miR-1224-5p Mediates Mitochondrial Damage to Affect Silica-Induced Pulmonary Fibrosis by Targeting BECN1

Abstract
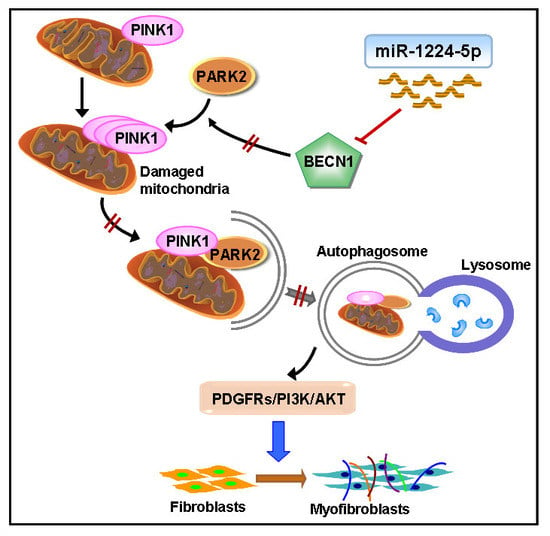
:
1. Introduction
2. Results
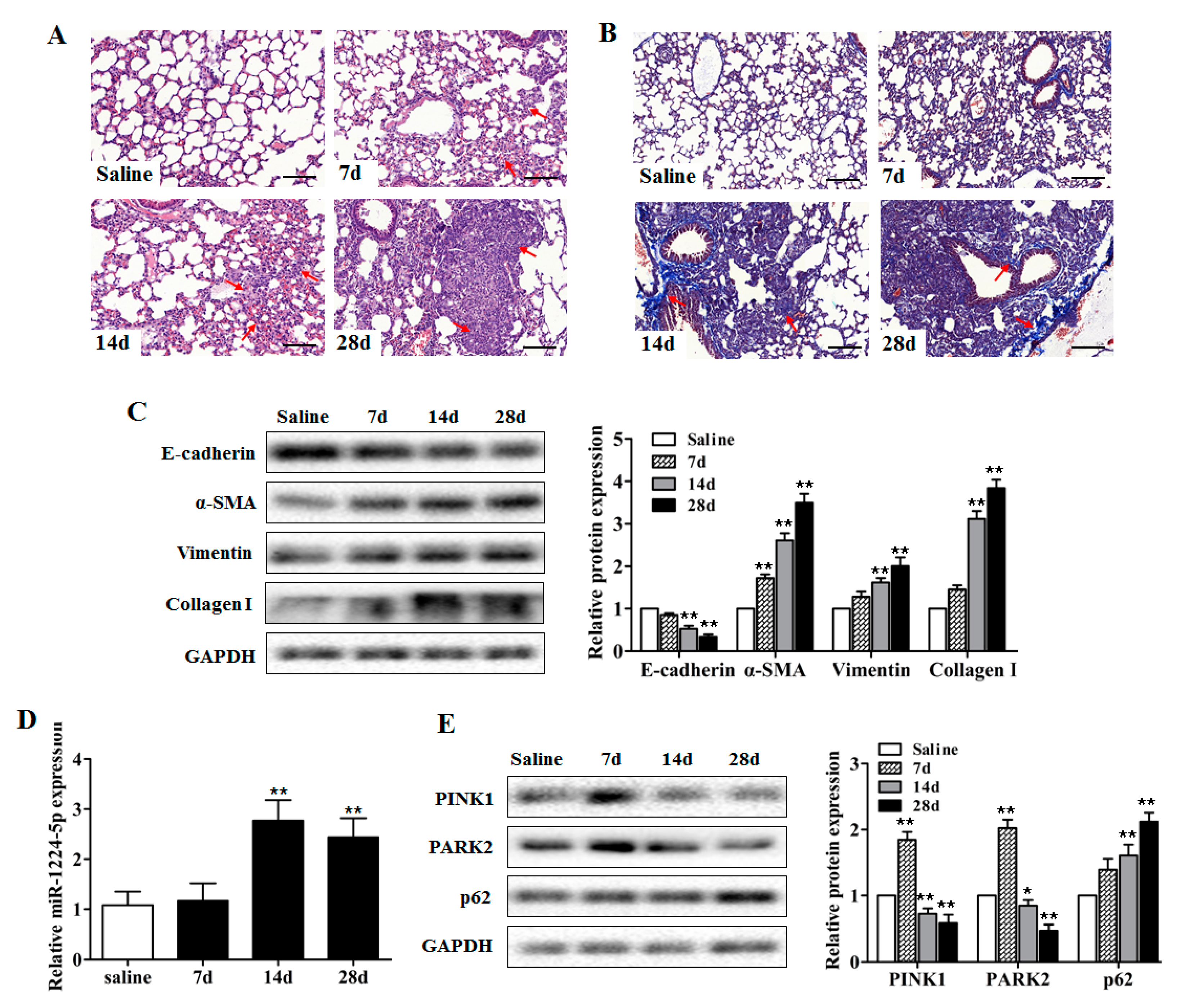
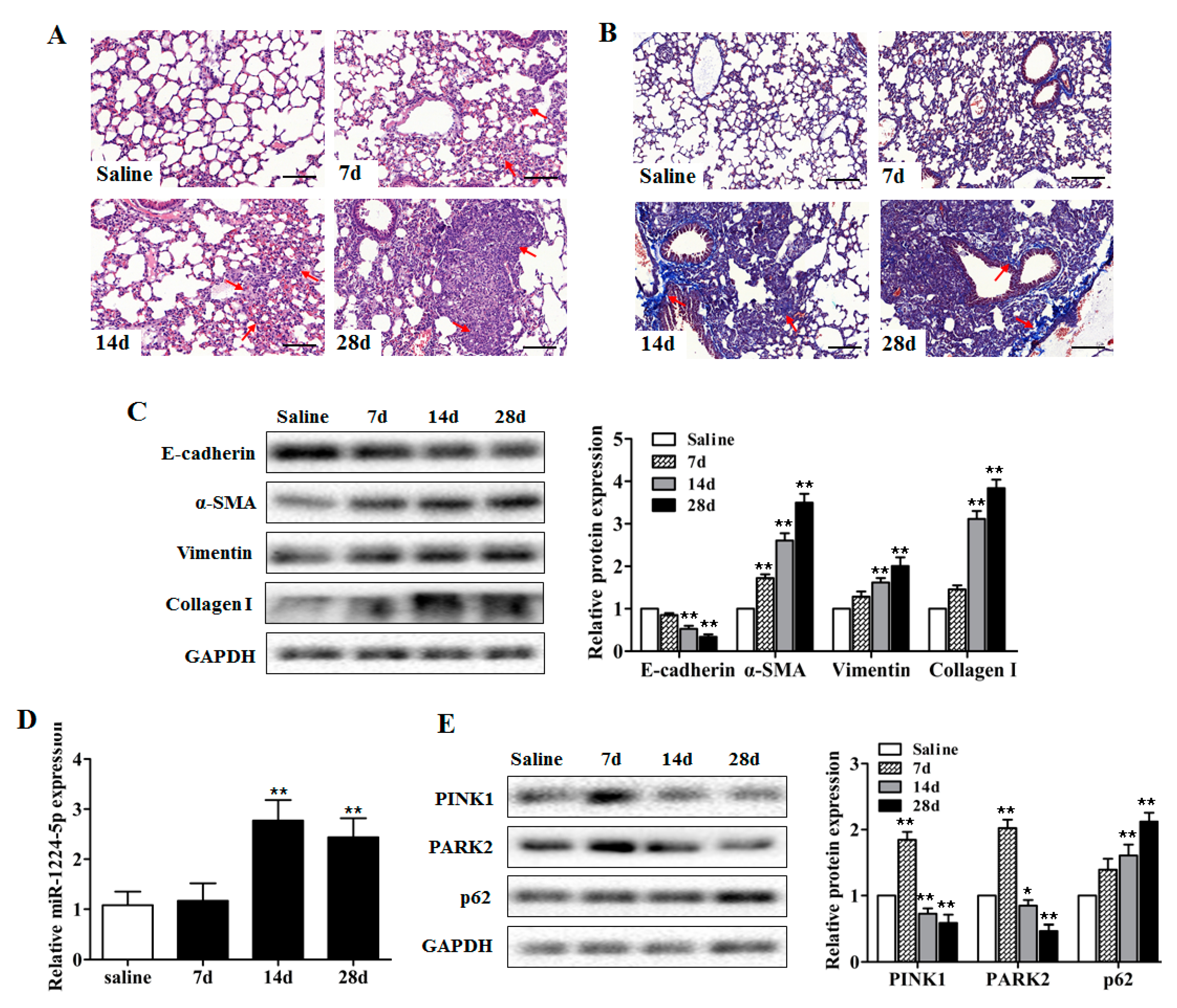
2.1. miR-1224-5p Is Increased and Mitophagy