenetic mechanisms, particularly DNA methylation and histone modifications, are the best known mechanisms underlying the repression and regulation of TEs [3,21]. In relation to human cancers, epigenetic alterations have also been continuously reported to play a significant role in the initiation of tumor development [22]. A growing number of studies have delineated that epigenetic mechanisms may also control TE reactivation with subsequent effects on carcinogenesis [2,21]. The hypermethylation of tumor suppressor genes accompanied by global hypomethylation occurs consecutively in human cancers [22,23]. Moreover, the global loss of methylation subsequently results in the reactivation of TEs [3,21,24]. In cancers, hypomethylation and TE activation are dynamic processes during tumor evolution and progression [25,26,27]. The reactivation of TEs could initiate oncogene activation [16,25], chromosomal breakages [28], and genomic instability [27] that further contribute to tumor initiation and progression [16,27]. To obtain a better understanding of this emerging area of research, we reviewed the current evidence on the deregulation of TEs by epigenetic mechanisms, especially DNA methylation and non-coding RNAs, and their potential contribution to the development of human cancers through genomic instability, chromosomal aberrations, and oncogenic activation.
2. Widespread Epigenetic Deregulation of Repetitive Elements in Cancer
TEs have been suggested to play an essential role in maintaining genome integrity [29] and therefore, dysregulated TE activity may result in genomic instability and subsequent carcinogenesis [5,30]. TEs and host regulatory factors cooperate in controlling TE activity through overlapping multiple epigenetic mechanisms [31]. TEs are able to independently recruit silencing signals to form robust building blocks of inactivation not only at the level of a single gene, but also across large chromosomal regions [3,31]. In addition, self-control mechanisms by TEs will initiate the repression of retrotransposition adverse effects while maintaining their ability to replicate. The interdependency of host regulatory controls is mediated through the transcription of truncated suppressor clones, the presence of splice sites and hidden polyadenylation signals, and transposon-mediated autoregulation (as reviewed in [32]). Several other host regulatory defense mechanisms are also involved in restricting TE activity. However, the primary regulators controlling TE transcription are epigenetic mechanisms, mainly DNA methylation and histone modifications; that effectively suppress TEs while maintaining their presence in the human genome [3,31]. In the context of cancer, the loss of DNA methylation causing the reactivation of LINE1 has been reported in colorectal cancer [26], hepatocellular carcinoma [33], and breast cancer [34]. Figure 1 describes the effects of the loss of methylation on TE reactivation in a cancer cell.
The hypomethylation of LINE1 is reported in both solid tumors and leukemia, and is frequently correlated with unfavorable clinical outcomes [26,35]. In addition, the hypomethylation of LINE1 and Alu in circulating blood has potential value for cancer diagnoses [36]. Exposure to DNA demethylation agents including azacytidine and decitabine leads to LINE1 demethylation, as well as subsequent activation, and is followed by oncogene activation [16]. The human endogenous retroviruses (HERVs) are also related to carcinogenesis. Increased levels of HERV RNA and reverse transcriptase enzymes have been reported in lymphomas and breast cancers [37]. In addition, HERV-like-viruses have also been associated with melanomas, especially those with metastases [37]. Ovarian, colon, and testicular cancers express higher levels of envelope genes of HERV [37].
Heterochromatin formation has also been associated with the regulation of TE silencing in cooperation with DNA methylation and small RNAs [31]. Histone tail modifications affect the binding of protein and transcription factors. TE-associated nucleosomes are commonly methylated at the histone 3 lysine 9 (H3K9) representing signals for transcriptional silencing and inactive chromatin [38]. Mutations of H3K9 methyltransferases including SUV39 cause TE upregulation [39,40]. In addition, the deregulation of DNA methyltransferase during embryonic development leads to increased TE activity and is associated with some developmental disorders [31,41]. Specific patterns of DNA sequences and non-coding RNAs are implicated in the direct de novo and maintenance DNA methylation of specific genomic loci, including repetitive sequences [22,31]. Chromatin structure-modifying proteins implicated in chromatin remodeling and condensation are also involved in TE silencing [31,42]. Several TEs, particularly SINEs, contain binding sites for CTCF that function as an insulator and consecutively regulate chromatin condensation [43,44]. A recent study by Pugacheva et al. showed the specific binding of CTCF and BORIS to DNA repeats in which BORIS preferentially bound to SVA repeats (SINEs, VNTRs, Alus), suggesting a potential role of BORIS in the regulation of active TEs in cancer cells [45]. DDM1, LSH1, SETDB1, and KDM1A are among numerous chromatin remodeling proteins that regulate and maintain TE inactivation [46]. Especially in mammals, DNA methylation, along with chromatic remodeling factors and non-coding RNAs, are responsible for keeping TEs in a dormant state [42]. However, recent evidence suggests that different mechanisms are involved in regulating TEs during embryogenesis, mainly through using histone modifying enzymes such as KDM1A and SETDB1 [21,46]. Upon epigenetic silencing, TEs are kept inactive to allow their embedded regulatory regions to be exapted. Mutations and the deregulation of chromatin remodeling genes are closely related with human cancers of different etiologies [47,48]. However, their direct effects on the reactivation of repetitive elements in cancers to initiate genomic instability and oncogenic activation need to be clarified [49,50].
3. Transcriptional Deregulation
TEs are able to modulate gene transcription by insertion into transcriptional regulatory regions [3]. TE insertions can induce functional defects of regulatory regions, including promoters, enhancers, or silencers [3]. TE integrations are able to create new exons that interfere with the biological functions of the host gene product. The insertion of TE within exons might alter the open reading frame (ORF) and initiate missense or nonsense mutations that can consecutively destroy transcription factor binding sites [3]. By contrast, insertion into gene regulatory and coding regions can introduce new splice sites [51], perturb the canonical splice sites [52], or create new polyadenylation signals [10]. Insertions of TE into introns are also able to create alternatively spliced exons that are biologically functional [52,53]. For instance, Alu sequences are commonly found to create new exons in mature transcripts upon insertion [51,53]. Sorek et al. reported that more than 80% of Alu insertions in the exons caused either a frame-shift or premature stop codon [54]. In addition, TE insertions in 3′UTRs and introns affect mRNA stability, localization, and translation [11], causing a reduction of gene expression. Transcription factors that are commonly involved in carcinogenesis, including FOXA1 [55], SP1 [56], GATA [57], P53 [58], and retinoic acid receptors [59], have binding sites in Alu elements [58]. In addition, p53 binding sites have been found to interact with several LTR- and non LTR elements. It is predicted that around 400,000 p53 binding sites of the human genome are associated with the Alu element [58]. The deregulation of transcription factor p53 affects the expression of many other genes and is suggested to be involved in more than half of human cancers [60]. Mutations of the TP53 gene cause an elevated activity of RNA polymerase III [61]. Levels of Alu RNA expression are increased in many cancers, including hepatocellular carcinoma and lung cancer [61]. It seems that the p53 binding to Alu sequences accompanied by transcriptional repression can trigger the deregulation of multiple genes [58]. It has recently been shown that p53 is able to restrain retrotransposon activity through direct interactions with the PIWI-interacting RNA pathway [62].
TE insertions within the genome are considered to be non-random events, as the insertions might affect transcriptional regulation [3,63]. It has been documented that TE integrations caused significant alterations in several cis-regulatory elements of gene expression [63]. A comprehensive analysis using ChIP-seq has revealed that almost 20% of transcription factor binding sites are embedded within TEs [63]. TEs are able to divide the genome into transcriptionally active or inactive regions by influencing the extent of heterochromatin formation [64]. The effects of retrotransposition on gene transcription in human cancer are summarized in Figure 2. Cancer-associated genes that are transcriptionally modulated by TE insertions are recapitulated in Table 1.
TEs also interact with non-coding RNAs, including microRNAs and lncRNAs, to modulate transcription, as discussed below. TEs use other mechanisms to influence gene expression, including the spreading of repressive chromatin signals to adjacent regions in order to silence the nearby genes [85]. In addition, TEs disrupt gene expression through chromosomal integration that can result in shorter 3′UTR and polyadenylation signal disruption [86]. The insertion of an Alu element into the NF-1 gene has been shown to cause deletion and reading frame-shift, leading to the development of neurofibromatosis [87].
4. Genomic Instability and Chromosomal Rearrangements
The vast majority of TE insertions target particular genomic loci that are specifically recognized by endonuclease domains causing alterations of adjacent gene expression and significant genomic deletions [88]. Under certain conditions, random TE integrations can lead to insertional mutagenesis [2]. Genomic variations affected by TE mobility include the loss of heterozygocity insertion, deletion, duplication, translocation, and inversion [2,15,89]. Gene mutations caused by retrotransposon insertions are found in around 0.3% of total mutations [2]. Several mechanisms leading to TE-mediated genomic instability in cancer have been reported. First, the insertion of inverted TEs in the human genome can induce DNA damage [90]. Because TEs are naturally very mobile, there is a high chance for insertion in an inverse orientation. Several adjacent Alu and short sequence repeats that are inversely inserted during DNA replication tend to form a secondary hairpin structure that is susceptible to double-strand breaks (DSBs) [90,91]. However, the arrangement of inverted Alu elements in the human genome is relatively rare. In some parts, there is a tendency for the direct orientation of newly adjacent Alu insertion to prevent secondary hairpin formation [92]. Despite the unusual nature of closely inverted Alu insertion, the fact that unregulated Alu insertions frequently occur in cancer has increased the probability of de novo inverted insertions [93].
The second mechanism of TE-mediated genomic instability is the induction of unstable microsatellite seeding after TE insertions [15]. Mutations in the poly-A tail are able to initiate the seeding of large tracts of unstable microsatellite repetitive sequences [15]. If located in fragile sites of the human genomes, the unstable microsatellites can induce carcinogenesis [15]. In addition, the expansion of intronic microsatellite sequences is able to disrupt gene transcription, as shown in Friedrich’s ataxia [94]. Microsatellite instability is an important feature of several colorectal, stomach, endometrium, ovary, urinary tract, skin, and brain cancers [95]. In addition, the incorporation of TEs into introns can produce unstable long repeats in the microsatellites, causing these regions to become highly unstable and vulnerable to DNA damage [15,95].
TE-mediated genomic instability is also associated with alterations in the overall transcription in cancer. As previously mentioned, TE mobility can induce aberrant and recombinant gene transcription [58]. The interplay between the replication fork and transcription factors has significantly increased the chances of DNA damage [95]. The TE-mediated introduction of novel alternative promoters is capable of elevating the overall levels of stress-associated gene expression that may further increase DNA damage rates [89,96]. The majority of cancers represent epigenetic reprogramming with global widespread hypomethylation that can increase the transcriptional activity and mobility of these TEs, thus increasing the probability of transcriptional stress-associated DNA damage. Transcriptional stress and DNA damage are the major contributors of genomic instability in cancers [97].
