
Old Maids: Aging and Its Impact on Microglia Function

Abstract
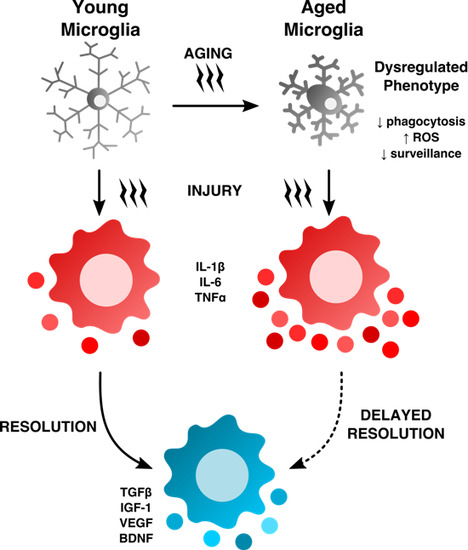
:
1. Introduction and Overview
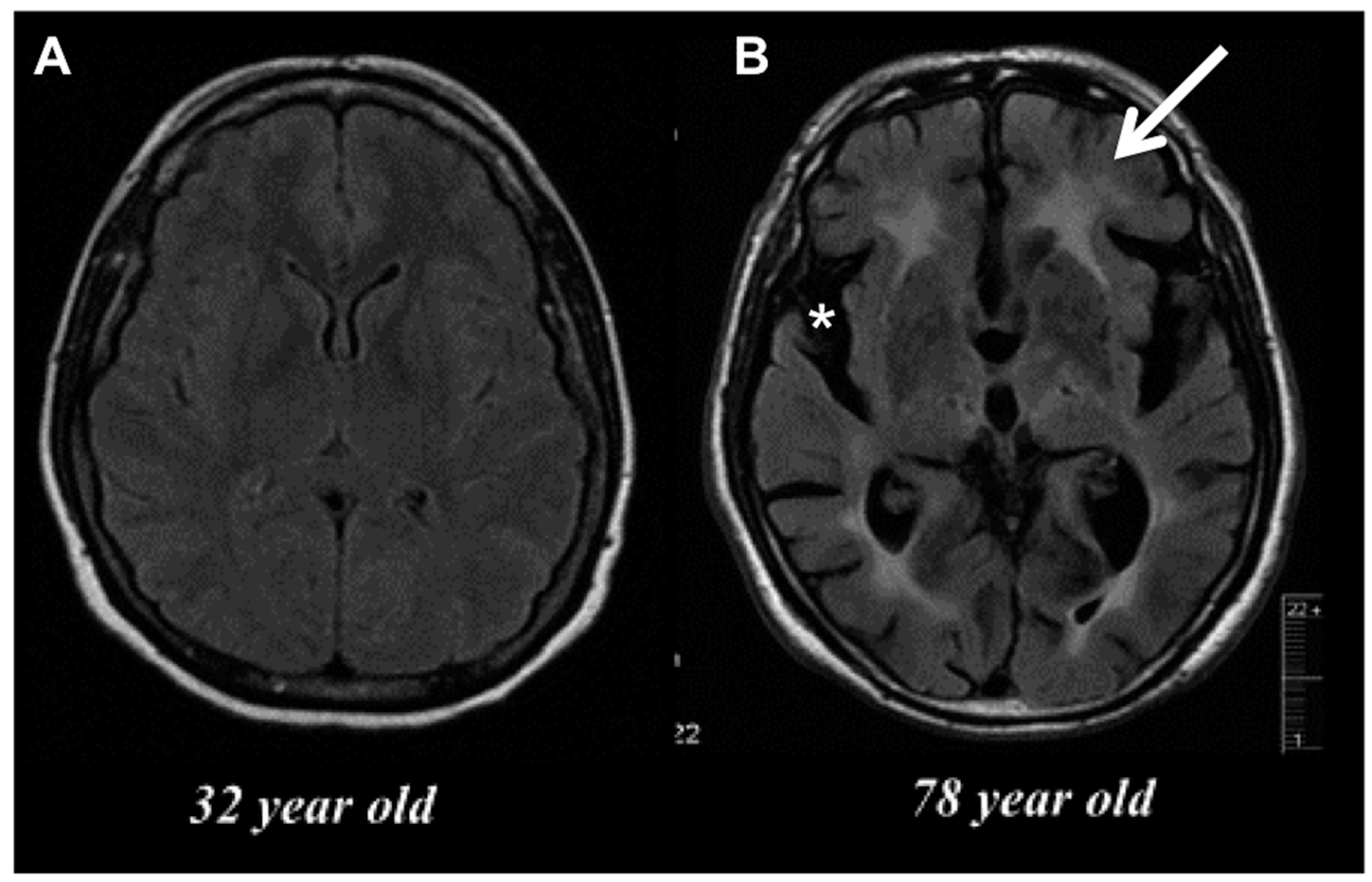
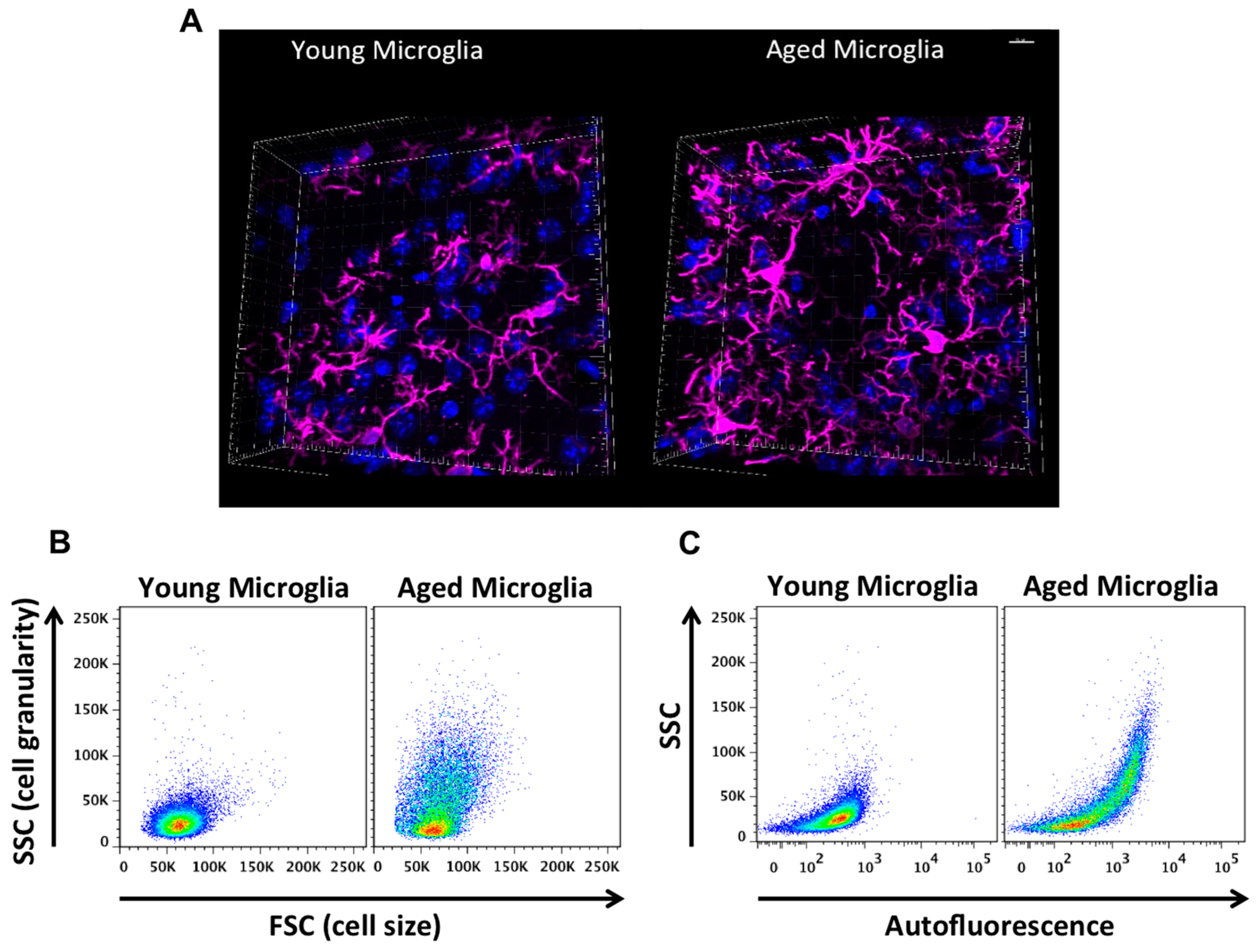
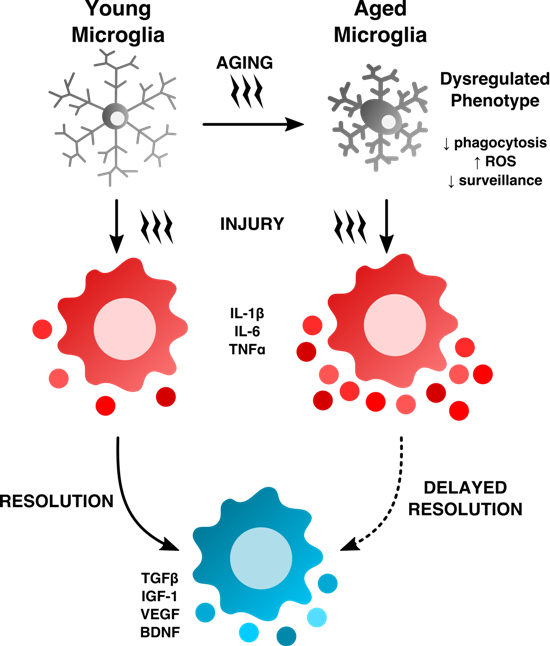
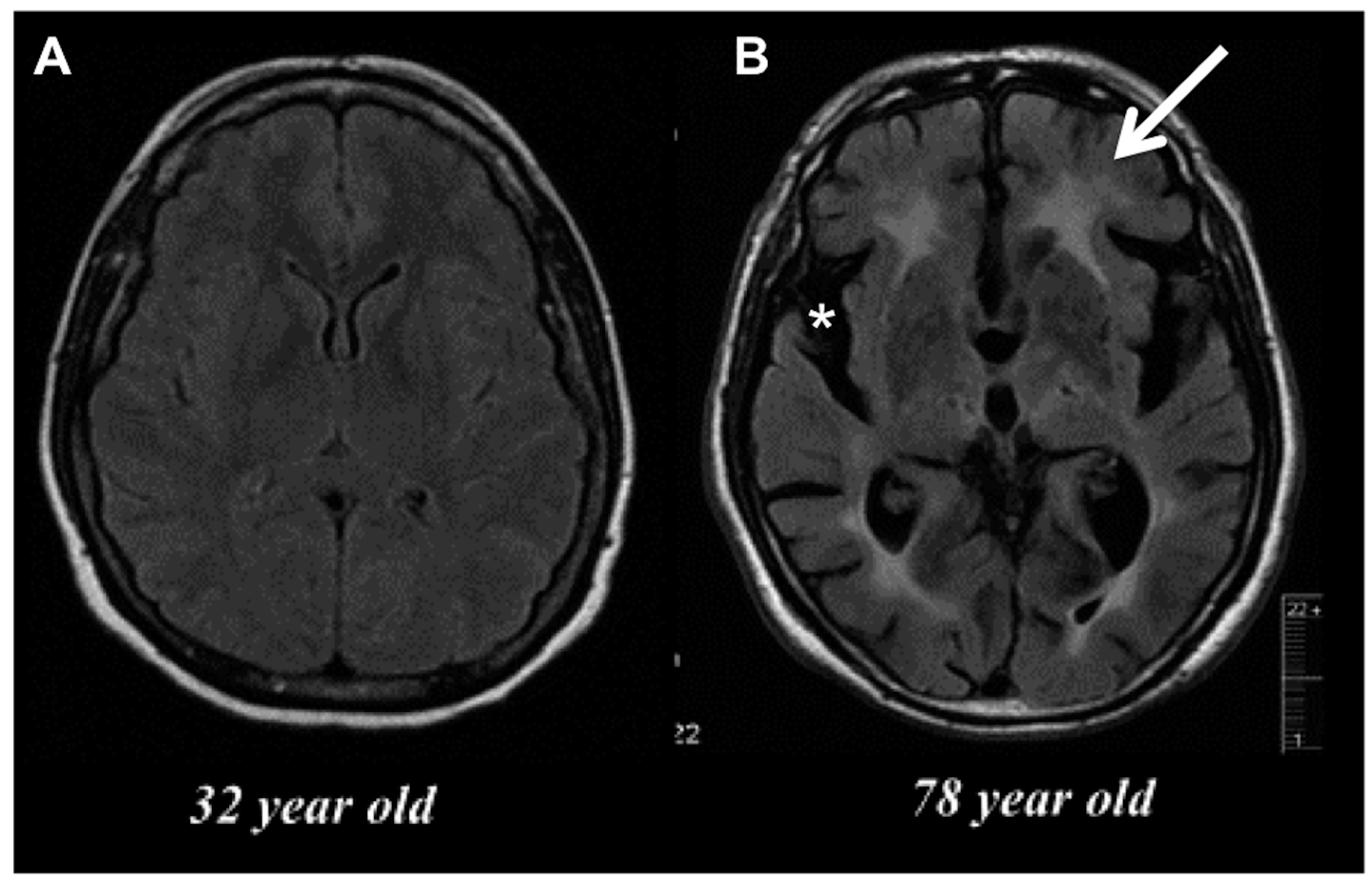
2. Age-Related Changes in Microglia Phenotype
3. Strategies to Investigate Functional Characteristics of Microglia
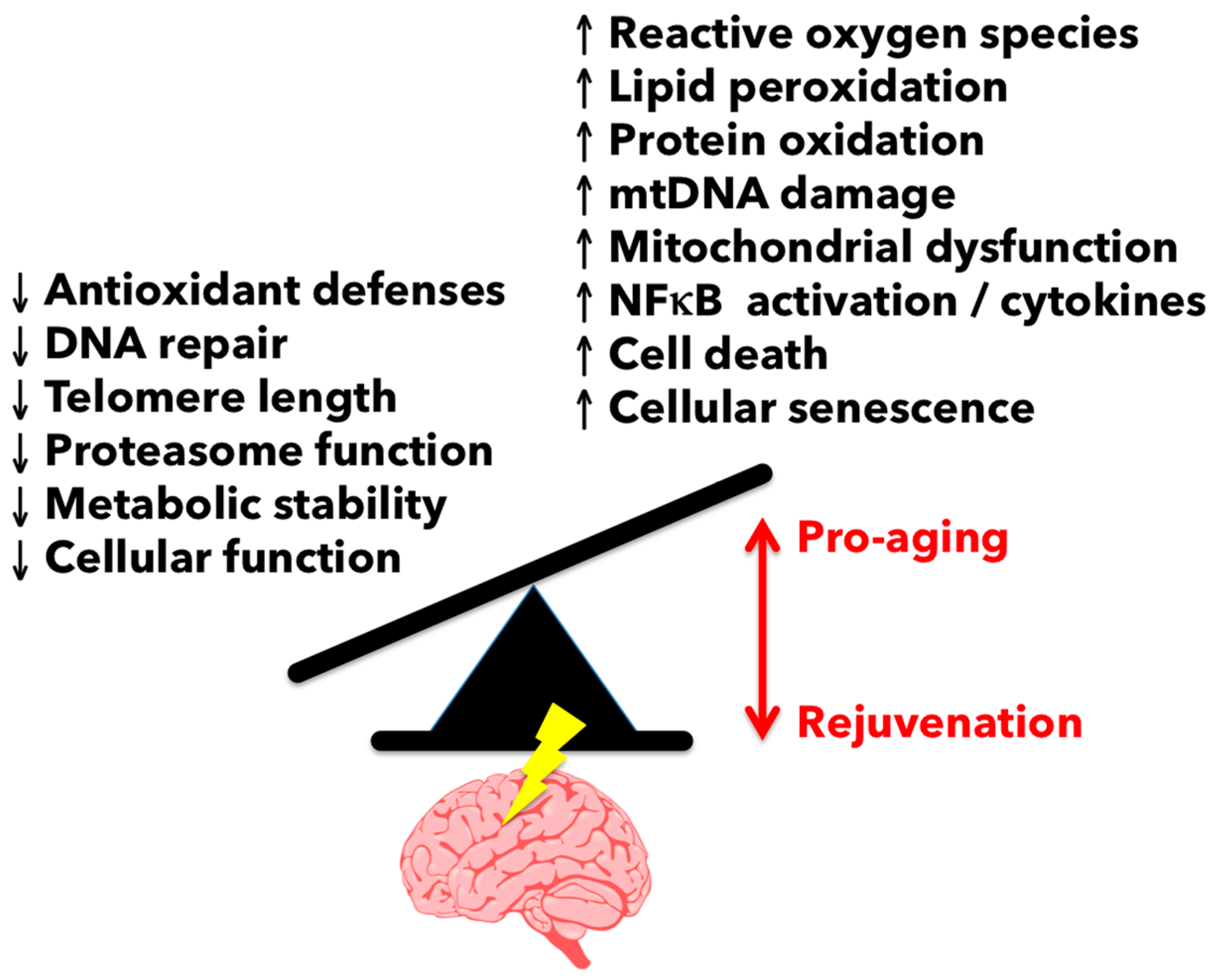
3.1. Reactive Oxygen Species-Mediated Damage in the Aging Brain
3.2. Neuronal–Glial Interactions and Immunoinhibitory Signaling in the Aging Brain
3.3. CD200/CD200R1
3.4. CX3CL1/CX3CR1
3.5. Phagocytosis in the Aging Brain
3.6. Microglial Depletion and Implications to Aging
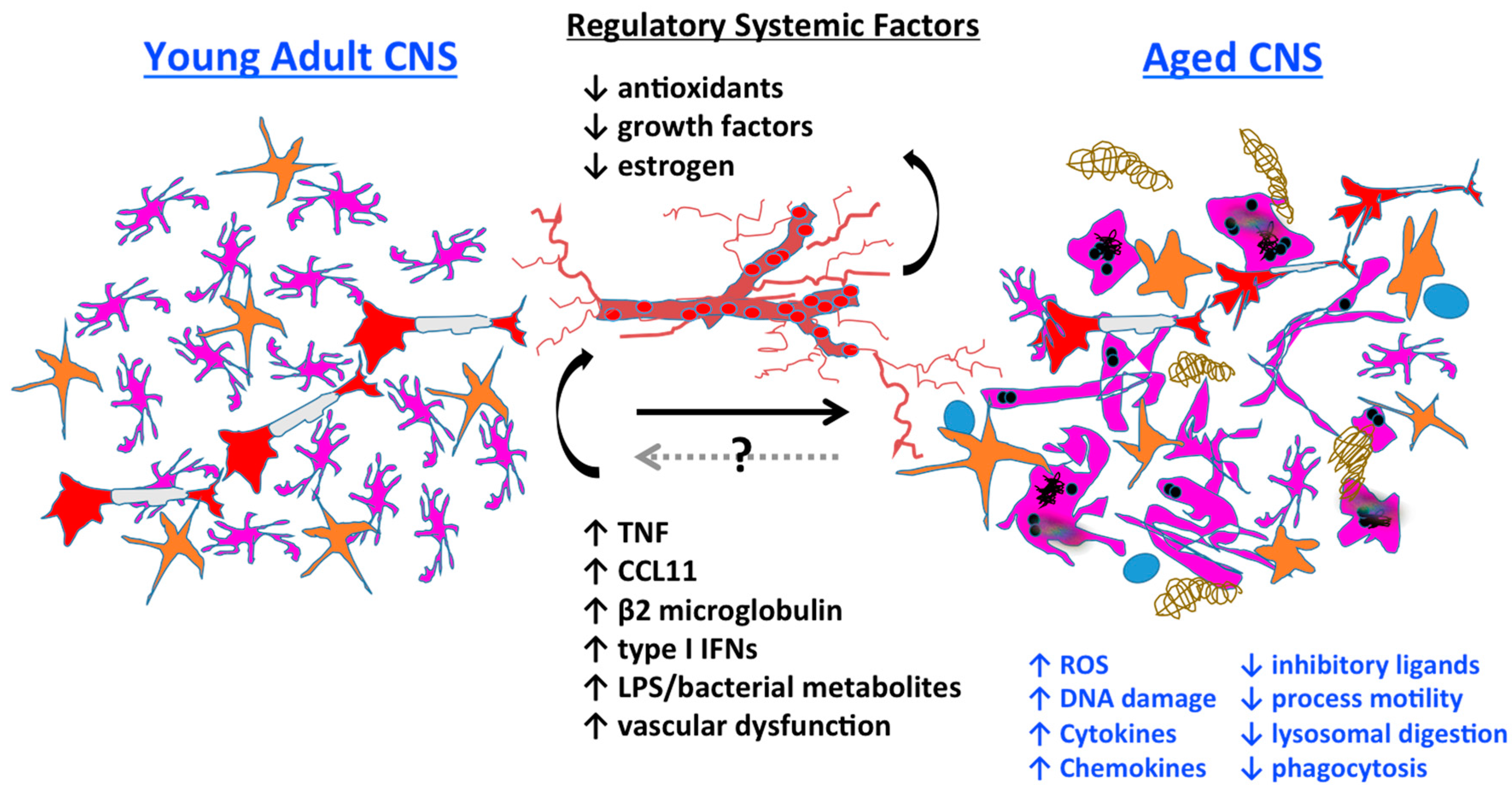
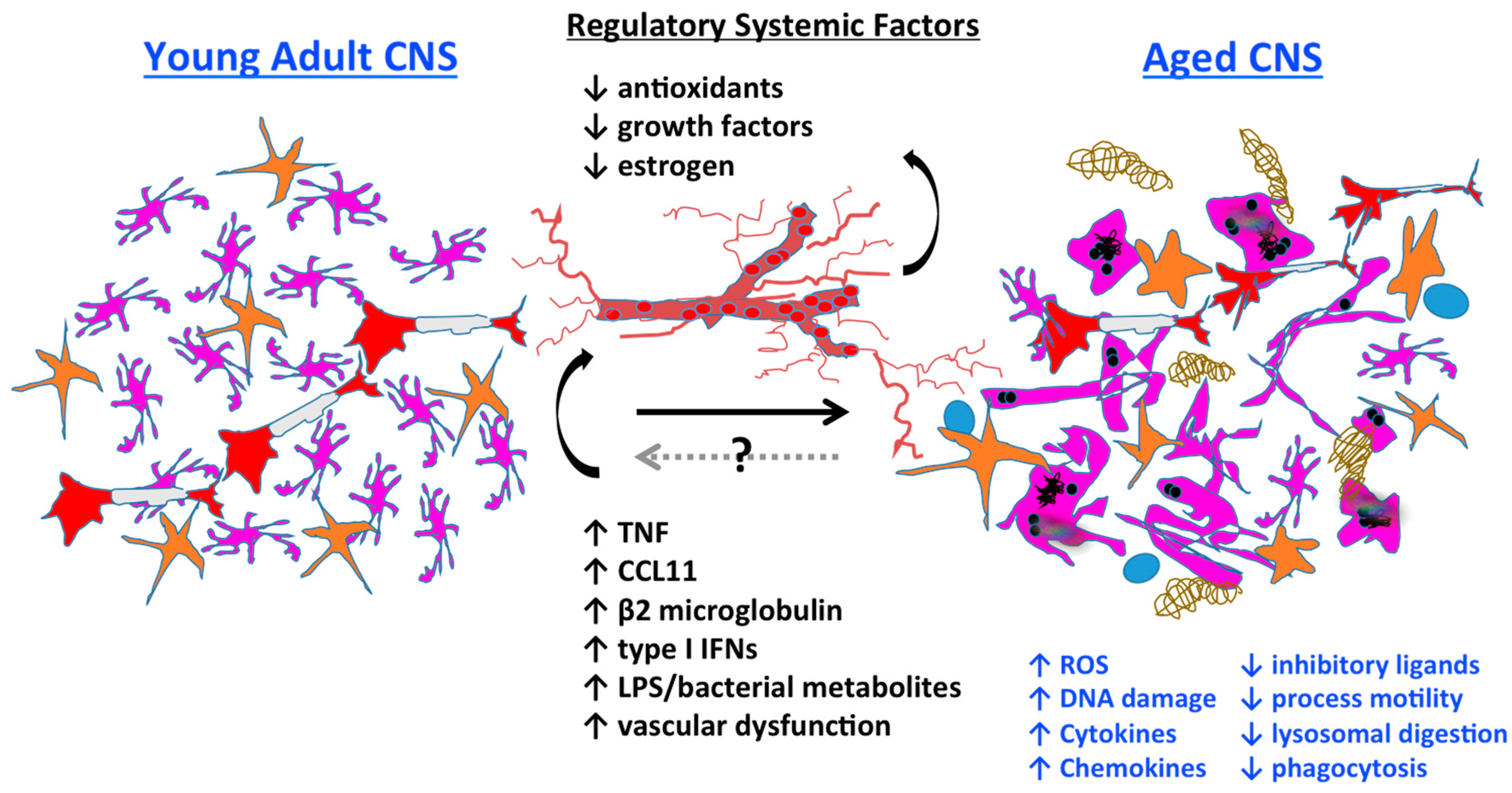
3.7. Systemic Regulation of Microglia Aging
4. The Role of Aging on the Microglial Response to Brain Injury and Disease
4.1. Aging Exacerbates Lipopolysaccharide-Induced Pro-Inflammatory Microglial Response
4.2. Aged Microglia Contribute to Enhanced Pathology Following Traumatic Brain Injury (TBI)
4.3. Aged Microglia Contribute to Worse Recovery and Functional Outcomes Following Stroke
4.4. The Role of Aged Microglia in Alzheimer’s Disease
5. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
References
- He, W.; Goodkind, D.; Kowal, P.R. An Aging World: 2015; U.S. Census Bureau, Ed.; U.S. Government Publishing Office: Washington, DC, USA, 2015; p. 165.
- Eshkoor, S.A.; Hamid, T.A.; Mun, C.Y.; Ng, C.K. Mild cognitive impairment and its management in older people. Clin. Interv. Aging 2015, 10, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Ovbiagele, B.; Goldstein, L.B.; Higashida, R.T.; Howard, V.J.; Johnston, S.C.; Khavjou, O.A.; Lackland, D.T.; Lichtman, J.H.; Mohl, S.; Sacco, R.L.; et al. Forecasting the future of stroke in the United States: A policy statement from the American Heart Association and American Stroke Association. Stroke 2013, 44, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.; Villeda, S.A. Aging and brain rejuvenation as systemic events. J. Neurochem. 2015, 132, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, S.; Capri, M.; Valensin, S.; Tieri, P.; Monti, D.; Ottaviani, E.; Franceschi, C. Inflamm-aging, cytokines and aging: State of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr. Pharm. Des. 2006, 12, 3161–3171. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Franceschi, C.; Monti, D.; Ginaldi, L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005, 579, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Miyamoto, A.; Nabekura, J. Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013, 36, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Hefendehl, J.K.; Neher, J.J.; Suhs, R.B.; Kohsaka, S.; Skodras, A.; Jucker, M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 2014, 13, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Wu, Z. Microglia-aging: Roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav. Brain Res. 2009, 201, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, S.T.; Graham, D.I. Aging-associated changes in human brain. J. Neuropathol. Exp. Neurol. 1997, 56, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.A.; Woulfe, J. Lipofuscin and aging: A matter of toxic waste. Sci. Aging Knowl. Environ. 2005, 2005, re1. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Sheng, H.; Xu, Y.F.; Lin, W.L.; Innes, A.E.; Gass, J.; Yu, X.; Wuertzer, C.A.; Hou, H.; Chiba, S.; et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 2010, 177, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Patel, A.R.; Pan, S.; Crapser, J.; Hammond, M.; Jellison, E.; McCullough, L.D. Age- and location-related changes in microglial function. Neurobiol. Aging 2015, 36, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.C.; Carrara, R.C.; Simoes, D.L.; Saggioro, F.P.; Carlotti, C.G., Jr.; Covas, D.T.; Neder, L. Sudan Black B treatment reduces autofluorescence and improves resolution of in situ hybridization specific fluorescent signals of brain sections. Histol. Histopathol. 2010, 25, 1017–1024. [Google Scholar] [PubMed]