is Impaired in Mouse Lung Tissues in a Model of Silica-Induced Pulmonary Fibrosis
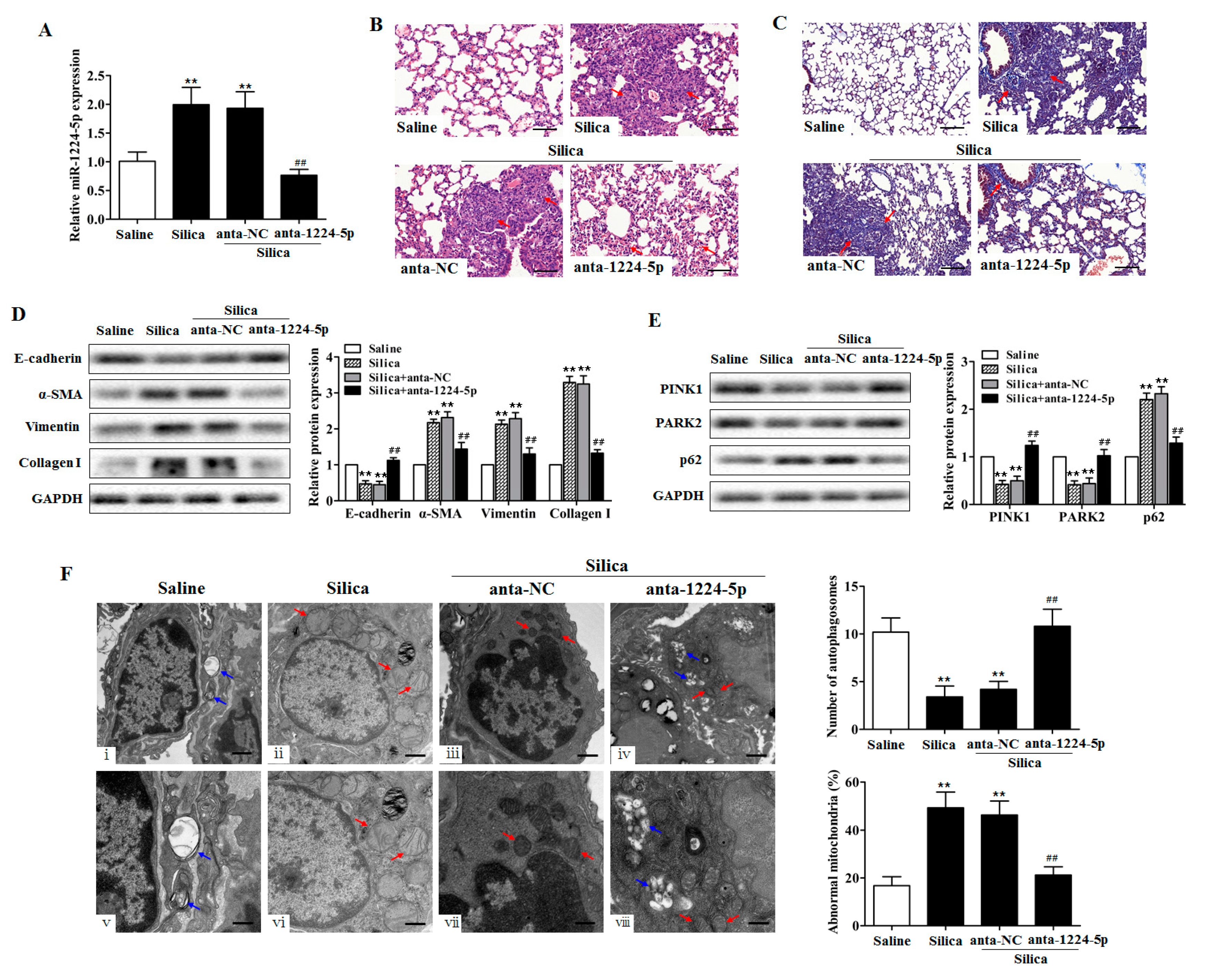
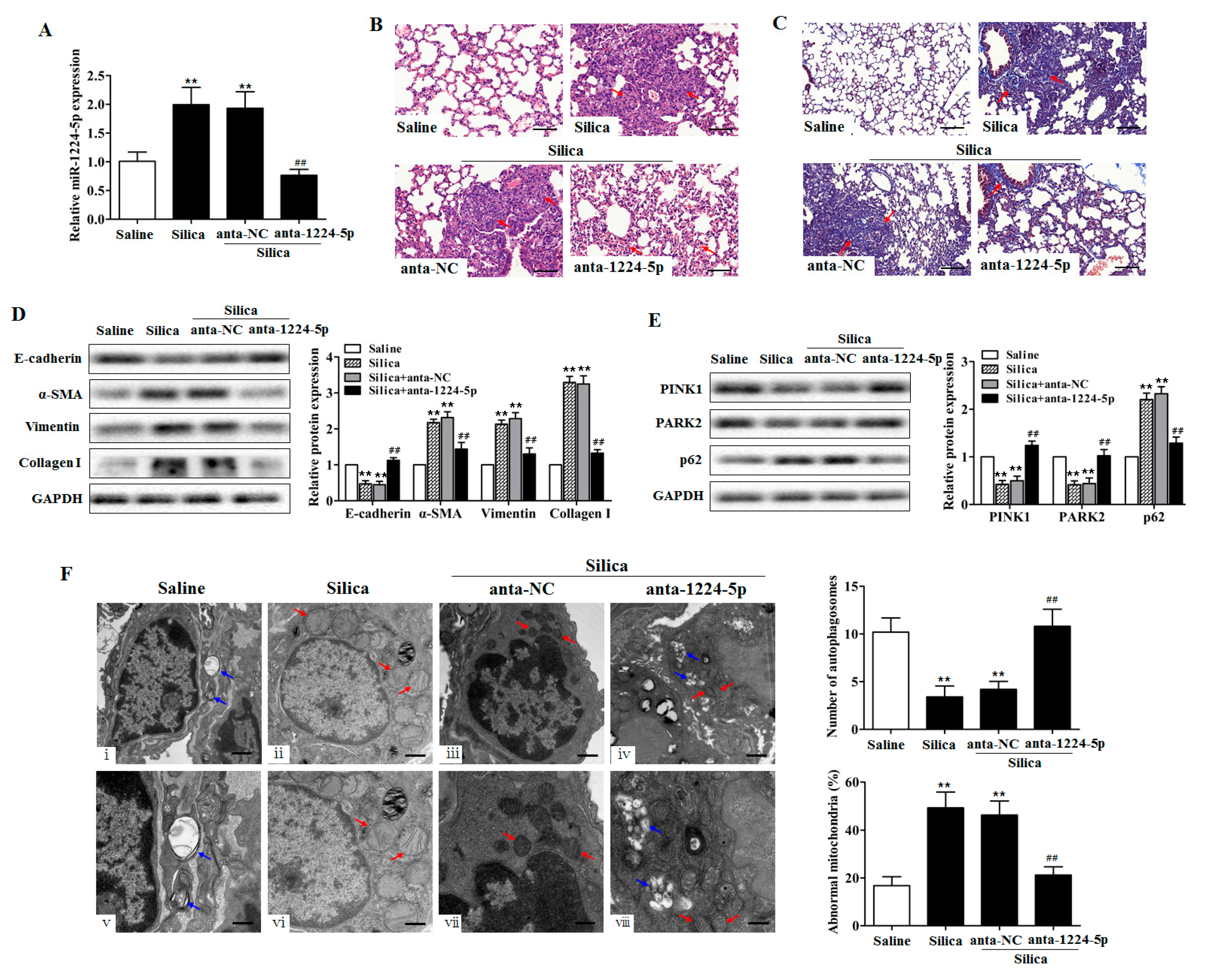