The induction of genomic instability by TE insertion will induce host DNA repair signaling. The recruitment of the DNA repair machinery to the TE insertion loci is accompanied by cell cycle arrest affecting the relative efficiency of TE mobility. However, TE insertion also targets genes that are involved in the DNA repair pathway. The breast cancer susceptibility gene BRCA2 has been affected by multiple TE insertions in cancers [12,98]. In addition, the APC gene is also disrupted by multiple TE insertions (Alu and LINE1) and is suggested to play a role in colorectal carcinogenesis [13,99]. Voineagu et al. have suggested that inverted repeat sequences might emerge as fragile sites inducing genomic instability [90]. LINE1 endonucleases are eminent sources of DSBs that subsequently affect genomic instability [100]. In addition, LINE1 transposition causes DNA breakage and, even after DNA repair, this event still produces the deletion of a few base pairs [101,102]. In addition, genotoxic stress by a low dose of radiation results in LINE1 activation and is associated with the initiation of carcinogenesis [103]. TE-associated genes that lead to deletions or variations of a few base pairs (genomic instability) in human cancer are summarized in Table 2.
TE insertions have also been associated with chromosomal rearrangement. Alu sequences are frequently found in several breakage points of chromosomal translocations and are closely associated with cancer [120]. TE insertion-mediated deletions and chromosomal rearrangement were reported upon pathogenic LINE1 and Alu insertions [121,122]. TE-mediated insertions can cause deletions, ranging from a few to several thousand nucleotides in size [122]. Novel LINE1 insertions often target endogenous elements, suggesting a preference for specific incorporation sites [5,121]. Chromosomal rearrangements frequently occur in rather prone regions during TE insertions. Subsequently, host DNA repair systems will try to eliminate nascent TE insertions to limit the impacts on genome instability and chromosomal rearrangement [101,123].
TE-associated chromosomal rearrangements are also mediated by segmental duplications that are relatively common in the human genome [124]. Segmental duplications have been established as significant factors in genome evolution, as well as in the development of human cancer. A comprehensive analysis of the human genome has revealed segmental duplications with an increased density of Alu repeats within or near the duplication intersections [124]. In humans, it is believed that an outburst of Alu repeats can initiate segmental duplications [124,125]. However, the exact mechanisms by which Alu and other TE species induce segmental duplications remain unclear. Homologous repair is able to generate duplications by arranging replicons tandemly. However, the mechanism underlying large duplication events with a distance of more than 1 Mb remains elusive. The relatively frequent Alu element insertions at the junction of the segmental duplication, accompanied by the involvement of evolutionarily young and identical elements, suggest that homology sequences guide these events [124,125]. TE-associated genes that are affected by chromosomal aberrations in human cancers are listed in Table 3.
5. Inactivation of Tumor Suppressor Genes and Activation of Oncogenes
Random integrations of TEs into specific sites of the human genome increase the chance for insertional mutagenesis followed by the activation of signaling pathways leading to carcinogenesis [2,15]. LINE1, Alu, and SVA are among the most common TEs that frequently induce insertional mutagenesis [2]. The activation of oncogenic drivers is also mediated by the deleterious insertion of tumor suppressor genes and the disruption of regulatory sequences. The deregulation of gene expression, splicing-induced truncated proteins, and destabilized mRNAs all contribute to oncogenic activation [3,134]. In addition, genomic instability, including chromosomal breakages and DNA recombination that can be induced by mobile TE insertions, reinforces mutation rates and carcinogenesis [13,89,127].
Non-allelic homologous recombination causing deletions or duplications in the presence of Alu is abundantly found in tumors with TP53 mutations [58]. The close proximity of several Alu insertions tends to impose an inverted orientation, leading to the loss of p53 functions [58]. Alu sequences are also involved in mismatch repair (MMR) by disrupting MLH1 and MLH2 genes [92]. In addition, rearrangements due to Alu insertion and the presence of Alu in the MLH1 and MLH2 proteins are associated with hereditary non-polyposis colorectal cancer [92]. Recombination events due to the high density of Alu elements within the BRCA1 gene are associated with the important deregulation of genomic integrity in breast cancer [12,98]. Alu repeats have also contributed significantly to chromosomal translocations, including BCR/ABL rearrangement in chronic myelogeneous leukemia [126]. The recombination of Alu has also caused myoblastosis (MYB) duplication to T-cell acute lymphoblastic leukemia [132]. Chromosomal rearrangements in mixed-lineage leukemia (MLL) have also been reported due to Alu recombination [135]. Especially in partial duplication events, TEs are usually inserted near the translocation breakpoints. MYB and MLL duplications have also been found in healthy controls, whereas leukemogenesis is induced by TE insertions during blood cell differentiation [136]. Additionally, Alu rearrangements have been reported in the tumor suppressor gene von Hipple-Lindau (VHL) [133]. Compared to oncogenes, tumor suppressor genes contain more Alu sequences [93].
6. Transposable Elements and Non-Coding RNAs
The interconnection between TEs and non-coding RNAs (ncRNAs) has been recently delineated, especially in the biogenesis of small ncRNAs, including microRNAs that are associated with TEs. MicroRNAs are small non-coding RNAs that regulate post-transcriptional gene expression and modulate several important oncogenic pathways establishing a dynamic network of cell homeostasis (reviewed in [137,138]). A large number of microRNAs originate from loci flanked from two related TEs in one genomic locus that is easily transcribed and processed into hairpin RNA structures following common microRNA biogenesis [139,140]. Both bioinformatics and genome-wide screening have identified a significant number of TE-based microRNA (up to 15% of total microRNAs) [140,141]. Several types of TEs are involved in microRNA biogenesis, especially DNA transposons, LINEs, and SINEs [142]. The first example of TE-associated microRNA is hsa-mir-548 that derives from inverted-repeat transposable elements [143]. The deregulation of TE is implicated in the disruption of microRNA biogenesis. More importantly, TE-associated ncRNAs are involved in several important regulatory networks and associated with some diseases. The involvement of lncRNAs in cancers and their potential applications in clinical management of cancer have been comprehensively reviewed in [144,145].
It has also been indicated that Alu insertions are able to create a targeting site for mRNA decay by forming stable RNA/mRNA interactions through intra- or intermolecular base pairing with 3′UTRs and complementary binding with RNA targets [146]. Hundreds of long non-coding RNAs (lncRNAs, up tp 80%) contain endogenous insertions of TEs in human cells that serve as regulatory signals for cell growth and proliferation [147]. Numerous long circular ncRNAs are found in fibroblasts and act in a similar manner to “sponges” for miRNAs, in which intronic Alu sequences that flank exon and the alternative formation of inverted Alu pairs are suggested to greatly contribute to RNA circularization [148,149]. Therefore, the deregulation of TE might interrupt ncRNA functions and is associated with the initiation of carcinogenesis. Alu and LINE-embedded sequences also have regulatory roles for the transcription of lnc-RNAs and the stability of mRNA products [150]. Since lncRNAs containing repeated sequences are also important in the regulation of other epigenetic mechanisms, including genomic imprinting and chromatin remodeling, the deregulation of this network is suggested to play a role in the development of human cancer [143,150,151]. Carrieri et al. have delineated two functionally important regions in the antisense lncRNAs, i.e., 5′-end and embedded SINEB2 and Alu elements that can interact with targeted mRNAs to inhibit the translation. [152]. They also identified 31 antisense lncRNAs containing similar SINE/Alu elements that might interact with 3′UTR mRNAs, suggesting an important role of TE-embedded regions in the post-transcriptional regulation of gene expression [152].
Reciprocally, small non-coding RNAs including microRNAs have also been reported to regulate genomic stability through direct or indirect transcriptional and post-transcriptional TE repression [150]. As cytoplasmic ncRNAs, microRNAs indirectly control DNA repair and genome stability [153]. siRNAs mediate the trans enrolment of Ago complexes to subsequently regulate genome instability through transcriptional repression and repetitive DNA element recombination [153]. In addition, the microRNA biogenesis machinery has been inferred as an important regulator of heterochomatin formation and transcriptional silencing. The AGO1 protein is reported to be involved in transcriptional gene silencing through histone H3K9 methylation [154]. An initial report suggested that promoter DNA methylation causing transcriptional silencing can be stimulated by a complementary siRNA to direct DNA methylation into specific loci, including repetitive sequences [155]. Another mechanism of ncRNA deregulation is the insertion into microRNA’s regulatory regions. Alu sequences occupy a large portion of microRNA genes, as well as 3′UTR mRNA targets [156]. In general, tumors are able to avoid miRNA-mediated regulation, causing a further enhancement of genomic instability and mutability because of TE reactivation.
LncRNAs have been associated with myriads of important signaling pathways, most of which require an interaction with proteins, as well as transcription factors. TE-associated lncRNAs are reported to bind to some important transcription factors for cell proliferation, including p53, sp1, and NF-Y [157]. In response to cellular stress, Alu RNAs are able to bind to RNA polymerase II to regulate the expression of some responsive genes, as well as to promote evolution through exonization and alternative splicing [158]. Analysis using mass spectrometry has revealed that Alu-derived piRNAs are able to bind some nuclear proteins, suggesting their potential roles in DNA repair, chromatin reprogramming, and cell proliferation [159]. Therefore, TE mobility within lncRNA transcripts reflects evolutionary and versatile functions in regulating cellular homeostasis.
The PIWI-piRNA axis has also been implicated in the silencing of transposable elements and contributes to the development of human cancers [160]. Transcribed mostly from a cluster, piRNAs are small non-coding RNAs that interact with PIWI proteins that are abundantly found in germ cells [161]. PIWI proteins interact with DNA methylation and chomatin modifications to silence TEs while maintaining the ability of germ stem cells for self-renewing [162]. In addition, the piRNA pathway interacts with the p53 protein to restrict mobile element insertions and regulate chromatin repressive marks in the 5′ of LINE1 sequences [62]. One of the most important roles of the piRNA-PIWI protein complex is to protect germ cells from transposon insertions [160,163]. In cells that are previously exposed to TEs, piRNAs containing a complementary TE sequence will induce degradation through Piwi proteins [164]. In the first encounter to TEs, piRNA transcripts that are complementary to the TE RNA can induce degradation through the piRNA-Piwi complex [165]. The piRNA-Piwi axis has also been associated with the maintenance of genomic imprinting through DNA methylation in the Rasgfr locus, in which the differentially methylated region contains the LINE1 sequence [166]. Correspondingly, the deregulation of PIWI proteins and transposable elements has been reported in gliomas, sarcomas, and adenocarcinomas [160].