- Moussaud, S.; Draheim, H.J. A new method to isolate microglia from adult mice and culture them for an extended period of time. J. Neurosci. Methods 2010, 187, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Tichauer, J.; Eugenin-von Bernhardi, L. Proliferating culture of aged microglia for the study of neurodegenerative diseases. J. Neurosci. Methods 2011, 202, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcao, A.S.; Fernandes, A.; Brites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell. Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Ruiz, C.R.; Lebson, L.; Selenica, M.L.; Rizer, J.; Hunt, J.B., Jr.; Rojiani, R.; Reid, P.; Kammath, S.; Nash, K.; et al. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 2013, 34, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Park, E.J.; Jou, I.; Kim, J.H.; Joe, E.H. Reactive oxygen species mediate Aβ(25–35)-induced activation of BV-2 microglia. Neuroreport 2001, 12, 1449–1452. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Polster, B.M. NADPH oxidase- and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Scheff, S.W. NADPH-oxidase activation and cognition in Alzheimer disease progression. Free Radic. Biol. Med. 2011, 51, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Levesque, S.; Surace, M.J.; Block, M.L. Chronic apocynin treatment attenuates β amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS ONE 2011, 6, e20153. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Zhou, M.L.; Johnson, A.W.; Singh, I.; Liao, F.; Vellimana, A.K.; Nelson, J.W.; Milner, E.; Cirrito, J.R.; Basak, J.; et al. Contribution of reactive oxygen species to cerebral amyloid angiopathy, vasomotor dysfunction, and microhemorrhage in aged Tg2576 mice. Proc. Natl. Acad. Sci. USA 2015, 112, E881–E890. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Hong, J.S.; Crews, F.T. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Bailey, W.M.; McVicar, A.L.; Gensel, J.C. Age increases reactive oxygen species production in macrophages and potentiates oxidative damage after spinal cord injury. Neurobiol. Aging 2016, 47, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Njie, E.G.; Boelen, E.; Stassen, F.R.; Steinbusch, H.W.; Borchelt, D.R.; Streit, W.J. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol. Aging 2012, 33, 195.e1–195.e12. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yu, J.; Zhu, A.; Nakanishi, H. Nutrients, Microglia Aging, and Brain Aging. Oxid. Med. Cell. Longev. 2016, 2016, 7498528. [Google Scholar] [CrossRef] [PubMed]
- Denieffe, S.; Kelly, R.J.; McDonald, C.; Lyons, A.; Lynch, M.A. Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain Behav. Immun. 2013, 34, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; Lyons, A.; Denieffe, S.; Browne, T.C.; Cox, F.F.; Lynch, M.A. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: A role for Toll-like receptor activation. J. Biol. Chem. 2011, 286, 34722–34732. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Crapser, J.; Patel, A.R.; Verma, R.; Grenier, J.M.; Chauhan, A.; Jellison, E.R.; McCullough, L.D. Age-Associated Resident Memory CD8 T Cells in the Central Nervous System Are Primed To Potentiate Inflammation after Ischemic Brain Injury. J. Immunol. (Baltimore, Md.: 1950) 2016, 196, 3318–3330. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, K.; Gonzalez, P.; Acarin, L. The immune inhibitory complex CD200/CD200R is developmentally regulated in the mouse brain. J. Comp. Neurol. 2012, 520, 2657–2675. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Zhang, S.; Yan, Z.Q.; Zhao, Y.X.; Zhou, H.Y.; Wang, Y.; Lu, G.Q.; Zhang, J.D. Impaired CD200-CD200R-mediated microglia silencing enhances midbrain dopaminergic neurodegeneration: Roles of aging, superoxide, NADPH oxidase, and p38 MAPK. Free Radic. Biol. Med. 2011, 50, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Dalsing-Hernandez, J.E.; Campbell, N.A.; Lue, L.F. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: A potential mechanism leading to chronic inflammation. Exp. Neurol. 2009, 215, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; McQuillan, K.; Deighan, B.F.; O’Reilly, J.A.; Downer, E.J.; Murphy, A.C.; Watson, M.; Piazza, A.; O’Connell, F.; Griffin, R.; et al. Decreased neuronal CD200 expression in IL-4-deficient mice results in increased neuroinflammation in response to lipopolysaccharide. Brain Behav. Immun. 2009, 23, 1020–1027. [Google Scholar] [CrossRef] [PubMed]
- Cox, F.F.; Carney, D.; Miller, A.M.; Lynch, M.A. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav. Immun. 2012, 26, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Downer, E.J.; Costello, D.A.; Murphy, N.; Lynch, M.A. Dok2 mediates the CD200Fc attenuation of Abeta-induced changes in glia. J. Neuroinflamm. 2012, 9, 107. [Google Scholar] [CrossRef] [PubMed]
- Varnum, M.M.; Kiyota, T.; Ingraham, K.L.; Ikezu, S.; Ikezu, T. The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2995–3007. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Minogue, A.M.; Jones, R.S.; Fitzpatrick, O.; Noonan, J.; Campbell, V.A.; Lynch, M.A. Analysis of the Impact of CD200 on Phagocytosis. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, V.; Benavides, J.; Vige, X.; Carter, C.; Taupin, V. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Lyons, A.; Lynch, A.M.; Downer, E.J.; Hanley, R.; O’Sullivan, J.B.; Smith, A.; Lynch, M.A. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attentuates microglial activation in vivo and in vitro. J. Neurochem. 2009, 110, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Fenn, A.M.; Smith, K.M.; Lovett-Racke, A.E.; Guerau-de-Arellano, M.; Whitacre, C.C.; Godbout, J.P. Increased micro-RNA 29b in the aged brain correlates with the reduction of insulin-like growth factor-1 and fractalkine ligand. Neurobiol. Aging 2013, 34, 2748–2758. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.S.; Yang, X.; Chen, Z.G.; Lu, M.O.; Morris, C.; Winblad, B.; Zhu, J. Decreased fractalkine and increased IP-10 expression in aged brain of APP(swe) transgenic mice. Neurochem. Res. 2008, 33, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Wynne, A.M.; Henry, C.J.; Huang, Y.; Cleland, A.; Godbout, J.P. Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav. Immun. 2010, 24, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bie, B.; Yang, H.; Xu, J.J.; Brown, D.L.; Naguib, M. Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol. Aging 2013, 34, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xu, G.; Jay, T.R.; Bhatta, S.; Kim, K.W.; Jung, S.; Landreth, G.E.; Ransohoff, R.M.; Lamb, B.T. Opposing effects of membrane-anchored CX3CL1 on amyloid and tau pathologies via the p38 MAPK pathway. J. Neurosci. 2014, 34, 12538–12546. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces β-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Sun, B.; Zhou, Y.; Kauppinen, T.M.; Halabisky, B.; Wes, P.; Ransohoff, R.M.; Gan, L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 2011, 286, 32713–32722. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Lee, D.C.; Hunt, J.B., Jr.; Morganti, J.M.; Selenica, M.L.; Moran, P.; Reid, P.; Brownlow, M.; Guang-Yu Yang, C.; Savalia, M.; et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol. Aging 2013, 34, 1540–1548. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef] [PubMed]