2.2. Down-Regulated miR-1224-5p Attenuates Silica-Induced Pulmonary Fibrosis and Restores Mitophagy In Vivo
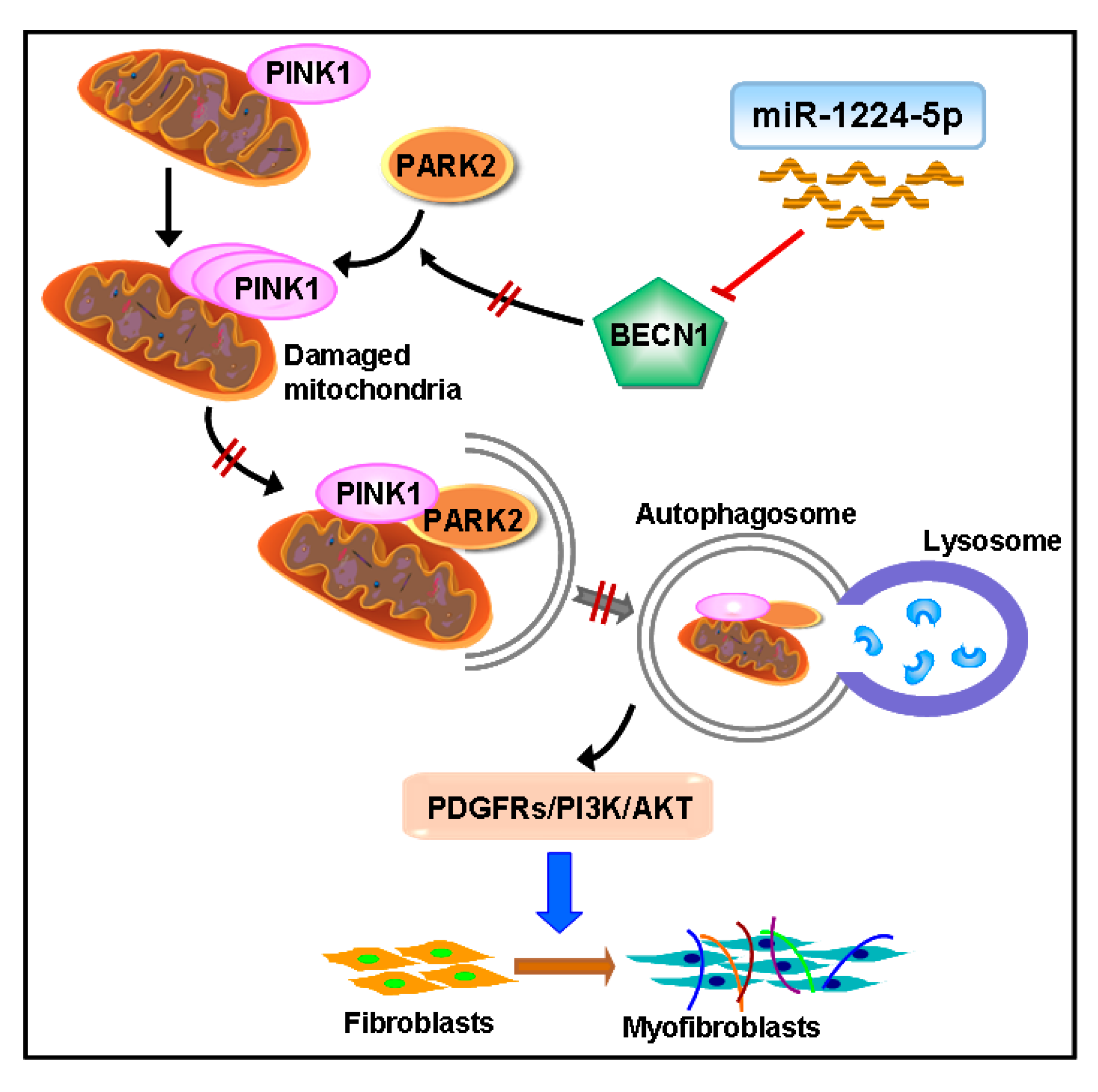