7. Conclusions
The above compiled results and observations suggest an important role of TEs in transcriptional control, genomic instability, chromosomal rearrangements, non-coding RNA regulation, and oncogenic activation. Although a direct association between TE insertion and cancer initiation still has to be clarified, the deregulation of TEs is closely linked to cancers of various etiologies. Bioinformatic screens have revealed mutagenenic TE insertions in different types of cancers. TE insertions also function as a reservoir of endogenous gene regulatory factors that have been co-opted by the human genome to control gene expression, as well as cellular phenotypes. After co-option or exaptation, insertions of TEs can provide novel transcription factor binding sites in the promoters, enhancers, or insulators, leading to cancer specific activation. TEs flanking ncRNA genes are also involved in ncRNA biogenesis. In addition, repetitive sequences within TEs are important loci for the complementary binding of ncRNAs acting as a reservoir for ncRNA inactivation. Therefore, the deregulation of TEs affects genomic stability, transcription, and non-coding RNA regulation, leading to cancer development and progression (please see Figure 3). However, the mechanisms for TE-mediated oncogenic activation need to be further investigated to reveal new insights into carcinogenesis and identify novel targets for therapeutic interventions. Clarification of the specific epigenetic regulation of TEs in such studies may raise the possibility of applying epigenetic agents in TE-driven cancers such as demethylase and bromodomain inhibitors; that are now in preclinical and clinical trials for solid tumors and hematological malignancies.
Author Contribution
Sumadi Lukman Anwar and Ulrich Lehmann conceived and designed the review; Sumadi Lukman Anwar, Wahyu Wulaningsih, and Ulrich Lehmann contributed to the writing of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., 3rd; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Cancer genome atlas research network landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-J.; Xue, H.-Y.; Qi, X.; Xu, J.; Ma, S.-J. LINE-1 in cancer: Multifaceted functions and potential clinical implications. Genet. Med. 2016, 18, 431–439. [Google Scholar]
- Criscione, S.W.; Zhang, Y.; Thompson, W.; Sedivy, J.M.; Neretti, N. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genom. 2014, 15, 583. [Google Scholar] [CrossRef] [PubMed]
- Bodega, B.; Orlando, V. Repetitive elements dynamics in cell identity programming, maintenance and disease. Curr. Opin. Cell Biol. 2014, 31, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Van de Lagemaat, L.N.; Landry, J.R.; Mager, D.L.; Medstrand, P. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions. Trends Genet. 2003, 19, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Tress, M.L.; Valencia, A. Alternative splicing and co-option of transposable elements: The case of TMPO/LAP2α and ZNF451 in mammals. Bioinformatics 2015, 31, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Roy-Engel, A.M.; El-Sawy, M.; Farooq, L.; Odom, G.L.; Perepelitsa-Belancio, V.; Bruch, H.; Oyeniran, O.O.; Deininger, P.L. Human retroelements may introduce intragenic polyadenylation signals. Cytogenet. Genome Res. 2005, 110, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, T.; Huang, S. 3′-UTR-located inverted Alu repeats facilitate mRNA translational repression and stress granule accumulation. Nucleus 2012, 3, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Teugels, E.; de Brakeleer, S.; Goelen, G.; Lissens, W.; Sermijn, E.; de Grève, J. De novo Alu element insertions targeted to a sequence common to the BRCA1 and BRCA2 genes. Hum. Mutat. 2005, 26, 284. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B.; Horii, A.; Miyoshi, Y.; Nakamura, Y. Disruption of the APC Gene by a retrotransposal insertion of LI sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar]
- Rodríguez-Martín, C.; Cidre, F.; Fernández-Teijeiro, A.; Gómez-Mariano, G.; de La Vega, L.; Ramos, P.; Zaballos, A.; Monzón, S.; Alonso, J. Familial retinoblastoma due to intronic LINE-1 insertion causes aberrant and noncanonical mRNA splicing of the RB1 gene. J. Hum. Genet. 2016, 61, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Ade, C.; Roy-Engel, A.M.; Deininger, P.L. Alu elements: An intrinsic source of human genome instability. Curr. Opin. Virol. 2013, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, T.; Lee, W.; Obenauf, A.C.; Ran, L.; Murali, R.; Zhang, Q.F.; Wong, E.W.P.; Hu, W.; Scott, S.N.; Shah, R.H.; et al. Alternative transcription initiation leads to expression of a novel ALK isoform in cancer. Nature 2015, 526, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Lock, F.E.; Rebollo, R.; Miceli-Royer, K.; Gagnier, L.; Kuah, S.; Babaian, A.; Sistiaga-Poveda, M.; Lai, C.B.; Nemirovsky, O.; Serrano, I.; et al. Distinct isoform of FABP7 revealed by screening for retroelement-activated genes in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E3534–E3543. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Pellegrino, E.; Mereu, E.; Kwee, I.; Agnelli, L.; Bergaggio, E.; Garaffo, G.; Vitale, N.; Caputo, M.; Machiorlatti, R.; et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood 2016, 127, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, P.; Richardson, S.R.; Mager, D.L.; Faulkner, G.J. Transposable elements in the mammalian embryo: Pioneers surviving through stealth and service. Genome Biol. 2016, 17, 100. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Hong, C.; Zhang, B.; Lowdon, R.F.; Xing, X.; Li, D.; Zhou, X.; Lee, H.J.; Maire, C.L.; Ligon, K.L.; et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat. Genet. 2013, 45, 836–841. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Molecular origins of cancer epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, Z.; Tanay, A. Hypomethylation marks enhancers within transposable elements. Nat. Genet. 2013, 45, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Swets, M.; Zaalberg, A.; Boot, A.; van Wezel, T.; Frouws, M.; Bastiaannet, E.; Gelderblom, H.; van de Velde, C.; Kuppen, P. Tumor LINE-1 methylation level in association with survival of patients with stage II colon cancer. Int. J. Mol. Sci. 2016, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Daskalos, A.; Nikolaidis, G.; Xinarianos, G.; Savvari, P.; Cassidy, A.; Zakopoulou, R.; Kotsinas, A.; Gorgoulis, V.; Field, J.K.; Liloglou, T. Hypomethylation of retrotransposable elements correlates with genomic instability in non-small cell lung cancer. Int. J. Cancer 2009, 124, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nau, M.M.; Zucman-Rossi, J.; Powell, J.I.; Allegra, C.J.; Wright, J.J. LINE-1 element insertion at the t(11;22) translocation breakpoint of a desmoplastic small round cell tumor. Genes Chromosomes Cancer 1997, 18, 232–239. [Google Scholar] [CrossRef]
- Hedges, D.J.; Deininger, P.L. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 616, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Ayarpadikannan, S.; Kim, H.-S. The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genom. Inf. 2014, 12, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.L.; Moran, J.V. Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Baba, Y.; Ishimoto, T.; Chikamoto, A.; Kosumi, K.; Hayashi, H.; Nitta, H.; Hashimoto, D.; Beppu, T.; Baba, H. LINE-1 methylation level and patient prognosis in a database of 208 hepatocellular carcinomas. Ann. Surg. Oncol. 2014, 1, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Terry, M.B.; Delgado-Cruzata, L.; Vin-Raviv, N.; Wu, H.C.; Santella, R.M. DNA methylation in white blood cells: Association with risk factors in epidemiologic studies. Epigenetics 2011, 6, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Loss of DNA methylation at imprinted loci is a frequent event in hepatocellular carcinoma and identifies patients with shortened survival. Clin. Epigenet. 2015, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gammon, M.D.; Hernandez-Vargas, H.; Herceg, Z.; Wetmur, J.G.; Teitelbaum, S.L.; Bradshaw, P.T.; Neugut, A.I.; Santella, R.M.; Chen, J. DNA methylation in peripheral blood measured by LUMA is associated with breast cancer in a population-based study. FASEB J. 2012, 26, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G. Endogenous retroviruses and the development of cancer. J. Immunol. 2014, 192, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Penke, T.J.R.; McKay, D.J.; Strahl, B.D.; Gregory Matera, A.; Duronio, R.J. Direct interrogation of the role of H3K9 in metazoan heterochromatin function. Genes Dev. 2016, 30, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Fallis, A. An epigenetic switch ensures transponson repressioin upon dynamic loss of DNA methylation in embryonic stem cells. eLife 2013, 53, 1689–1699. [Google Scholar]