- Floden, A.M.; Combs, C.K. Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J. Alzheimer’s Dis. JAD 2011, 25, 279–293. [Google Scholar] [PubMed]
- Lynch, A.M.; Murphy, K.J.; Deighan, B.F.; O’Reilly, J.A.; Gun’ko, Y.K.; Cowley, T.R.; Gonzalez-Reyes, R.E.; Lynch, M.A. The impact of glial activation in the aging brain. Aging Dis. 2010, 1, 262–278. [Google Scholar] [PubMed]
- Hendrickx, D.A.; Schuurman, K.G.; van Draanen, M.; Hamann, J.; Huitinga, I. Enhanced uptake of multiple sclerosis-derived myelin by THP-1 macrophages and primary human microglia. J. Neuroinflamm. 2014, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Gitik, M.; Liraz-Zaltsman, S.; Oldenborg, P.A.; Reichert, F.; Rotshenker, S. Myelin down-regulates myelin phagocytosis by microglia and macrophages through interactions between CD47 on myelin and SIRPα (signal regulatory protein-α) on phagocytes. J. Neuroinflamm. 2011, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. β-Amyloid stimulation of microglia and monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [PubMed]
- Bliederhaeuser, C.; Grozdanov, V.; Speidel, A.; Zondler, L.; Ruf, W.P.; Bayer, H.; Kiechle, M.; Feiler, M.S.; Freischmidt, A.; Brenner, D.; et al. Age-dependent defects of α-synuclein oligomer uptake in microglia and monocytes. Acta. Neuropathol. 2016, 131, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Tichauer, J.E.; Flores, B.; Soler, B.; Eugenin-von Bernhardi, L.; Ramirez, G.; von Bernhardi, R. Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Li, A.; Ceballos-Diaz, C.; Eddy, J.A.; Funk, C.C.; Moore, B.; DiNunno, N.; Rosario, A.M.; Cruz, P.E.; Verbeeck, C.; et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron 2015, 85, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Kan, M.J.; Lee, J.E.; Wilson, J.G.; Everhart, A.L.; Brown, C.M.; Hoofnagle, A.N.; Jansen, M.; Vitek, M.P.; Gunn, M.D.; Colton, C.A. Arginine deprivation and immune suppression in a mouse model of Alzheimer’s disease. J. Neurosci. 2015, 35, 5969–5982. [Google Scholar] [CrossRef] [PubMed]
- Dagher, N.N.; Najafi, A.R.; Kayala, K.M.; Elmore, M.R.; White, T.E.; Medeiros, R.; West, B.L.; Green, K.N. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflamm. 2015, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.A.; Spangenberg, E.E.; Yamate-Morgan, H.; Lee, R.J.; Arora, R.P.; Hernandez, M.X.; Tenner, A.J.; West, B.L.; Green, K.N. Elimination of Microglia Improves Functional Outcomes Following Extensive Neuronal Loss in the Hippocampus. J. Neurosci. 2015, 35, 9977–9989. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jopson, T.D.; Paladini, M.S.; Liu, S.; West, B.L.; Gupta, N.; Rosi, S. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflamm. 2016, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci. Rep. 2016, 6, 31545. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Soto, I.; Graham, L.C.; Richter, H.J.; Simeone, S.N.; Radell, J.E.; Grabowska, W.; Funkhouser, W.K.; Howell, M.C.; Howell, G.R. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015, 13, e1002279. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. beta2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.C.; Harder, J.M.; Soto, I.; de Vries, W.N.; John, S.W.; Howell, G.R. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 21568. [Google Scholar] [CrossRef] [PubMed]