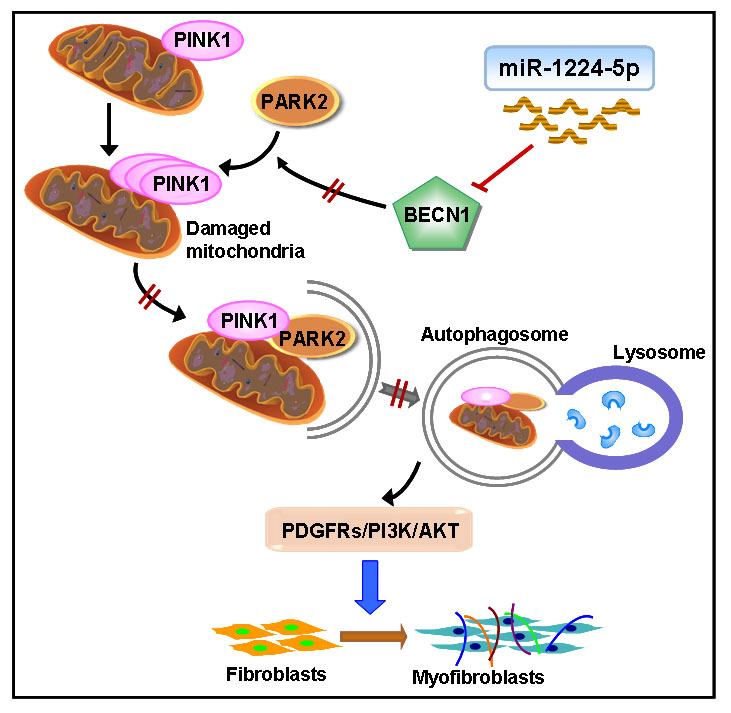
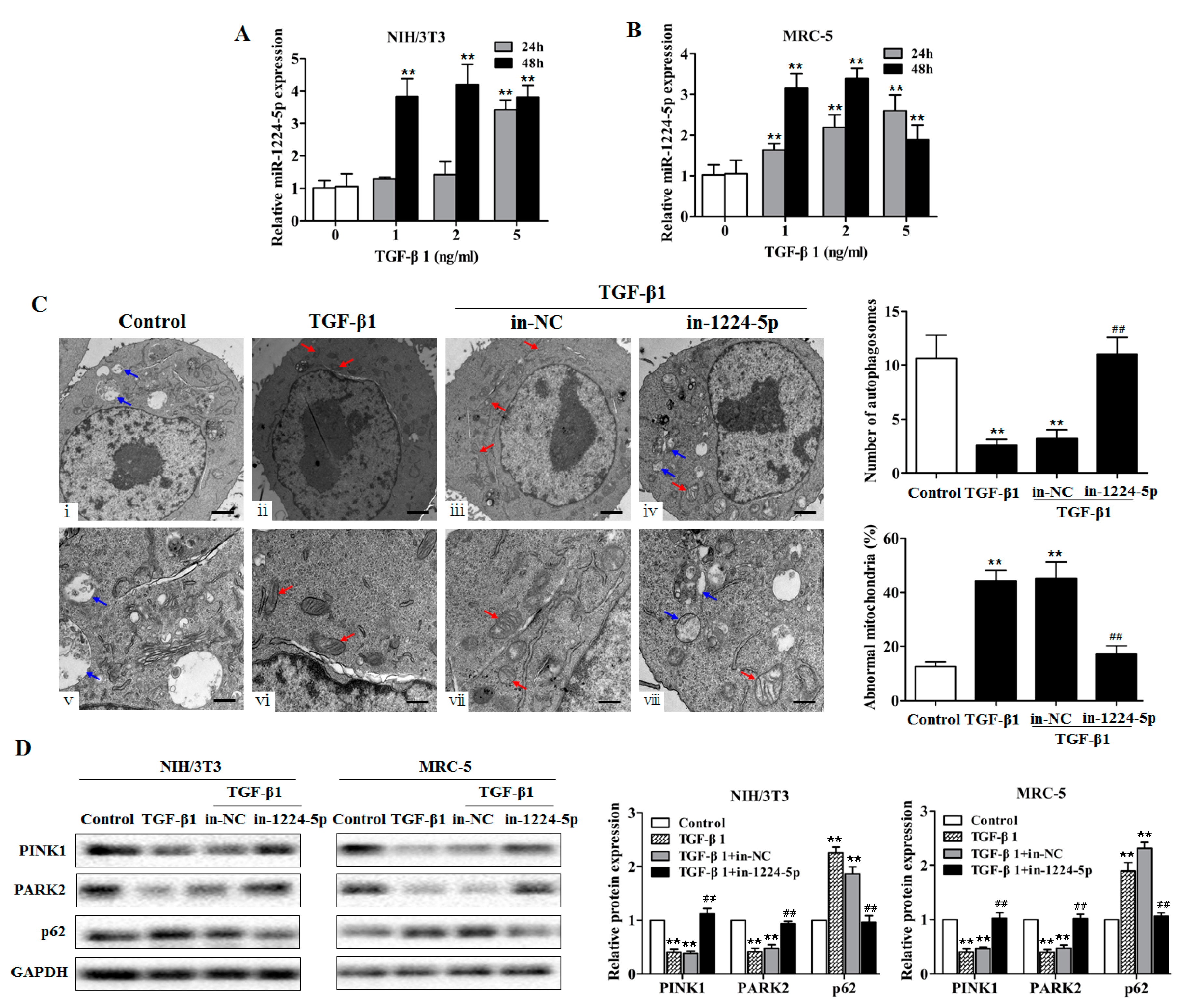
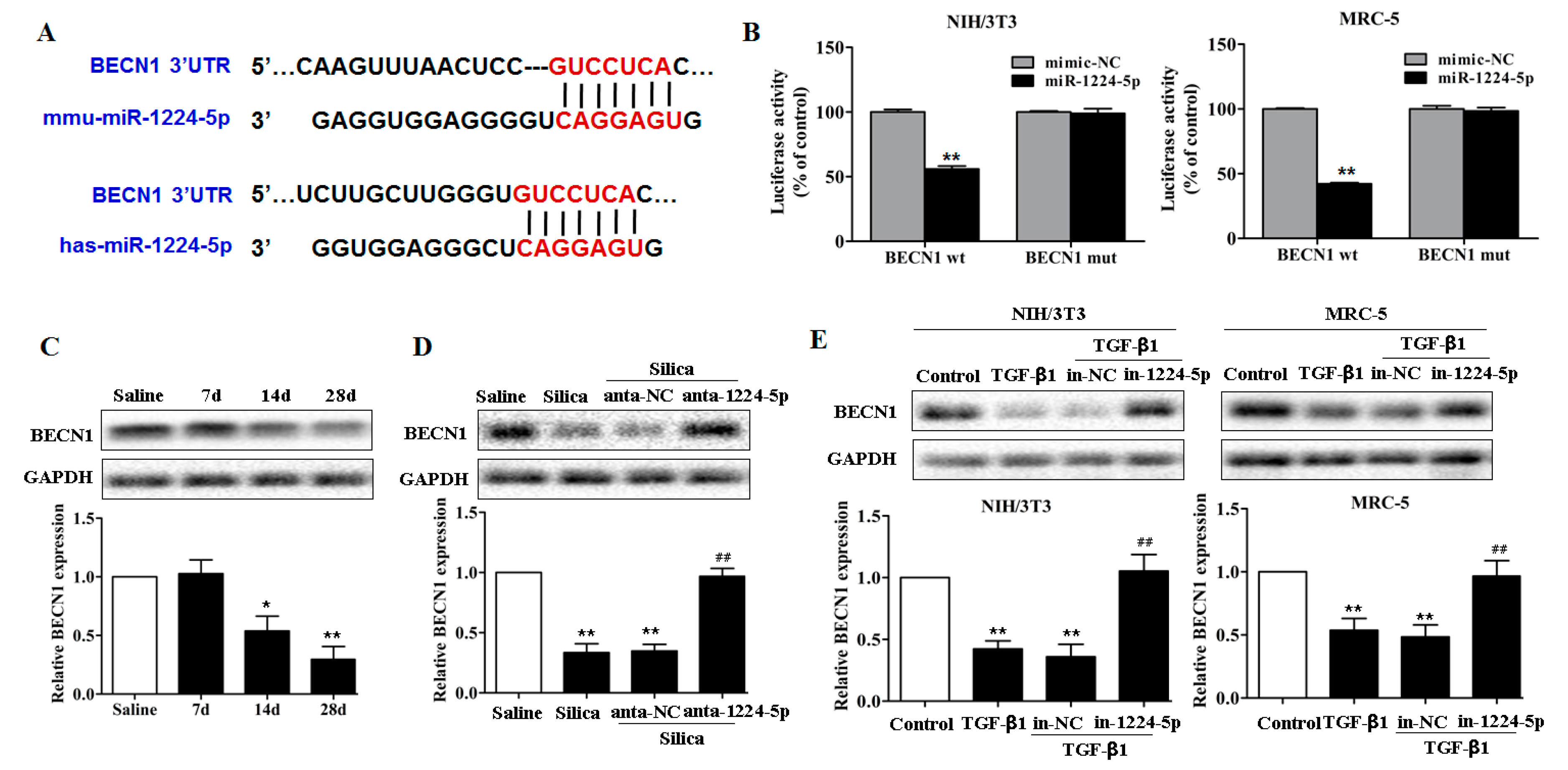