- Bulut-Karslioglu, A.; DeLaRosa-Velázquez, I.A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galán, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 2014, 55, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-J.; Liu, Z.-P. Histone methylations in heart development, congenital and adult heart diseases. Epigenomics 2015, 7, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Rigal, M.; Mathieu, O. A “mille-feuille” of silencing: Epigenetic control of transposable elements. Biochim. Biophys. Acta Gene Regul. Mech. 2011, 1809, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Gonalves, Â.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Teplyakov, E.; Wu, Q.; Li, J.; Chen, C.; Meng, C.; Liu, J.; Robinson, S.; Loukinov, D.; Boukaba, A.; et al. The cancer-associated CTCFL/BORIS protein targets multiple classes of genomic repeats, with a distinct binding and functional preference for humanoid-specific SVA transposable elements. Epigenet. Chromatin 2016, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Gifford, W.D.; Pfaff, S.L.; MacFarlan, T.S. Transposable elements as genetic regulatory substrates in early development. Trends Cell Biol. 2013, 23, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Jene-Sanz, A.; Lopez-Bigas, N. The mutational landscape of chromatin regulatory factors across 4623 tumor samples. Genome Biol. 2013, 14, r106. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenet. Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Hur, S.K.; Park, E.J.; Han, J.E.; Kim, Y.A.; Kim, J.D.; Kang, D.; Kwon, J. Roles of human INO80 chromatin remodeling enzyme in DNA replication and chromosome segregation suppress genome instability. Cell. Mol. Life Sci. 2010, 67, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Sorek, R.; Shomron, N.; Ast, G. The birth of an alternatively spliced exon: 3’ Splice-site selection in Alu exons. Science 2003, 300, 1288–1291. [Google Scholar] [CrossRef] [PubMed]
- Lev-Maor, G.; Ram, O.; Kim, E.; Sela, N.; Goren, A.; Levanon, E.Y.; Ast, G. Intronic Alus influence alternative splicing. PLoS Genet. 2008, 4, e1000204. [Google Scholar] [CrossRef] [PubMed]
- Sela, N.; Mersch, B.; Hotz-Wagenblatt, A.; Ast, G. Characteristics of transposable element exonization within human and mouse. PLoS ONE 2010, 5, e10907. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Ast, G.; Graur, D. Alu-containing exons are alternatively spliced. Genome Res. 2002, 12, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Allton, K.; Duncan, S.A.; Shen, L.; Barton, M.C. Foxa1 functions as a pioneer transcription factor at transposable elements to activate Afp during differentiation of embryonic stem cells. J. Biol. Chem. 2010, 285, 16135–16144. [Google Scholar] [CrossRef] [PubMed]
- Javier Piedrafita, F.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar]
- Hambor, J.E.; Mennone, J.; Coon, M.E.; Hanke, J.H.; Kavathas, P. Identification and characterization of an Alu-containing, T-cell-specific enhancer located in the last intron of the human CD8α gene. Mol. Cell. Biol. 1993, 13, 7056–7070. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Sirotin, M.V.; Zhurkin, V.B. Impact of Alu repeats on the evolution of human p53 binding sites. Biol. Direct 2011, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Vansant, G.; Reynolds, W.F. The consensus sequence of a major Alu subfamily contains a functional retinoic acid response element. Proc. Natl. Acad. Sci. USA 1995, 92, 8229–8233. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.B.; Wang, H.Y.; Lu, H.Y.; Xiong, J.; Li, H.H.; Qiu, X.H.; Liu, H.Q. Increased level of polymerase III transcribed Alu RNA in hepatocellular carcinoma tissue. Mol. Carcinog. 2005, 42, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.; Jones, A.E.; D’Brot, A.; Lu, W.J.; Kurtz, P.; Moran, J.V.; Rakheja, D.; Chen, K.S.; Hammer, R.E.; Comerford, S.A.; et al. p53 genes function to restrain mobile elements. Genes Dev. 2016, 30, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Van Bortle, K.; Corces, V.G. The role of chromatin insulators in nuclear architecture and genome function. Curr. Opin. Genet. Dev. 2013, 23, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Farivar, S.; Mager, D.L. C-GATE—Catalogue of genes affected by transposable elements. Mob. DNA 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Valles, I.; Pajares, M.J.; Segura, V.; Guruceaga, E.; Gomez-Roman, J.; Blanco, D.; Tamura, A.; Montuenga, L.M.; Pio, R. Identification of novel deregulated RNA metabolism-related genes in non-small cell lung cancer. PLoS ONE 2012, 7, e42086. [Google Scholar] [CrossRef] [PubMed]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered adenosine-to-inosine RNA editing in human cancer Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Chang, J.W.O.; Park, J.K.U.; Hwang, S.G. Increased aldehyde reductase expression mediates acquired radioresistance of laryngeal cancer cells via modulating p53. Cancer Biol. Ther. 2012, 13, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, M.; Zhong, D.; Tong, N.; Chu, H.; Sheng, X.; Zhang, Z. ADH1C Ile350Val polymorphism and cancer risk: Evidence from 35 case-control studies. PLoS ONE 2012, 7, e37227. [Google Scholar] [CrossRef] [PubMed]
- Zamanian-Daryoush, M.; di Donato, J.A. Apolipoprotein A–I and cancer. Front. Pharmacol. 2015, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Flor, I.; Bullerdiek, J. The dark side of a success story: MicroRNAs of the C19MC cluster in human tumours. J. Pathol. 2012, 227, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Bekku, S.; Mochizuki, H.; Yamamoto, T.; Ueno, H.; Takayama, E.; Tadakuma, T. Expression of carbonic anhydrase I or II and correlation to clinical aspects of colorectal cancer. Hepatogastroenterology 2000, 47, 998–1001. [Google Scholar] [PubMed]
- Dorr, C.; Janik, C.; Weg, M.; Been, R.A.; Bader, J.; Kang, R.; Ng, B.; Foran, L.; Landman, S.R.; O’Sullivan, M.G.; et al. Transposon mutagenesis screen identifies potential lung cancer drivers and CUL3 as a tumor suppressor. Mol. Cancer Res. 2015, 13, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Jair, K.W.; Yen, R.W.; Lengauer, C.; Herman, J.G.; Kinzler, K.W.; Vogelstein, B.; Baylin, S.B.; Schuebel, K.E. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 2000, 404, 1003–1007. [Google Scholar] [PubMed]
- McAtee, C.O.; Barycki, J.J.; Simpson, M.A. Emerging roles for hyaluronidase in cancer metastasis and therapy. Adv. Cancer Res. 2014, 123, 1–34. [Google Scholar] [PubMed]
- Zhang, Y.; Romanish, M.T.; Mager, D.L. Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput. Biol. 2011, 7, e1002046. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Takimoto, K. Selective expression of HERG and Kv2 channels influences proliferation of uterine cancer cells. Int. J. Oncol. 2004, 25, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Romanish, M.T.; Nakamura, H.; Lai, B.C.; Wang, Y.; Mager, D.L. A novel protein isoform of the multicopy human NAIP gene derives from intragenic Alu SINE promoters. PLoS ONE 2009, 4, e5761. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Deregulation of RB1 expression by loss of imprinting in human hepatocellular carcinoma. J. Pathol. 2014, 233, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, E.; Sugioka-Sugiyama, R.; Mizuguchi, T.; Mehta, S.; Cui, B.; Grewal, S.I.S. A homolog of male sex-determining factor SRY cooperates with a transposon-derived CENP-B protein to control sex-specific directed recombination. Proc. Natl. Acad. Sci. USA 2011, 108, 18754–18759. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Watanabe, Y.; Inoue, J.-I.; Semba, K. Transcriptional activity of testis-determining factor SRY is modulated by the Wilms’ tumor 1 gene product, WT1. Oncogene 2003, 22, 7900–7904. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, M.; Luostari, K.; Tengstrom, M.; Ahonen, H.; Berdel, B.; Kataja, V.; Soini, Y.; Kosma, V.-M.; Mannermaa, A. Low expression levels of hepsin and TMPRSS3 are associated with poor breast cancer survival. BMC Cancer 2015, 15, 431. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, K.; Wang, Z.; Bachvarova, M.; Gregoire, J.; Renaud, M.C.; Plante, M.; Bachvarov, D. A novel genome-based approach correlates TMPRSS3 overexpression in ovarian cancer with DNA hypomethylation. Gynecol. Oncol. 2012, 125, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Cappadocia, L.; Pichler, A.; Lima, C.D. Structural basis for catalytic activation by the human ZNF451 SUMO E3 ligase. Nat. Struct. Mol. Biol. 2015, 22, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Rebollo, R.; Karimi, M.M.; Bilenky, M.; Gagnier, L.; Miceli-Royer, K.; Zhang, Y.; Goyal, P.; Keane, T.M.; Jones, S.; Hirst, M.; et al. Retrotransposon-induced heterochromatin spreading in the mouse revealed by insertional polymorphisms. PLoS Genet. 2011, 7, e1002301. [Google Scholar] [CrossRef] [PubMed]