- Kohman, R.A.; Bhattacharya, T.K.; Wojcik, E.; Rhodes, J.S. Exercise reduces activation of microglia isolated from hippocampus and brain of aged mice. J. Neuroinflamm. 2013, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Loncarevic-Vasiljkovic, N.; Pesic, V.; Todorovic, S.; Popic, J.; Smiljanic, K.; Milanovic, D.; Ruzdijic, S.; Kanazir, S. Caloric restriction suppresses microglial activation and prevents neuroapoptosis following cortical injury in rats. PLoS ONE 2012, 7, e37215. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, C.; Ronaghan, N.; Zaheer, R.; Dicay, M.; Le, T.; MacNaughton, W.K.; Surrette, M.G.; Swain, M.G. Probiotics Improve Inflammation-Associated Sickness Behavior by Altering Communication between the Peripheral Immune System and the Brain. J. Neurosci. 2015, 35, 10821–10830. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Buga, A.M.; Tica, A.A.; Albu, C.V. Perfusion deficits, inflammation and aging precipitate depressive behaviour. Biogerontology 2014, 15, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Bickford, P.C.; Flowers, A.; Grimmig, B. Aging leads to altered microglial function that reduces brain resiliency increasing vulnerability to neurodegenerative diseases. Exp. Gerontol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Blazer, D.G.; Yaffe, K.; Karlawish, J. Cognitive aging: A report from the Institute of Medicine. JAMA 2015, 313, 2121–2122. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.L. Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology 2015, 96, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Androsova, G.; Krause, R.; Winterer, G.; Schneider, R. Biomarkers of postoperative delirium and cognitive dysfunction. Front. Aging Neurosci. 2015, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Gleason, L.J.; Schmitt, E.M.; Kosar, C.M.; Tabloski, P.; Saczynski, J.S.; Robinson, T.; Cooper, Z.; Rogers, S.O., Jr.; Jones, R.N.; Marcantonio, E.R.; et al. Effect of Delirium and Other Major Complications on Outcomes After Elective Surgery in Older Adults. JAMA Surg. 2015, 150, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Inouye, S.K.; Westendorp, R.G.; Saczynski, J.S. Delirium in elderly people. Lancet 2014, 383, 911–922. [Google Scholar] [CrossRef]
- Vasunilashorn, S.M.; Ngo, L.; Inouye, S.K.; Libermann, T.A.; Jones, R.N.; Alsop, D.C.; Guess, J.; Jastrzebski, S.; McElhaney, J.E.; Kuchel, G.A.; et al. Cytokines and Postoperative Delirium in Older Patients Undergoing Major Elective Surgery. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1289–1295. [Google Scholar] [CrossRef] [PubMed]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1329–1331. [Google Scholar]
- Godbout, J.P.; Moreau, M.; Lestage, J.; Chen, J.; Sparkman, N.L.; O’Connor, J.; Castanon, N.; Kelley, K.W.; Dantzer, R.; Johnson, R.W. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Simone, M.J.; Tan, Z.S. The role of inflammation in the pathogenesis of delirium and dementia in older adults: A review. CNS Neurosci. Ther. 2011, 17, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.K.; Frank, M.G. The Alarmin HMGB1 Mediates Age-Induced Neuroinflammatory Priming. J. Neurosci. 2016, 36, 7946–7956. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Henry, C.J.; Dantzer, R.; Johnson, R.W.; Godbout, J.P. Exaggerated sickness behavior and brain proinflammatory cytokine expression in aged mice in response to intracerebroventricular lipopolysaccharide. Neurobiol. Aging 2008, 29, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Rosczyk, H.A.; Sparkman, N.L.; Johnson, R.W. Neuroinflammation and cognitive function in aged mice following minor surgery. Exp. Gerontol. 2008, 43, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Zhang, C.; Dong, Y.; Zhang, Y.; Nakazawa, H.; Kaneki, M.; Zheng, H.; Shen, Y.; Marcantonio, E.R.; Xie, Z. Battery of behavioral tests in mice to study postoperative delirium. Sci. Rep. 2016, 6, 29874. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Peng, M.; Dong, Y.; Zhang, Y.; Chen, M.; Yin, N.; Marcantonio, E.R.; Xie, Z. Surgery plus anesthesia induces loss of attention in mice. Front. Cell. Neurosci. 