2.3. miR-1224-5p Suppresses Mitophagy by Targeting BECN1
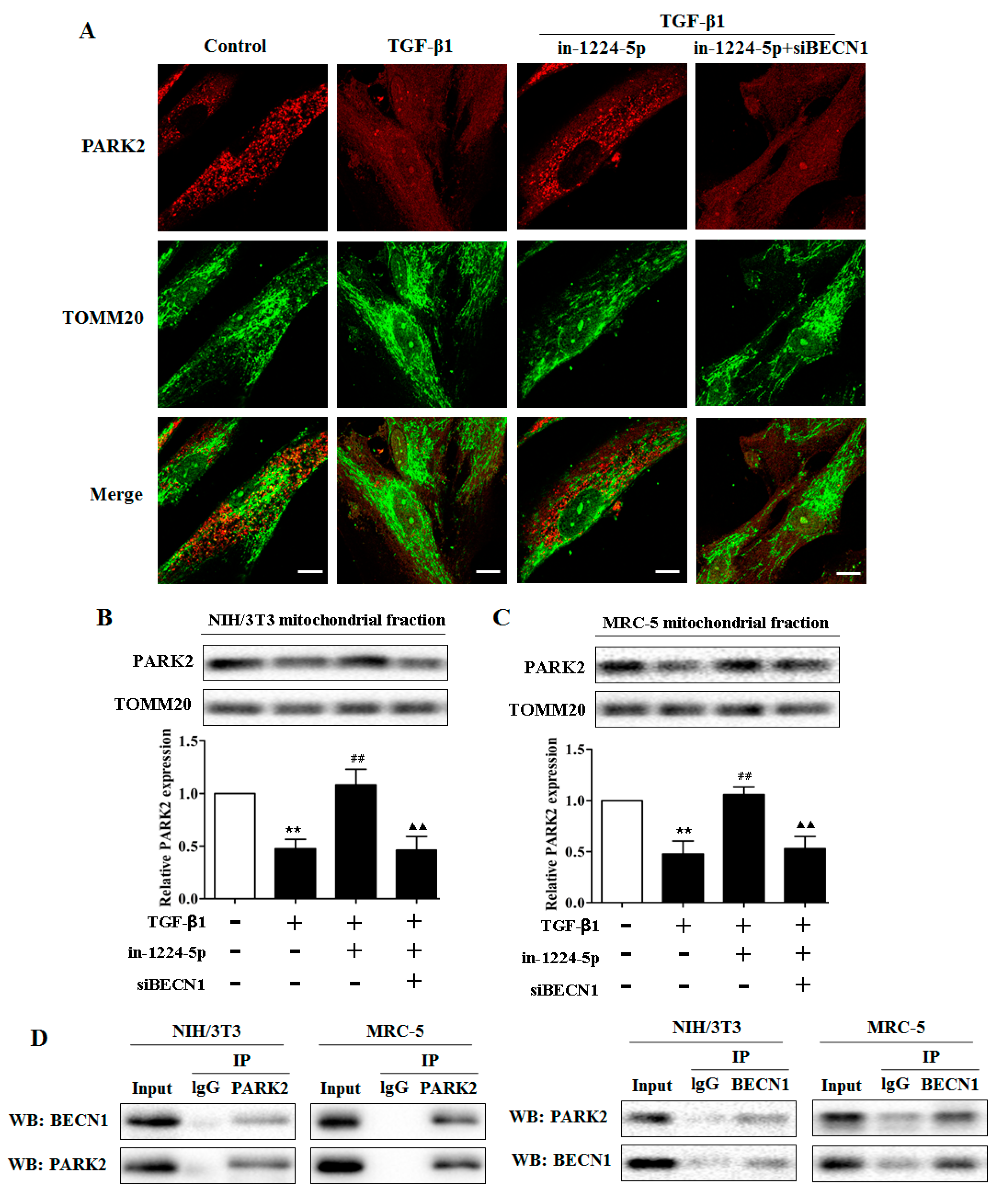
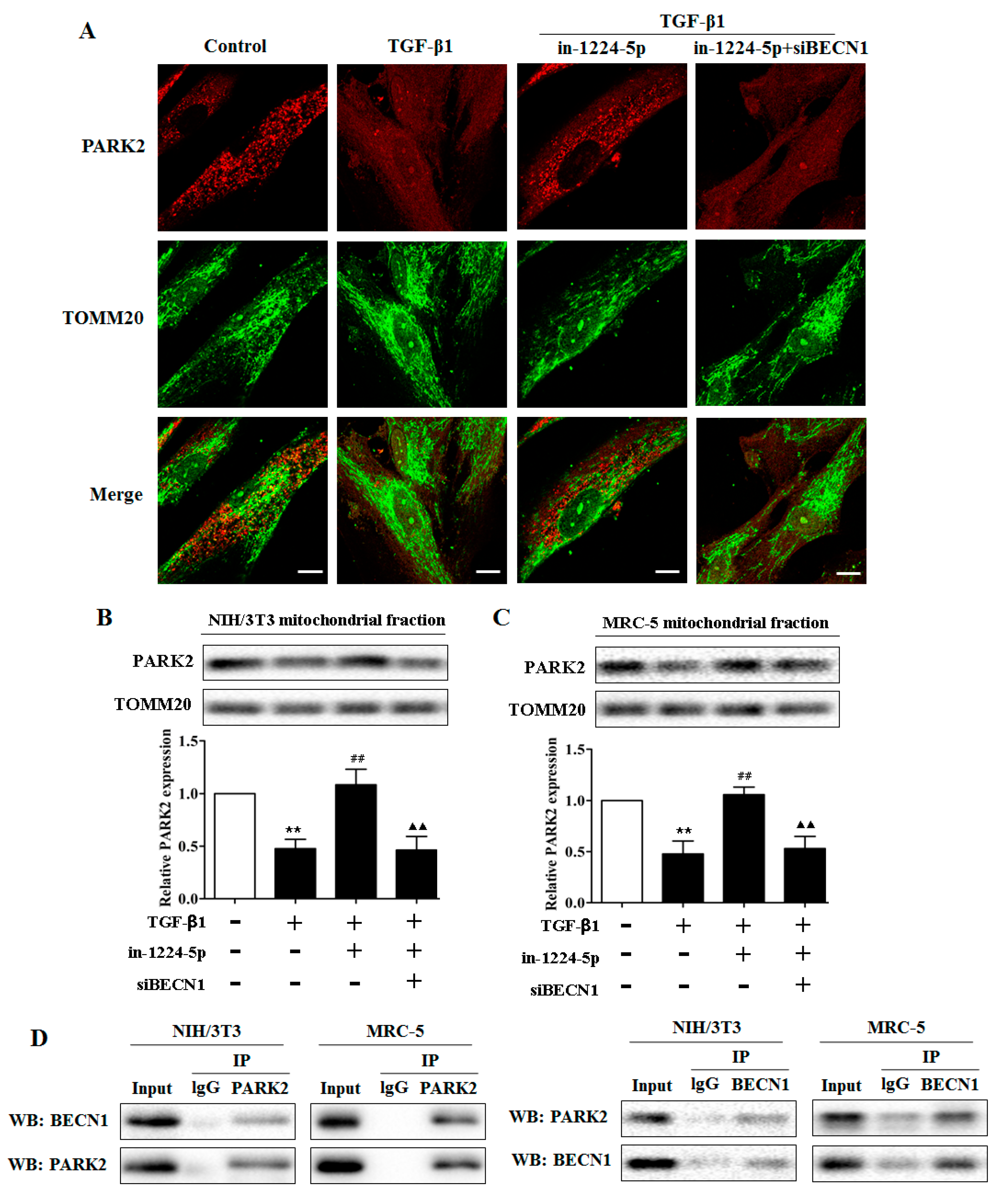
2.4. BECN1 Facilitates PARK2 Translocation to Mitochondria