- Mateo, L.; Ullastres, A.; González, J. A Transposable element insertion confers xenobiotic resistance in Drosophila. PLoS Genet. 2014, 10, e1004560. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Andersen, L.B.; Saulino, A.M.; Gregory, P.E.; Glover, T.W.; Collins, F.S. A de novo Alu insertion results in neurofibromatosis type. Nature 1991, 353, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Stenson, P.D.; Cooper, D.N.; Férec, C. A systematic analysis of LINE-1 endonuclease-dependent retrotranspositional events causing human genetic disease. Hum. Genet. 2005, 117, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Edwards, A.; Fan, W.; Deininger, P.; Zhang, K. Alu distribution and mutation types of cancer genes. BMC Genom. 2011, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Narayanan, V.; Lobachev, K.S.; Mirkin, S.M. Replication stalling at unstable inverted repeats: Interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 9936–9941. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, G.; Bacolla, A.; Zhao, J.; Spitser, S.; Vasquez, K.M. Short inverted repeats are hotspots for genetic instability: Relevance to cancer genomes. Cell Rep. 2015, 10, 1674–1680. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.S.; Stenger, J.E.; Kozyreva, O.G.; Jurka, J.; Gordenin, D.A.; Resnick, M.A. Inverted Alu repeats unstable in yeast are excluded from the human genome. EMBO J. 2000, 19, 3822–3830. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Hedges, D.J.; Herke, S.W.; Batzer, M.A. Estimating the retrotransposition rate of human Alu elements. Gene 2006, 373, 134–137. [Google Scholar] [CrossRef] [PubMed]
- De Biase, I.; Rasmussen, A.; Monticelli, A.; Al-Mahdawi, S.; Pook, M.; Cocozza, S.; Bidichandani, S.I. Somatic instability of the expanded GAA triplet-repeat sequence in Friedreich ataxia progresses throughout life. Genomics 2007, 90, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Carnevali, D.; Bollati, V.; Fustinoni, S.; Pellegrini, M.; Dieci, G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015, 43, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [PubMed]
- Lissens, W.; Teugels, E.; Brakeleer, S.D.; Gr, J.D. Systematic detection of pathogenic Alu element insertions in NGS-based diagnostic screens: The BRCA1/BRCA2 example. Hum. Mutat. 2013, 34, 785–791. [Google Scholar]
- Halling, K.C.; Lazzaro, C.R.; Honchel, R.; Bufill, J.A.; Powell, S.M.; Arndt, C.A.; Lindor, N.M. Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum. Hered. 1999, 49, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Belgnaoui, S.M.; Gosden, R.G.; Semmes, O.J.; Haoudi, A. Human LINE-1 retrotransposon induces DNA damage and apoptosis in cancer cells. Cancer Cell Int. 2006, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Gasior, S.L.; Wakeman, T.P.; Xu, B.; Deininger, P.L. The human LINE-1 retrotransposon creates DNA double-strand breaks. J. Mol. Biol. 2006, 357, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Tan, F.J.; Lai, D.C.; Celniker, S.E.; Hoskins, R.A.; Dunham, M.J.; Zheng, Y.; Koshland, D. Competitive repair by naturally dispersed repetitive DNA during non-allelic homologous recombination. PLoS Genet. 2010, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Luzhna, L.; Ilnytskyy, Y.; Kovalchuk, O. Mobilization of LINE-1 in irradiated mammary gland tissue may potentially contribute to low dose radiation-induced genomic instability. Genes Cancer 2015, 6, 71–81. [Google Scholar] [PubMed]
- Walsh, T.; Casadei, S.; Coats, K.H.; Swisher, E.; Stray, S.M.; Higgins, J.; Roach, K.C.; Mandell, J.; Lee, M.K.; Ciernikova, S.; et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006, 295, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, I.; Salomon, D.; Erne, B.; Schaeren-Wiemers, N.; Peles, E. Caspr3 and caspr4, two novel members of the caspr family are expressed in the nervous system and interact with PDZ domains. Mol. Cell. Neurosci. 2002, 20, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Huzarski, T.; Byrski, T.; Gronwald, J.; Górski, B.; Debniak, T.; Masojć, B.; Jakubowska, A.; Gliniewicz, B.; et al. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J. Med. Genet. 2006, 43, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.; Reynolds, L.E.; Robinson, S.D.; Lees, D.M.; Parsons, M.; Elia, G.; Hodivala-Dilke, K. Stromal claudin14-heterozygosity, but not deletion, increases tumour blood leakage without affecting tumour growth. PLoS ONE 2013, 8, e62516. [Google Scholar] [CrossRef] [PubMed]
- Knower, K.C.; To, S.Q.; Simpson, E.R.; Clyne, C.D. Epigenetic mechanisms regulating CYP19 transcription in human breast adipose fibroblasts. Mol. Cell. Endocrinol. 2010, 321, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, N.; Bastid, J.; Alberici, G.; Bensussan, A.; Eliaou, J.-F. CD39: A complementary target to immune checkpoints to counteract tumor-mediated immunosuppression. Oncoimmunology 2015, 4, e1003015. [Google Scholar] [CrossRef] [PubMed]
- Padró, M.; Cobler, L.; Garrido, M.; De Bolós, C. Down-regulation of FUT3 and FUT5 by shRNA alters Lewis antigens expression and reduces the adhesion capacities of gastric cancer cells. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Hankinson, S.E.; Colditz, G.A.; Hunter, D.J.; De Vivo, I. HSD17B1 gene polymorphisms and risk of endometrial and breast cancer. Cancer Epidemiol. Biomark. Prev. 2004, 13, 213–219. [Google Scholar] [CrossRef]
- Ihle, J.N.; Smith-White, B.; Sisson, B.; Parker, D.; Blair, D.G.; Schultz, A.; Kozak, C.; Lunsford, R.D.; Askew, D.; Weinstein, Y.; et al. Activation of the c-H-ras proto-oncogene by retrovirus insertion and chromosomal rearrangement in a Moloney leukemia virus-induced T-cell leukemia. J. Virol. 1989, 63, 2959–2966. [Google Scholar] [PubMed]
- Li, L.; McVety, S.; Younan, R.; Liang, P.; Du Sart, D.; Gordon, P.H.; Hutter, P.; Hogervorst, F.B.L.; Chong, G.; Foulkes, W.D. Distinct patterns of germ-line deletions in MLH1 and MSH2: The implication of Alu repetitive element in the genetic etiology of Lynch syndrome (HNPCC). Hum. Mutat. 2006, 27, 388. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Yamazaki, T. Nucleic acids research. Nucleic Acids Res. 1989, 17, 1–2. [Google Scholar]
- Katsanis, N.; Beales, P.L.; Woods, M.O.; Lewis, R.A.; Green, J.S.; Parfrey, P.S.; Ansley, S.J.; Davidson, W.S.; Lupski, J.R. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 67–70. [Google Scholar] [PubMed]
- Morse, B.; Rotherg, P.G.; South, V.J.; Spandorfer, J.M.; Astrin, S.M. Insertional mutagenesis of the myc locus by a LINE-1 sequence in a human breast carcinoma. Nature 1988, 333, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Pastan, I.; Hassan, R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014, 74, 2907–2912. [Google Scholar] [CrossRef] [PubMed]
- Rodić, N.; Zampella, J.G.; Cornish, T.C.; Wheelan, S.J.; Burns, K.H. Translocation junctions in TCF3-PBX1 acute lymphoblastic leukemia/lymphoma cluster near transposable elements. Mob. DNA 2013, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; He, Z.; Zhou, K.; Cheng, J.; Yao, H.; Lu, D.; Cai, R.; Jin, Y.; Dong, B.; Xu, Y.; Wang, Y. Essential role of trpc6 channels in G2/M phase transition and development of human glioma. J. Natl. Cancer Inst. 2010, 102, 1052–1068. [Google Scholar] [CrossRef] [PubMed]