2015, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, J.B.; Sparkman, N.L.; Chen, J.; Johnson, R.W. Cognitive and neuroinflammatory consequences of mild repeated stress are exacerbated in aged mice. Psychoneuroendocrinology 2008, 33, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; Capuron, L.; Irwin, M.R.; Miller, A.H.; Ollat, H.; Perry, V.H.; Rousey, S.; Yirmiya, R. Identification and treatment of symptoms associated with inflammation in medically ill patients. Psychoneuroendocrinology 2008, 33, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Coronado, V.G.; Thomas, K.E.; Sattin, R.W.; Johnson, R.L. The CDC traumatic brain injury surveillance system: Characteristics of persons aged 65 years and older hospitalized with a TBI. J. Head Trauma Rehabil. 2005, 20, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 3–13. [Google Scholar] [PubMed]
- Mosenthal, A.C.; Lavery, R.F.; Addis, M.; Kaul, S.; Ross, S.; Marburger, R.; Deitch, E.A.; Livingston, D.H. Isolated traumatic brain injury: Age is an independent predictor of mortality and early outcome. J. Trauma 2002, 52, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Stocchetti, N.; Paterno, R.; Citerio, G.; Beretta, L.; Colombo, A. Traumatic brain injury in an aging population. J. Neurotrauma 2012, 29, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Livingston, D.H.; Lavery, R.F.; Mosenthal, A.C.; Knudson, M.M.; Lee, S.; Morabito, D.; Manley, G.T.; Nathens, A.; Jurkovich, G.; Hoyt, D.B.; et al. Recovery at one year following isolated traumatic brain injury: A Western Trauma Association prospective multicenter trial. J. Trauma 2005, 59, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.A.; Malec, J.F.; Moessner, A.M.; Brown, A.W. Outcome after traumatic brain injury: Effects of aging on recovery. Arch. Phys. Med. Rehabil. 2005, 86, 1815–1823. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Kaup, A.; Barnes, D.E.; Yaffe, K. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurol. 2014, 71, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Liao, Y.E.; Yang, L.Y.; Wang, J.Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Meaney, D.F.; Cullen, D.K.; Smith, D.H. Animal models of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 115–128. [Google Scholar] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Stoica, B.A.; Sabirzhanov, B.; Burns, M.P.; Faden, A.I.; Loane, D.J. Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol. Aging 2013, 34, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.E.; Takizawa, B.T.; Strittmatter, S.M. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 1416–1423. [Google Scholar]
- Liu, G.; Ni, J.; Mao, L.; Yan, M.; Pang, T.; Liao, H. Expression of Nogo receptor 1 in microglia during development and following traumatic brain injury. Brain Res. 2015, 1627, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Sandu, R.E.; Buga, A.M.; Balseanu, A.T.; Moldovan, M.; Popa-Wagner, A. Twenty-four hours hypothermia has temporary efficacy in reducing brain infarction and inflammation in aged rats. Neurobiol. Aging 2016, 38, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Sandu, R.E.; Uzoni, A.; Ciobanu, O.; Moldovan, M.; Anghel, A.; Radu, E.; Coogan, A.N.; Popa-Wagner, A. Post-stroke gaseous hypothermia increases vascular density but not neurogenesis in the ischemic penumbra of aged rats. Restor. Neurol. Neurosci. 2016, 34, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Buga, A.M.; Di Napoli, M.; Popa-Wagner, A. Preclinical models of stroke in aged animals with or without comorbidities: Role of neuroinflammation. Biogerontology 2013, 14, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Crapser, J.; Ritzel, R.; Verma, R.; Venna, V.R.; Liu, F.; Chauhan, A.; Koellhoffer, E.; Patel, A.; Ricker, A.; Maas, K.; et al. Ischemic stroke induces gut permeability and enhances bacterial translocation leading to sepsis in aged mice. Aging 2016, 8, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Sieber, M.W.; Claus, R.A.; Witte, O.W.; Frahm, C. Attenuated inflammatory response in aged mice brains following stroke. PLoS ONE 2011, 6, e26288. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.A.; Jeong, S.I.; Kim, M.; Yoon, J.C.; Kim, H.S.; Park, E.M. Visceral adipose tissue inflammation is associated with age-related brain changes and ischemic brain damage in aged mice. Brain Behav. Immun. 2015, 50, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, J.; Hu, X.; Pu, H.; Shi, Y.; Hassan, S.H.; Xu, M.; Leak, R.K.; Stetler, R.A.; Gao, Y.; Chen, J. White matter injury and microglia/macrophage polarization are strongly linked with age-related long-term deficits in neurological function after stroke. Exp. Neurol. 2015, 272, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Moraga, A.; Pradillo, J.M.; Garcia-Culebras, A.; Palma-Tortosa, S.; Ballesteros, I.; Hernandez-Jimenez, M.; Moro, M.A.; Lizasoain, I. Aging increases microglial proliferation, delays cell migration, and decreases cortical neurogenesis after focal cerebral ischemia. J. Neuroinflamm. 2015, 12, 87. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Brody, M.; Liu, E.; Di, J.; Lu, M.; Margolin, R.A.; Werth, J.L.; Booth, K.; Shadman, A.; Brashear, H.R.; Novak, G. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Safety, Pharmacokinetics, and Biomarker Results of Subcutaneous Bapineuzumab in Patients with mild to moderate Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2016, 54, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Ivanoiu, A.; Pariente, J.; Booth, K.; Lobello, K.; Luscan, G.; Hua, L.; Lucas, P.; Styren, S.; Yang, L.; Li, D.; et al. Long-term safety and tolerability of bapineuzumab in patients with Alzheimer’s disease in two phase 3 extension studies. Alzheimer’s Res. Ther. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer’s Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; La Ferla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Butt, A.M.; Gardenal, E.; Parpura, V.; Verkhratsky, A. Complex and differential glial responses in Alzheimer’s disease and ageing. Curr. Alzheimer Res. 2016, 13, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Udeochu, J.C.; Shea, J.M.; Villeda, S.A. Microglia communication: Parallels between aging and Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2016, 7, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 1997, 18, 351–357. [Google Scholar] [CrossRef]
- Peters, F.; Collette, F.; Degueldre, C.; Sterpenich, V.; Majerus, S.; Salmon, E. The neural correlates of verbal short-term memory in Alzheimer’s disease: An fMRI study. Brain J. Neurol. 2009, 132, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, J.; Davis, E.G.; Rebeck, G.W. Aging reduces glial uptake and promotes extracellular accumulation of Abeta from a lentiviral vector. Front. Aging Neurosci. 2014, 6, 210. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Middeldorp, J.; Lehallier, B.; Villeda, S.A.; Miedema, S.S.; Evans, E.; Czirr, E.; Zhang, H.; Luo, J.; Stan, T.; Mosher, K.I.; et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016, 73, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Weber, A.; Rehli, M.; Peuker, A.; Muller, A.; Kastenberger, M.; Brockhoff, G.; Andreesen, R.; Kreutz, M. Expression of CD68 in non-myeloid cell types. Scand. J. Immunol. 2008, 67, 453–463. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
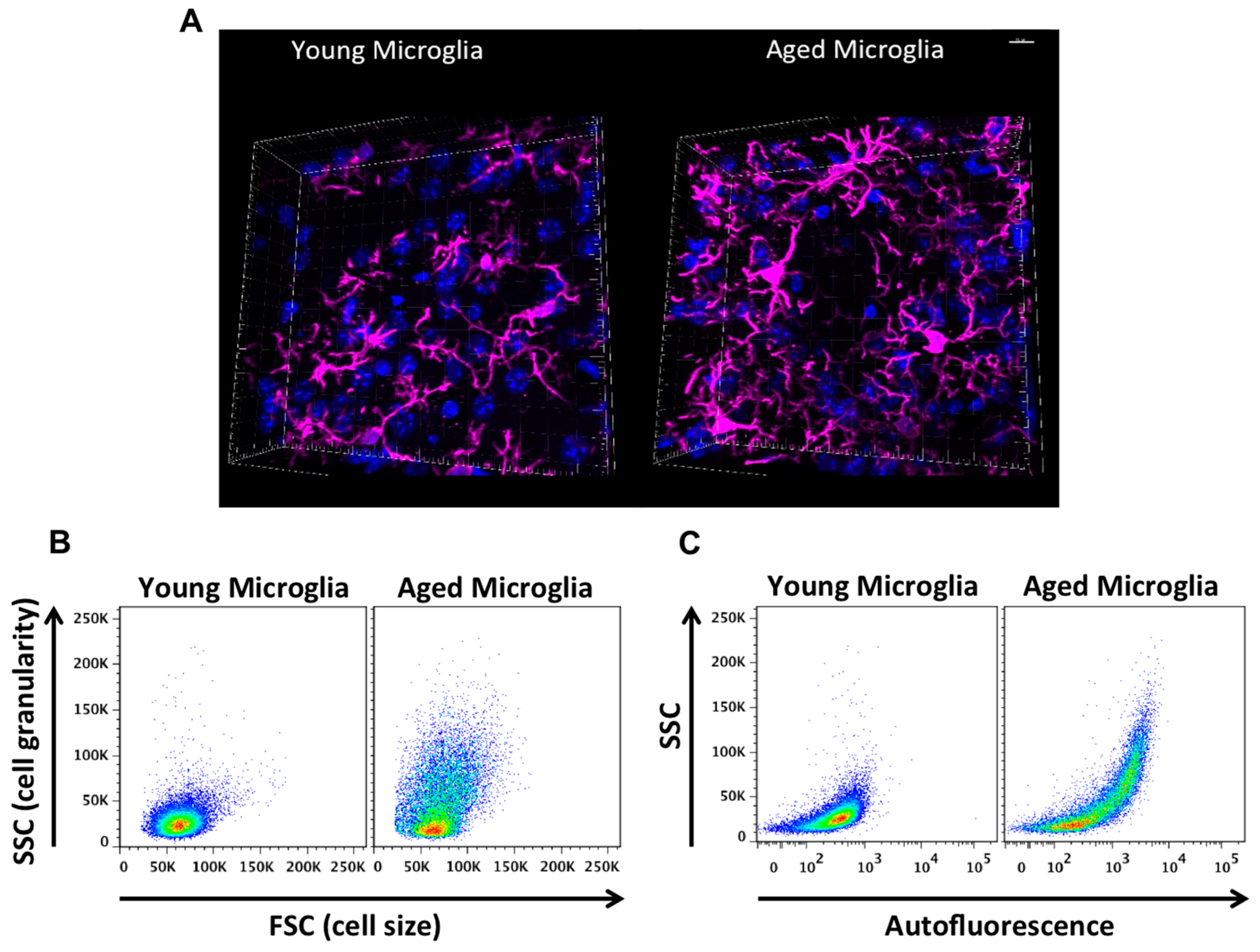{kind=link}
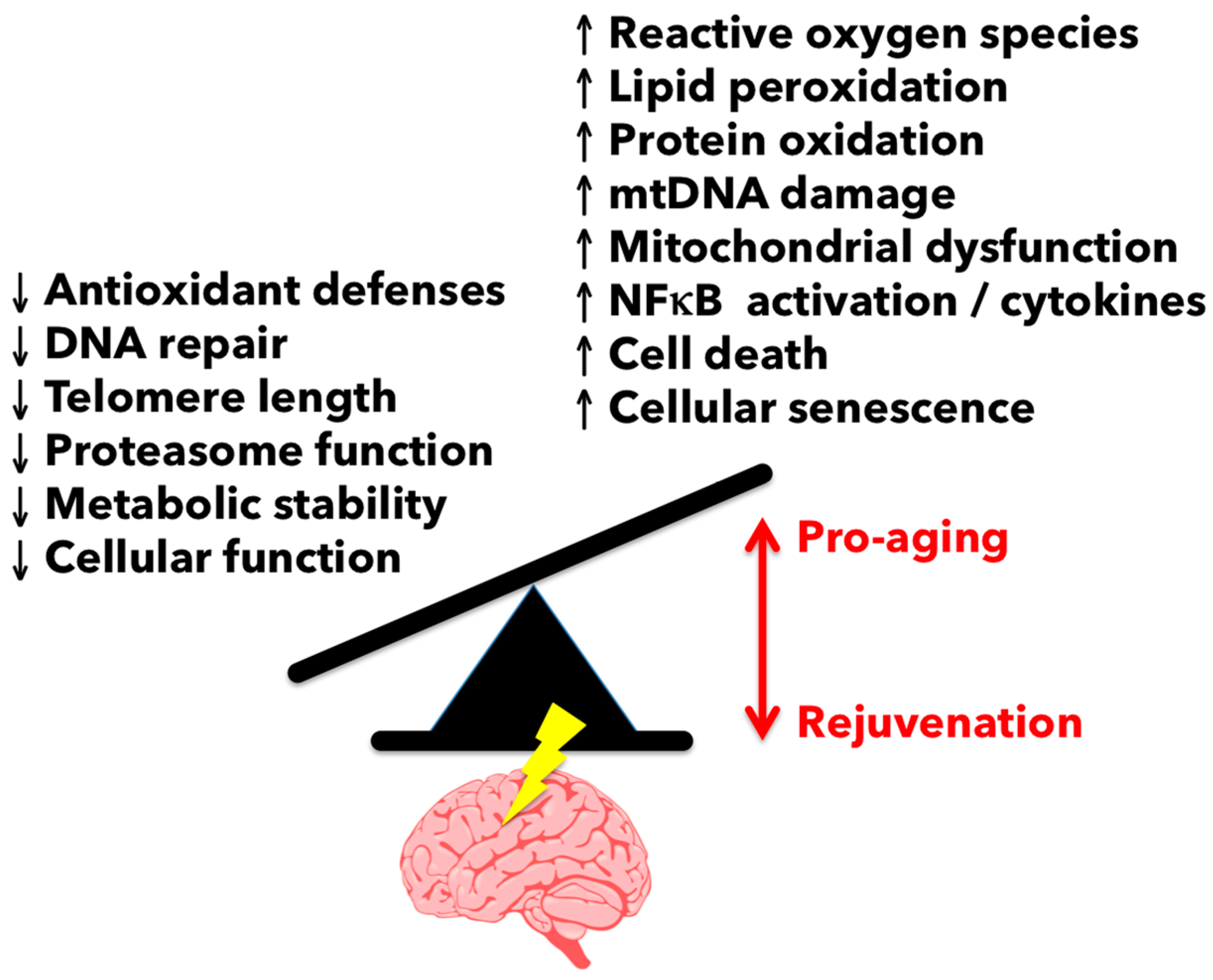{kind=link}
| Stressor | Study | Animals | Age(s) | Sex | Model | Notable Findings |
|---|---|---|---|---|---|---|
| Peripheral infection | [101] | BALB/c mice | Young 3–6 m Aged 20–24 m | Male | Lipopolysaccharide (LPS) i.p. injection | Exaggerated ↑ IL-1β, IL-6, lipid peroxidation in aged brain ↓ social behavior, food intake, weight loss in aged |