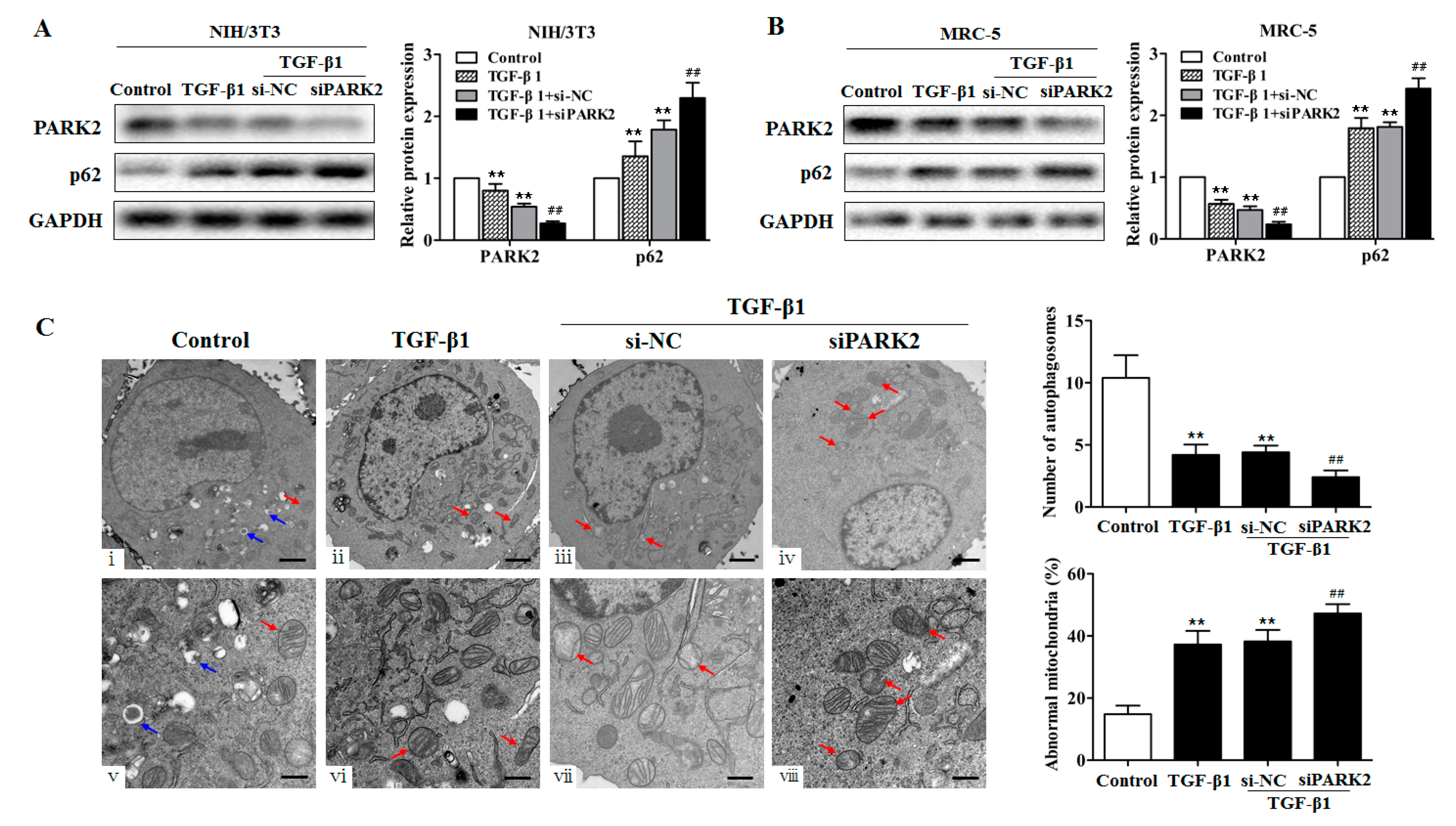
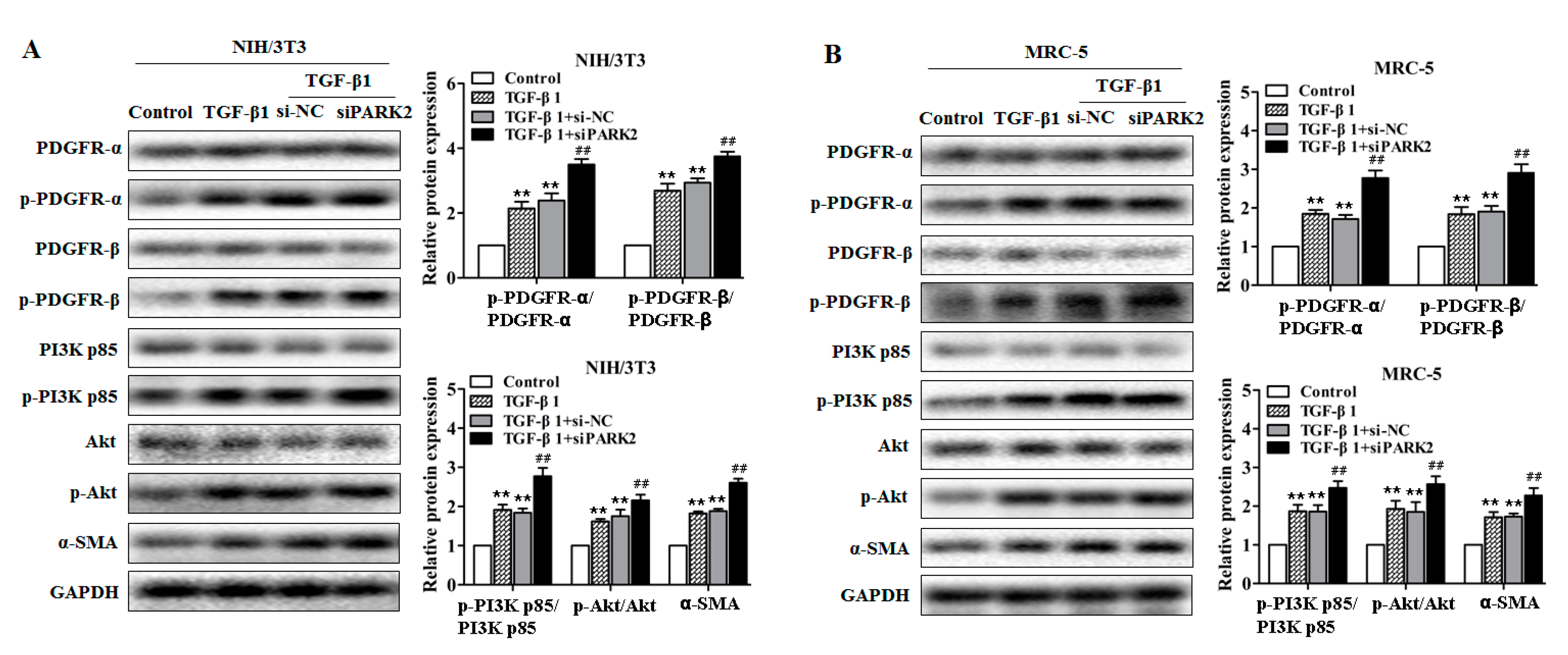
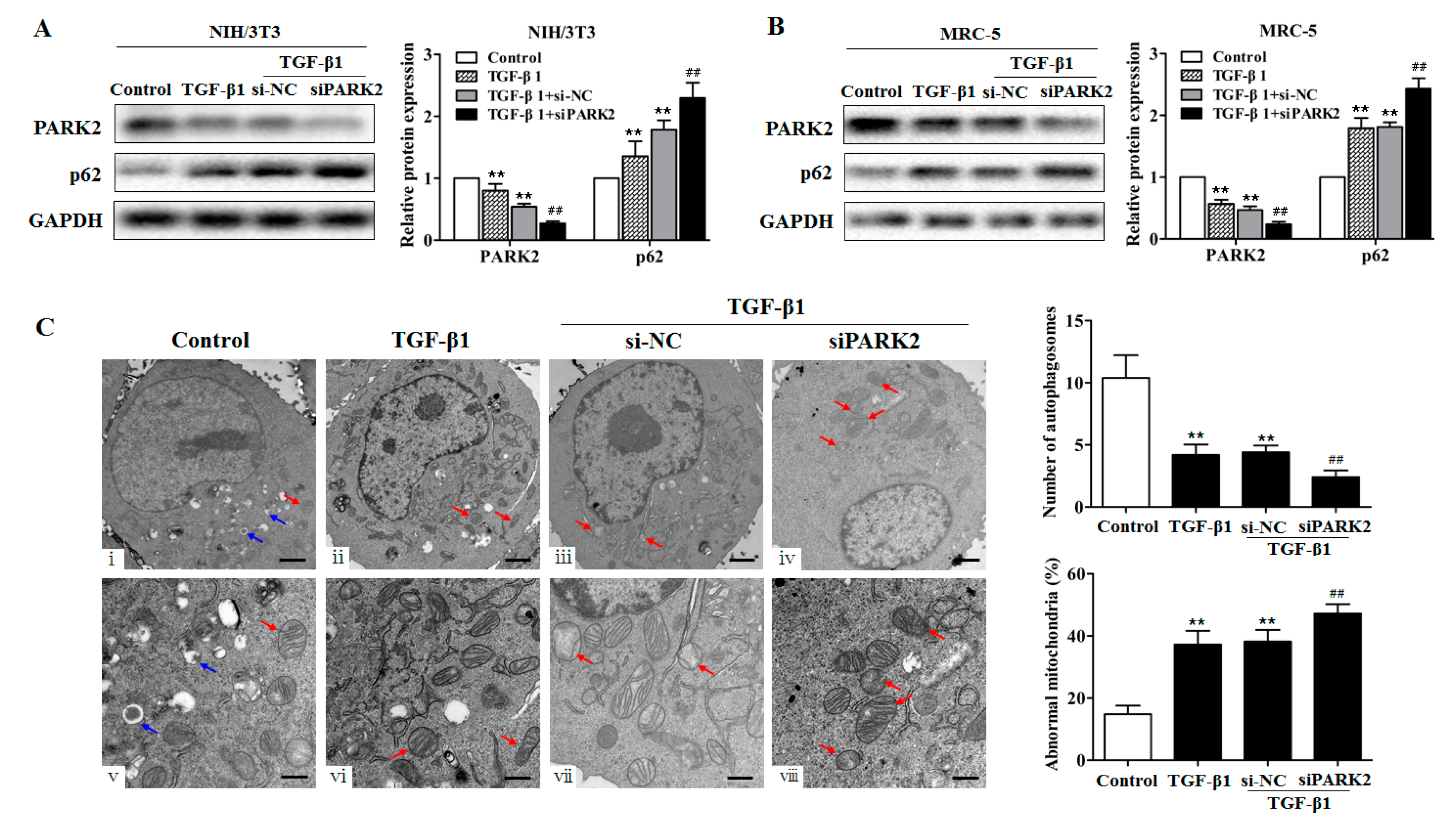