- Elliott, B.; Richardson, C.; Jasin, M. Chromosomal translocation mechanisms at intronic Alu elements in mammalian cells. Mol. Cell 2005, 17, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.-Y.; Chen, W.-Y.; Yeh, T.-S.; Sheen, I.-S.; Huang, S.-F. High-frequency Alu-mediated genomic recombination/deletion within the caspase-activated DNase gene in human hepatoma. Oncogene 2005, 24, 6584–6589. [Google Scholar] [CrossRef] [PubMed]
- Morrish, T.A.; Gilbert, N.; Myers, J.S.; Vincent, B.J.; Stamato, T.D.; Taccioli, G.E.; Batzer, M.A.; Moran, J.V. DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat. Genet. 2002, 31, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Liu, G.; Eichler, E.E. An Alu transposition model for the origin and expansion of human segmental duplications. Am. J. Hum. Genet. 2003, 73, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Kidd, J.M.; Marques-Bonet, T.; Aksay, G.; Antonacci, F.; Hormozdiari, F.; Kitzman, J.O.; Baker, C.; Malig, M.; Mutlu, O.; et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009, 41, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Jeffs, A.R.; Benjes, S.M.; Smith, T.L.; Sowerby, S.J.; Morris, C.M. The BCR gene recombines preferentially with Alu elements in complex BCR-ABL translocations of chronic myeloid leukaemia. Hum. Mol. Genet. 1998, 7, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Montagna, M.; Santacatterina, M.; Torri, A.; Menin, C.; Zullato, D.; Chieco-Bianchi, L.; D’andrea, E. Identification of a 3 kb Alu-mediated BRCA1 gene rearrangement in two breast/ovarian cancer families. Oncogene 1999, 18, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Sluiter, M.D.; van Rensburg, E.J. Large genomic rearrangements of the BRCA1 and BRCA2 genes: Review of the literature and report of a novel BRCA1 mutation. Breast Cancer Res. Treat. 2011, 125, 325–349. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, X.; Liu, T.; Liu, X.; Geng, J.; He, X.; Liu, Y.; Pang, D. Increased expression of mitotic arrest deficient-like 1 (MAD1L1) is associated with poor prognosis and insensitive to taxol treatment in breast cancer. Breast Cancer Res. Treat. 2013, 140, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Schichman, S.A.; Caligiuri, M.A.; Strout, M.P.; Carter, S.L.; Gu, Y.; Canaani, E.; Bloomfield, C.D.; Croce, C.M. ALL-1 Tandem duplication in acute myeloid leukemia with a normal karyotype involves homologous recombination between Alu elements. Cancer Res. 1994, 54, 4277–4280. [Google Scholar] [PubMed]
- O’Neil, J.; Tchinda, J.; Gutierrez, A.; Moreau, L.; Maser, R.S.; Wong, K.-K.; Li, W.; McKenna, K.; Liu, X.S.; Feng, B.; et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J. Exp. Med. 2007, 204, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Franke, G.; Bausch, B.; Hoffmann, M.M.; Cybulla, M.; Wilhelm, C.; Kohlhase, J.; Scherer, G.; Neumann, H.P.H. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: Molecular characterization and genotype-phenotype correlations in VHL patients. Hum. Mutat. 2009, 30, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Lin, L.; Cai, J.J.; Jiang, P.; Kenkel, E.J.; Stroik, M.R.; Sato, S.; Davidson, B.L.; Xing, Y. Widespread establishment and regulatory impact of Alu exons in human genes. Proc. Natl. Acad. Sci. USA 2011, 108, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- Strout, M.P.; Marcucci, G.; Bloomfield, C.D.; Caligiuri, M.A. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 1998, 95, 2390–2395. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhao, H.; Yi, Y.; Nakata, Y.; Kalota, A.; Gewirtz, A.M. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Investig. 2010, 120, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Mariño-Ramírez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef] [PubMed]
- Borchert, G.M.; Holton, N.W.; Williams, J.D.; Hernan, W.L.; Bishop, I.P.; Dembosky, J.A.; Elste, J.E.; Gregoire, N.S.; Kim, J.-A.; Koehler, W.W.; et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob. Genet. Elements 2011, 1, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.T.; Cooper, E.A.; Favreau, C.J.; Howell, J.S.; Lane, L.G.; Mills, J.E.; Newman, D.C.; Perry, T.J.; Russell, M.E.; Wallace, B.M.; et al. Continuing analysis of microRNA origins: Formation from transposable element insertions and noncoding RNA mutations. Mob. Genet. Elements 2013, 3, e27755. [Google Scholar] [CrossRef] [PubMed]
- Piriyapongsa, J.; Jordan, I.K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS ONE 2007, 2, e203. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, R.; Almeida, M.I.; Colombatti, A.; Calin, G.A. Long non-coding RNAs and cancer: A new frontier of translational research? Oncogene 2012, 31, 4577–4587. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, S.; Diederichs, S. From junk to master regulators of invasion: LncRNA functions in migration, EMT and metastasis. Int. J. Cancer 2016, 139, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Rinn, J.L. Transposable elements reveal a stem cell specific class of long noncoding RNAs. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Hadjiargyrou, M.; Delihas, N. The intertwining of transposable elements and non-coding RNAs. Int. J. Mol. Sci. 2013, 14, 13307–13328. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.L.; Krech, T.; Hasemeier, B.; Schipper, E.; Schweitzer, N.; Vogel, A.; Kreipe, H.; Lehmann, U. Loss of imprinting and allelic switching at the DLK1-MEG3 locus in human hepatocellular carcinoma. PLoS ONE 2012, 7, e49462. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Khanduja, J.S.; Calvo, I.A.; Joh, R.I.; Hill, I.T.; Motamedi, M. Nuclear noncoding RNAs and genome stability. Mol. Cell 2016, 63, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Villeneuve, L.; Morris, K.; Rossi, J. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat. Struct. Mol. Biol. 2006, 13, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, Y.; Pilpel, Y.; Oren, M. MicroRNAs and Alu elements in the p53–Mdm2–Mdm4 regulatory network. J. Mol. Cell Biol. 2014, 6, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Elkahloun, A.G.; Candotti, F.; Grajkowski, A.; Beaucage, S.L.; Petricoin, E.F.; Calvert, V.; Juhl, H.; Mills, F.; Mason, K.; et al. A novel function of RNAs arising from the long terminal repeat of human endogenous retrovirus 9 in cell cycle arrest. J. Virol. 2013, 87, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ponicsan, S.L.; Kugel, J.F.; Goodrich, J.A. Genomic gems: SINE RNAs regulate mRNA production. Curr. Opin. Genet. Dev. 2010, 20, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, B.; Lopez, M.F.; Wang, J.; Krastins, B.; Sarracino, D.; Tollervey, J.R.; Dobke, M.; Jordan, I.K.; Lunyak, V.V. Protein interactions with piALU RNA indicates putative participation of retroRNA in the cell cycle, DNA repair and chromatin assembly. Mob. Genet. Elements 2012, 2, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bamezai, S.; Rawat, V.P.S.; Buske, C. Concise review: The PIWI–piRNA axis: Pivotal beyond transposon silencing. Stem Cells 2012, 30, 2603–2611. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Tóth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013, 27, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Simonelig, M. Developmental functions of piRNAs and transposable elements: A Drosophila point-of-view. RNA Biol. 2011, 8, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.D.; Hannon, G.J. Small RNAs as guardians of the genome. Cell 2009, 136, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.J.; Toyoda, A.; et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332, 848–852. [Google Scholar] [CrossRef] [PubMed]
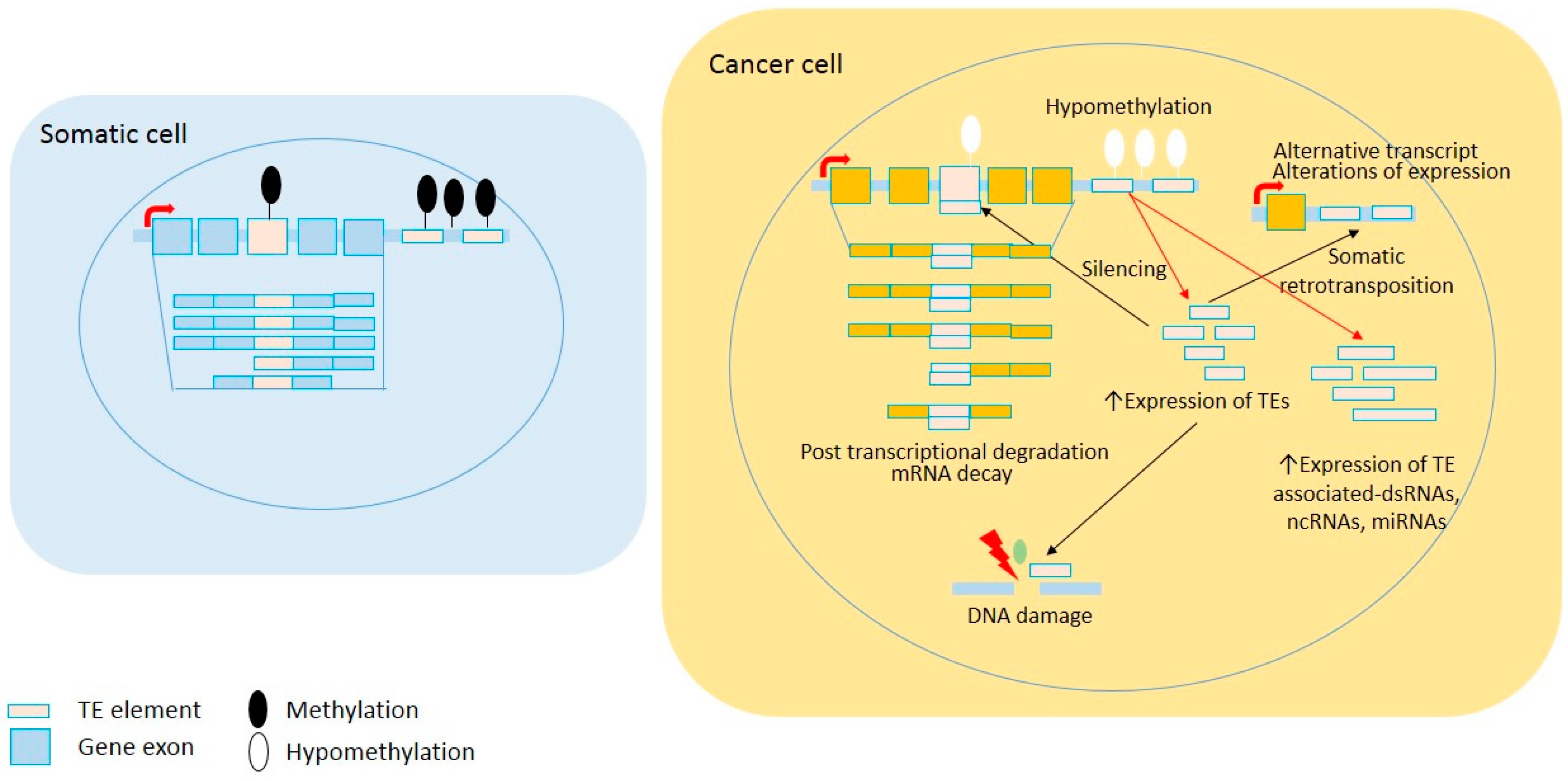
Figure 1.
Global loss of DNA methylation in cancer cells leads to TE reactivation. A common epigenetic feature in neoplastic cells is global demethylation, including within repeated sequences. Subsequently, TE reactivation can cause increasing somatic retrotransposition, non-coding RNA, and transcriptional deregulation. Red arrows show direct impacts of TE reactivation and black arrows show effects of retrotransposition.
Figure 1.
Global loss of DNA methylation in cancer cells leads to TE reactivation. A common epigenetic feature in neoplastic cells is global demethylation, including within repeated sequences. Subsequently, TE reactivation can cause increasing somatic retrotransposition, non-coding RNA, and transcriptional deregulation. Red arrows show direct impacts of TE reactivation and black arrows show effects of retrotransposition.
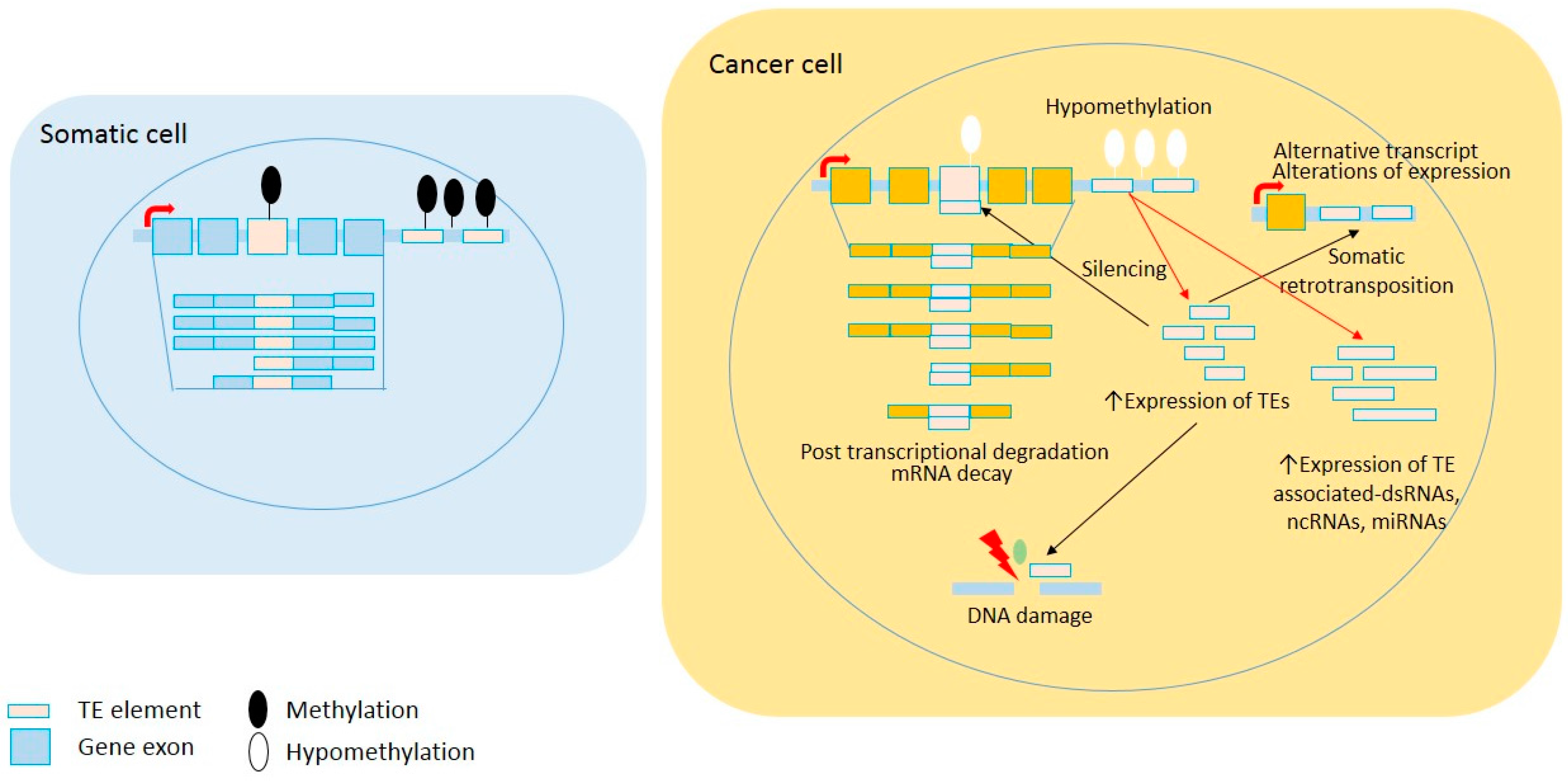
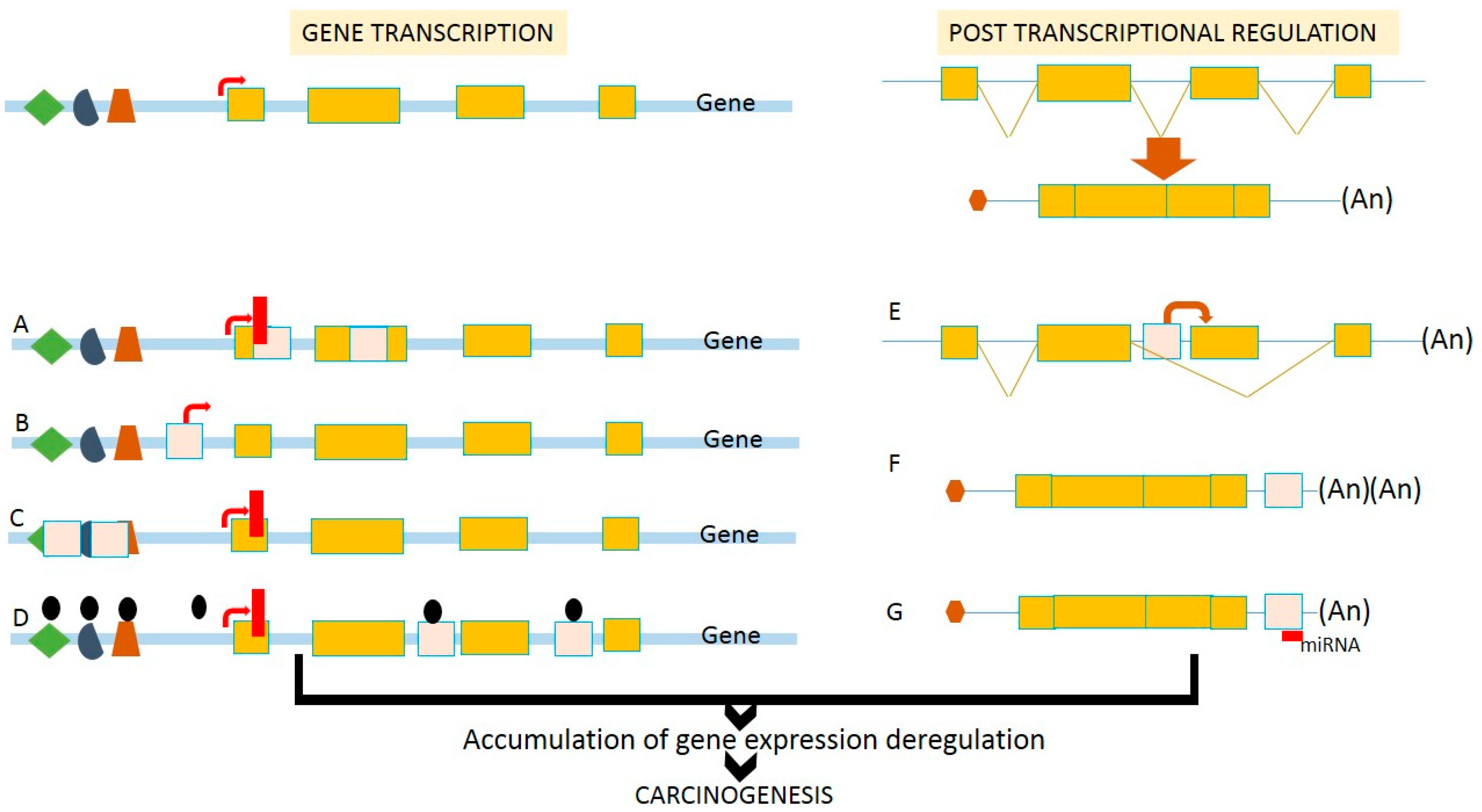
Figure 2.