| [104] | F344XBN rats | Young 3 m Aged 24 m | Male | Live E. coli i.p. injection | At baseline: ↑ hippocampal HMGB1 protein, mRNA in aged; ↑ HMGB1 protein in cerebrospinal fluid (CSF) of aged Following i.p. E. coli injection: Prolonged ↑ expression of IL-1β, IL-18, TNF in aged; prolonged sucrose anhedonia (depression) and ↓ juvenile social exploration in aged Inhibition of HMGB1: abrogated primed phenotype of aged brain to peripheral E. coli injection, restoring behavior to that of young animals | |
| Central innate immune activation | [105] | BALB/c mice | Young 3–4 m Aged 20–22 m | Male | LPS i.c.v. injection | Prolonged ↓ locomotor activity, social behavior, and food intake in aged ↑ cerebellar and hippocampal IL-1β, IL-6, and TNF expression in aged |
| Surgery | [106] | BALB/c mice | Young 4–6 m Aged 23–25 m | Male | 1.5 cm abdominal incision and gentle manipulation of internal organs for 1 min | Anesthetic and analgesics: no effect on hippocampal IL-1β, IL-6 and TNF mRNA expression Surgery: ↑ IL-1β expression in aged hippocampus; locomotor activity unchanged in in young or aged mice |
| [107] | C57Bl6/J mice | 4 m | Female | 0.5 cm abdominal incision | Surgery ↑ anxiety, ↓ special memory | |
| [108] | C57Bl6/J mice | 2 m–8 m | Female | Simple laparotomy | Surgery ↑ total alpha-synuclein and S100β in the cortex, ↓ attention | |
| Stress | [109] | BALB/c mice | Young 3–5 m Aged 22–24 m | Male | 30 min restraint stress daily for 4 days | Stress ↑ weight loss, exaggerated ↑ hippocampal and hypothalamic IL-1β mRNA expression in aged; exaggerated ↑ corticosterone in aged Higher hippocampal MHCII mRNA and immunohistochemistry staining in aged mice at baseline, and increased in aged mice following stress |
| Study | Strain | Age(s) | Model | Duration | Notable Findings |
|---|---|---|---|---|---|
| [150] | APP on C57Bl/6 background | Young 2–3 m Aged 16–20 m | Heterochronic parabiosis Aged APP—Young WT Aged APP—Aged APP Aged WT—Aged WT | 5 weeks | In the hippocampus: rejuvenation of synaptophysin and calbindin immunoreactivity; no change in total Aβ or Aβ-42 levels; no effect of CD68 immunoreactivity |
| Plasma transfer PBS Young plasma | Administration twice weekly for 4 weeks | In the hippocampus: rejuvenation of synaptophysin and calbindin immunoreactivity; no effect of CD68 immunoreactivity Improved memory, spatial learning memory with young plasma transfer | |||
| [151] | APPswe/PS1dE9 Tg | Young 3 m Tg 3 m | Heterochronic parabiosis Young Tg—Young WT Age-matched Tg Age-matched WT | 6 months | In heterochronic Tg parabionts: ↓ Aβ-40, Aβ-42, total Aβ, and Congo Red plaques in brain ↓ CAA vessel number and area Alleviation of neuronal degeneration and apoptosis |
| [152] | B6.CD45.1 5XFAD (CD45.2) | 4 or 8 m | Parabiosis B6.CD45.1–5XFAD | 4 weeks | No recruitment of CD45.1 WT monocytes to brains of 5XFAD parabionts Brain-resident microglia associate with amyloid plaques, not peripheral monocytes |
| B6.CD45.1 APP-PS1 (CD45.2) | 3.5 m | Parabiosis B6.CD45.1–APP-PS1 | 9 weeks | No recruitment of CD45.1 WT monocytes to brains of APP-PS1 parabionts |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. https://doi.org/10.3390/ijms18040769
Koellhoffer EC, McCullough LD, Ritzel RM. Old Maids: Aging and Its Impact on Microglia Function. International Journal of Molecular Sciences. 2017; 18(4):769. https://doi.org/10.3390/ijms18040769
Chicago/Turabian StyleKoellhoffer, Edward C., Louise D. McCullough, and Rodney M. Ritzel. 2017. "Old Maids: Aging and Its Impact on Microglia Function" International Journal of Molecular Sciences 18, no. 4: 769. https://doi.org/10.3390/ijms18040769
APA StyleKoellhoffer, E. C., McCullough, L. D., & Ritzel, R. M. (2017). Old Maids: Aging and Its Impact on Microglia Function. International Journal of Molecular Sciences, 18(4), 769. https://doi.org/10.3390/ijms18040769