Effects of retrotransposition on transcriptional deregulation. Insertion of TEs into (A) coding region can disturb or eliminate gene functions; (B) upstream of the gene loci can introduce a novel alternative promoter leading to the variation of protein products; (C) promoter region can disrupt cis-regulatory elements, as well as transcriptional start sites; (D) introns can introduce epigenetic remodeling events including DNA methylation and chromatin condensation, leading to gene silencing. At the post-transcriptional step, the introduction of TEs (E) in the introns can cause alternative splicing that causes various protein products and functions; (F) at the 3′UTR can introduce poly-adenylation sites leading to unstable mRNAs; and (G) at the 3′-UTR can create binding sites for miRNAs and other ncRNAs. Therefore, retrotransposition affects the efficiency of gene transcription and post-transcriptional regulation, and is associated with the deregulation of gene expression during human carcinogenesis.
Figure 2.
Effects of retrotransposition on transcriptional deregulation. Insertion of TEs into (A) coding region can disturb or eliminate gene functions; (B) upstream of the gene loci can introduce a novel alternative promoter leading to the variation of protein products; (C) promoter region can disrupt cis-regulatory elements, as well as transcriptional start sites; (D) introns can introduce epigenetic remodeling events including DNA methylation and chromatin condensation, leading to gene silencing. At the post-transcriptional step, the introduction of TEs (E) in the introns can cause alternative splicing that causes various protein products and functions; (F) at the 3′UTR can introduce poly-adenylation sites leading to unstable mRNAs; and (G) at the 3′-UTR can create binding sites for miRNAs and other ncRNAs. Therefore, retrotransposition affects the efficiency of gene transcription and post-transcriptional regulation, and is associated with the deregulation of gene expression during human carcinogenesis.

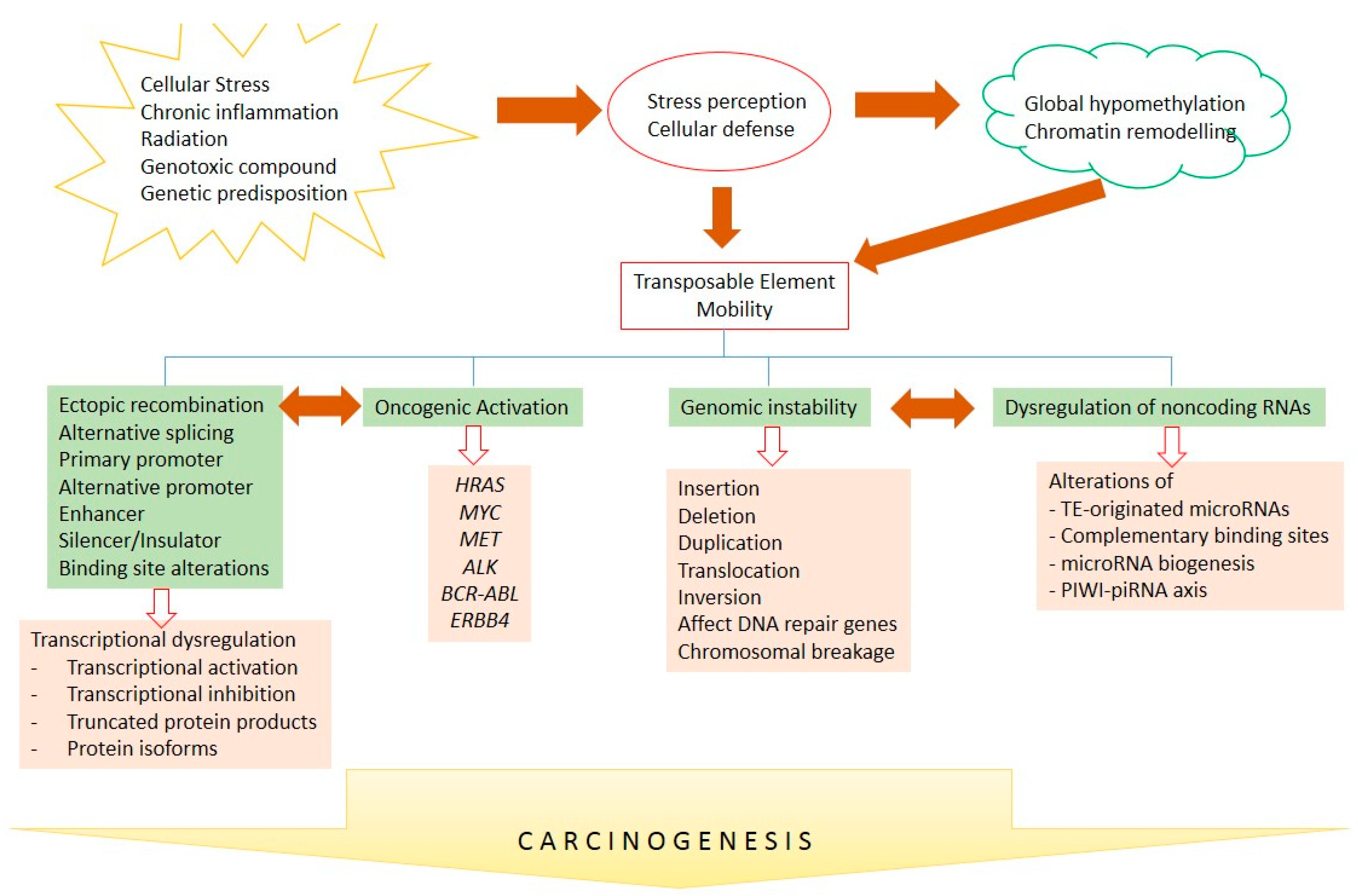
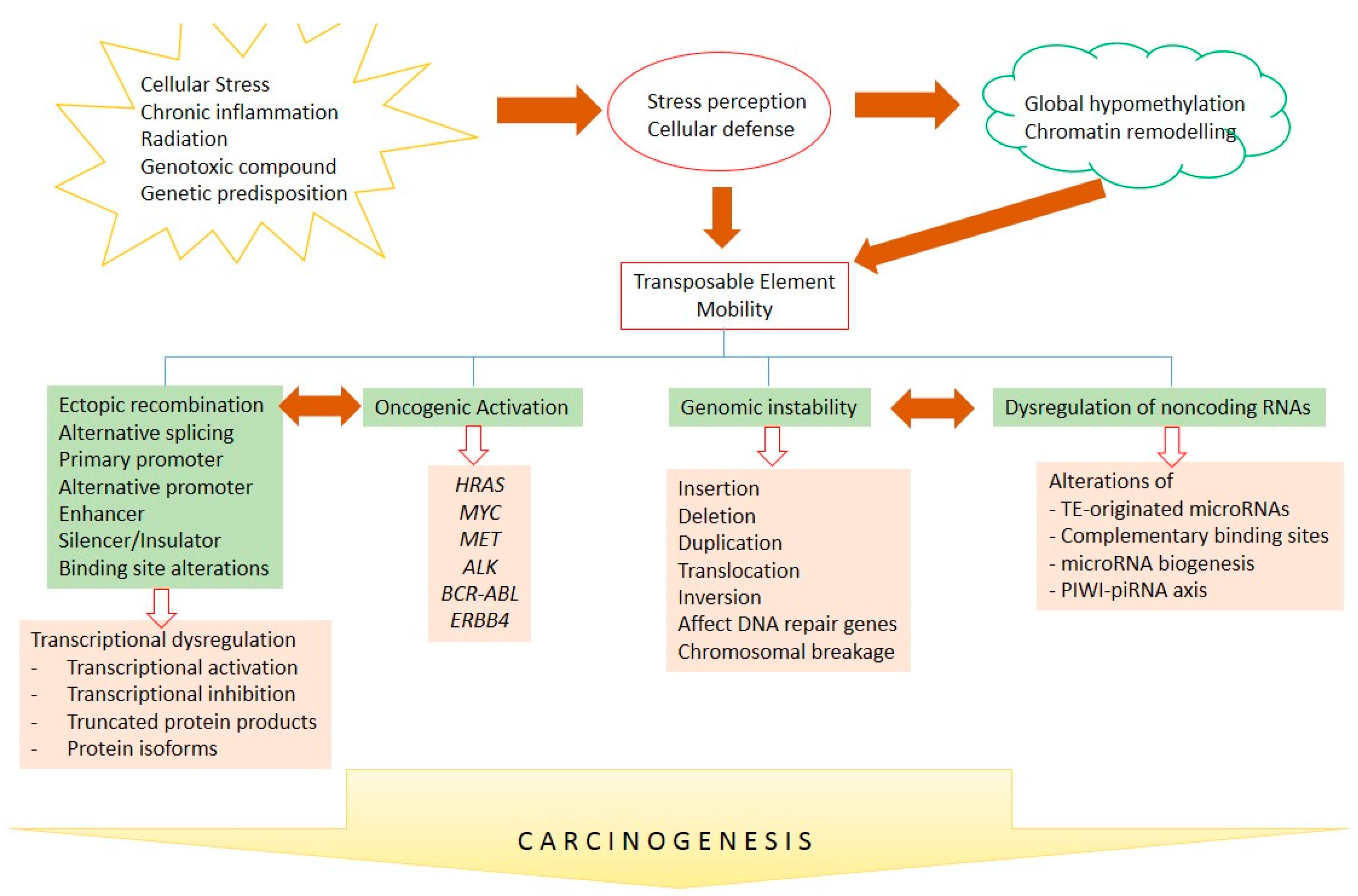
Figure 3.
TE-mediated carcinogenesis. Intra- and extracellular-mediated stresses lead to TE mobility through alterations of DNA methylation and chromatin remodeling. TE mobility might further induce and interconnect transcriptional deregulation, the activation of oncogenes, genomic instability, and ncRNA deregulation, to further contribute to human carcinogenesis. Arrows show causality and bidirectional arrows represent inter-correlation.
Figure 3.
TE-mediated carcinogenesis. Intra- and extracellular-mediated stresses lead to TE mobility through alterations of DNA methylation and chromatin remodeling. TE mobility might further induce and interconnect transcriptional deregulation, the activation of oncogenes, genomic instability, and ncRNA deregulation, to further contribute to human carcinogenesis. Arrows show causality and bidirectional arrows represent inter-correlation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
TE-associated transcriptional deregulation in human cancers.
| Locus or Genes | TE(s) | Mechanisms | Associated Cancers | References |
|---|---|---|---|---|
| ADARB1 | Alu | Alternative splicing | Lung, brain cancer | [65,66,67] |
| AKR1A1 | Alu | Alternative splicing | Head and neck cancer | [65,68] |
| ALS2CR8 | LINE1 | silencing | Colorectal cancer | [2] |
| ANKS1B_ | Alu | Deletion, silencing | Colorectal cancer | [2] |
| ANO9 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| ADH1C | LTR | Primary promoter | Cancers | [65,69] |
| ALK | LINE1, LTR | Alternative promoter | Melanoma, cancers | [17] |
| APOA | LINE1 | Enhancer | Cancers | [65,70] |
| APOC | LTR | Alternative promoter | Gastric cancer | [65] |
| ARNT | Alu | Alternative splicing | Lung cancer, metastasis of different cancers | [65] |
| ARHGEF12 | LINE1 | Alternative splicing | Ovarian cancer | [2] |
| ASMT | LINE1 | Alternative splicing | Glioma | [65] |
| B3GALNT2 | Alu | Alternative splicing | Breast cancer | [65] |
| B3GALT5 | Alu | Alternative promoter | Breast cancer | [65] |
| BAAT | LTR | Primary promoter | Lung cancer | [65] |
| BAHCC1 | Alu | Alternative splicing | Colorectal cancer | [65] |
| BBS7 | LINE1 | Primary promoter | Prostate cancer | [2] |
| BLVRA | Alu | Alternative splicing | Breast cancer | [65] |
| C19MC miRNA | Alu | POL III promoter | Hepatocellular, human cancers | [35,65,71] |
| CA1 | LTR | Primary promoter | Colorectal cancer | [8,72] |
| CASPR4 | LTR | Alternative promoter | Breast cancer, nasopharyngeal cancer | [65] |
| CD8A | LTR | Enhancer | Colorectal, pancreatic cancer | [65] |
| CDH12, CDH20 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| CHRM3 | LINE1 | Alternative promoter | Endometrial cancer | [65] |
| CHRNA1 | Alu | Alternative splicing | Testicular cancer | [65] |
| CLDN14 | LTR | Alternative promoter | Hepatocellular carcinoma | [65] |
| COL11A1, COL9A1 | LINE1 | Alternative splicing | Colorectal, prostate cancer | [2] |
| COX7B2 | LINE1 | Alternative splicing | Prostate cancer | [2] |
| CTNNA2 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| CWF19L1 | Alu | Alternative splicing | Breast cancer | [65] |
| CYB561D1 | Alu | Alternative splicing | Lung cancer | [65] |
| CYP19A1 | LTR | Alternative promoter | Breast cancer | [65] |
| CUL3 | LTR | Alternative splicing | Lung cancer | [73] |
| DAPK1 | ERV1 | Alternative splicing | Multiple myeloma | [2] |
| DBC1 | LINE1 | Deletion, Alternative promoter | Colorectal cancer | [2] |
| DHRS2 | LTR | Alternative promoter | Breast cancer | [65] |
| DNMT1 | Alu | Alternative splicing | Different cancers | [65,74] |
| DSCR4, DSCR8 | LTR | Primary promoter | Colorectal cancer | [65] |
| EPHA6 | LINE1 | Alternative transcript | Colorectal cancer | [2] |
| ERBB4 | LTR | Alternative promoter | Lymphoma, colorectal cancer | [2,19] |
| EBR | LTR | Alternative promoter | Bladder cancer | [65] |
| FABP7 | LTR | Alternative promoter | Lymphoma | [18] |
| FLT4/VEGFR3 | LTR | Alternative splicing | Different cancers | [65] |
| FMO1 | LINE1 | Silencer | Lung cancer | [65] |
| FOXP2 | LINE1 | Alternative promoter | Ovarian cancer | [2] |
| FUT5 | LINE1, Alu | Alternative splicing | Colorectal cancer | [65] |
| GABRG3 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| GBP5 | LTR | Primary promoter | Breast cancer | [65] |
| HHLA2, HHLA3 | LTR | Polyadenylation signal | Bladder cancer | [65] |
| HINFP/ZNF743 | Alu | Alternative splicing | Lung cancer | [65] |
| HYAL-4 | LINE1, Alu | Primary promoter | Cancers | [8,75] |
| IFNγ | Alu | Binding sites | Cancers | [65] |
| KCNH6 | Alu, LTR | Alternative splicing | Endometrial cancer | [76,77] |
| KDR | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| MCTP2 | LINE1 | Deletion, Alternative promoter | Colorectal cancer | [2] |
| MET | LINE1 | Alternative splicing | Bladder cancer | [16] |
| MKKS | LTR | Alternative promoter | Colorectal cancer | [65] |
| MSLN | LTR | Primary promoter | Pancreatic cancer | [65] |
| NAIP | Alu | Alternative promoter | Cancer | [78] |
| NFKBID | Alu | Alternative splicing | Colorectal cancer | [65] |
| NOS3, NOSIP | LR | Alternative promoter | Different cancers | [65] |
| NPAS3 | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| NRXN3 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| RB1 | LTR | Alternative promoter | Hepatocellular carcinoma, retinoblastoma | [14,79] |
| ROBO2 | LINE1 | Alternative splicing | Colorectal cancer | [2] |
| SLCC44A5, SLC35F1 | LINE1 | Alternative promoter, slencing | Colorectal cancer | [2] |
| SRY | LTR | Alternative transcript | Wilm’s tumor | [80,81] |
| STXBP5L | LINE1 | Alternative promoter | Colorectal cancer | [2] |
| TMED7 | Alu | Alternative promoter | Colorectal cancer | [2] |
| TMEM16J, TMEM56 | Alu | Alternative splicing, silencing | Colorectal cancer | [2] |
| TMPRSS3 | Alu, LTR | Alternative transcript | Breast, ovarian cancer | [8,82,83] |
| TP53 | Alu | Binding sites | Cancers, Pancreatic cancer | [58] |
| P63 | LTR | Primary promoter | Breast cancer | [65] |
| PDZK1 | Alu | Alternative splicing | Lung cancer | [65] |
| PODXL | Alu | Alternative splicing | Pancreatic cancer | [65] |
| PTN | LTR | Alternative promoter | Different cancers | [65] |
| ZNF451 | LTR | Alternative splicing | Hematological cancer | [9,84] |
| ZNF177, ZNF257, ZNF418 | LINE1, Alu | Alternative splicing | Different cancers | [65] |
Table 2.
TE insertion-associated loss or gain of a few base pairs and mutagenesis in human cancers.
| Locus or Genes | TE(s) | Associated Cancers | References |
|---|---|---|---|
| APC | Alu | Colon cancer | [13,99] |
| BRCA1 | Alu | Breast, ovarian cancer | [12,98,104] |
| BRCA2 | Alu | Breast, ovarian cancer | [12,98,104] |
| CASPR4 | LTR | Brain cancers | [8,105] |
| CHEK2 | Alu | Breast, ovarian, prostate cancer | [104,106] |
| CLDN14 | LTR | Melanoma | [8,107] |
| CYP19 | LTR | Breast cancer | [8,108] |
| ENTPD1 | LTR | Melanoma | [8,109] |
| FUT5 | LINE1, Alu | Gastric cancer | [8,110] |
| HSD17B1 | LTR | Breast, endometrial cancers | [111] |
| HRAS | LTR | T-cell leukemia | [112] |
| MLH1 | Alu | Colorectal cancer | [2,113] |
| MLVI2 | Alu | Leukemia | [114] |
| MKKS | LINE2, LTR | Embryonic cancers | [8,115] |
| MLH2 | Alu | Colorectal cancer | [113] |
| MYC | LINE1 | Breast cancer | [116] |
| NF1 | Alu | Neurofibromatosis type I | [87] |
| MSLN | LTR | Human cancers | [8,117] |
| RB1 | Alu, LTR | Retinoblastoma, hepatocellular cancer | [14,79] |
| TCF3-PBX1 | LTR | ALL | [118] |
| TRPC6 | LTR | Breast, prostate, gastric cancers, glioma | [76,119] |
Table 3.
TE-mediated chromosomal structure defects in human cancers.
| Locus of Genes | TE(s) | Chromosomal Defects | Associated Cancer | References |
|---|---|---|---|---|
| BCR-ABL | Alu | Chromosomal translocation | CML | [126] |
| BRCA1 | Alu | Chromosomal deletion, duplication, insertion | Breast, ovarian cancer | [127] |
| BRCA2 | Alu | Chromosomal deletion, duplication, insertion | Breast, ovarian cancer | [12,128] |
| CAD | Alu | Chromosomal deletion | Hepatocellular carcinoma | [122] |
| CDH1 | Alu | Chromosomal deletion | Diffuse gastric cancer | [129] |
| EWSR1-ETV | Alu, LINE1 | Chromosomal translocation | Ewing sarcoma | [28] |
| MAD1L1 | LTR | Chromosomal instability | Breast cancer | [8,130] |
| MLL1 | Alu | Chromosomal duplication | AML | [131] |
| MYB | Alu | Chromosomal duplication | T-ALL | [132] |
| VHL | Alu | Chromosomal deletion | von Hippel Lindau disease | [133] |
| WT1 | LINE1 | Chromosomal translocation | Sarcoma | [28] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Anwar, S.L.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. https://doi.org/10.3390/ijms18050974
AMA Style
Anwar SL, Wulaningsih W, Lehmann U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. International Journal of Molecular Sciences. 2017; 18(5):974. https://doi.org/10.3390/ijms18050974
Chicago/Turabian StyleAnwar, Sumadi Lukman, Wahyu Wulaningsih, and Ulrich Lehmann. 2017. "Transposable Elements in Human Cancer: Causes and Consequences of Deregulation" International Journal of Molecular Sciences 18, no. 5: 974. https://doi.org/10.3390/ijms18050974
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.