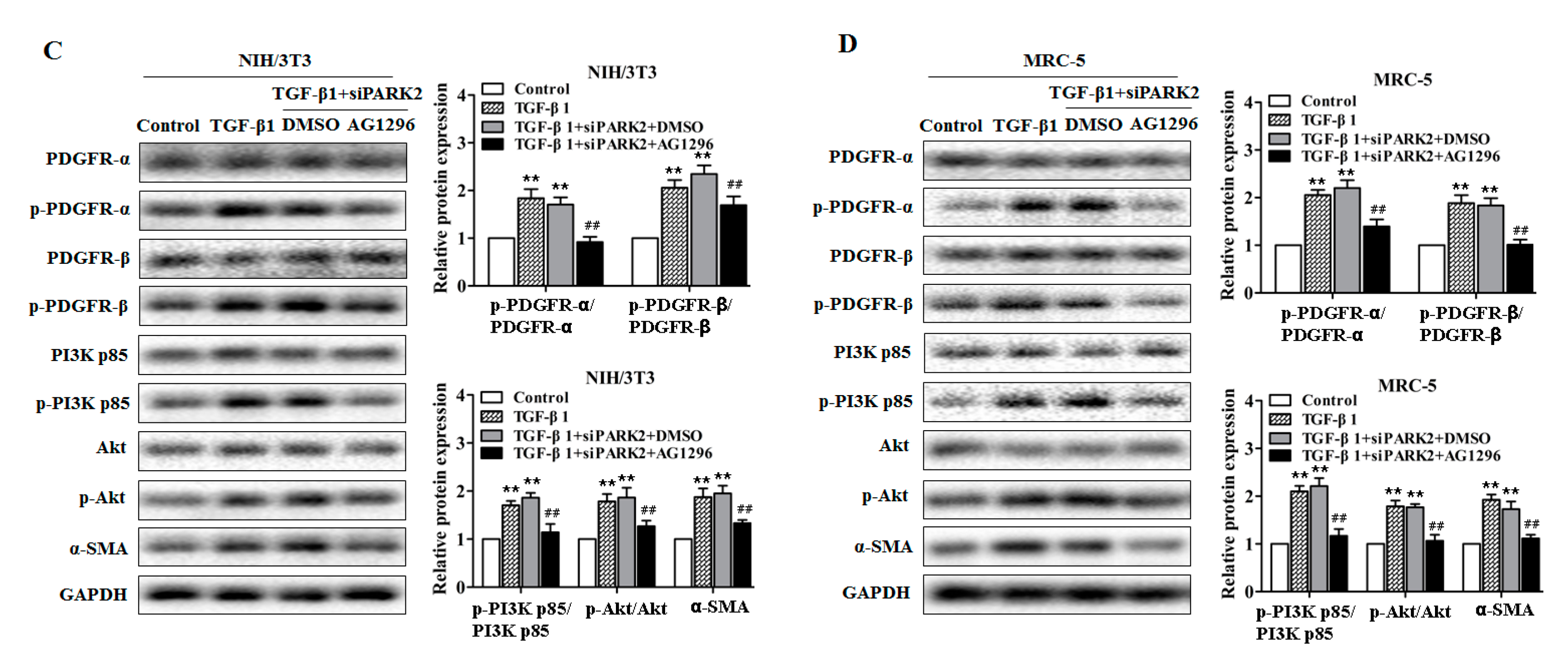
2.5. PARK2 Knockdown Suppresses Mitophagy, Activates PDGFRs/PI3K/AKT Signaling Pathway and α-SMA Expression in Fibroblasts
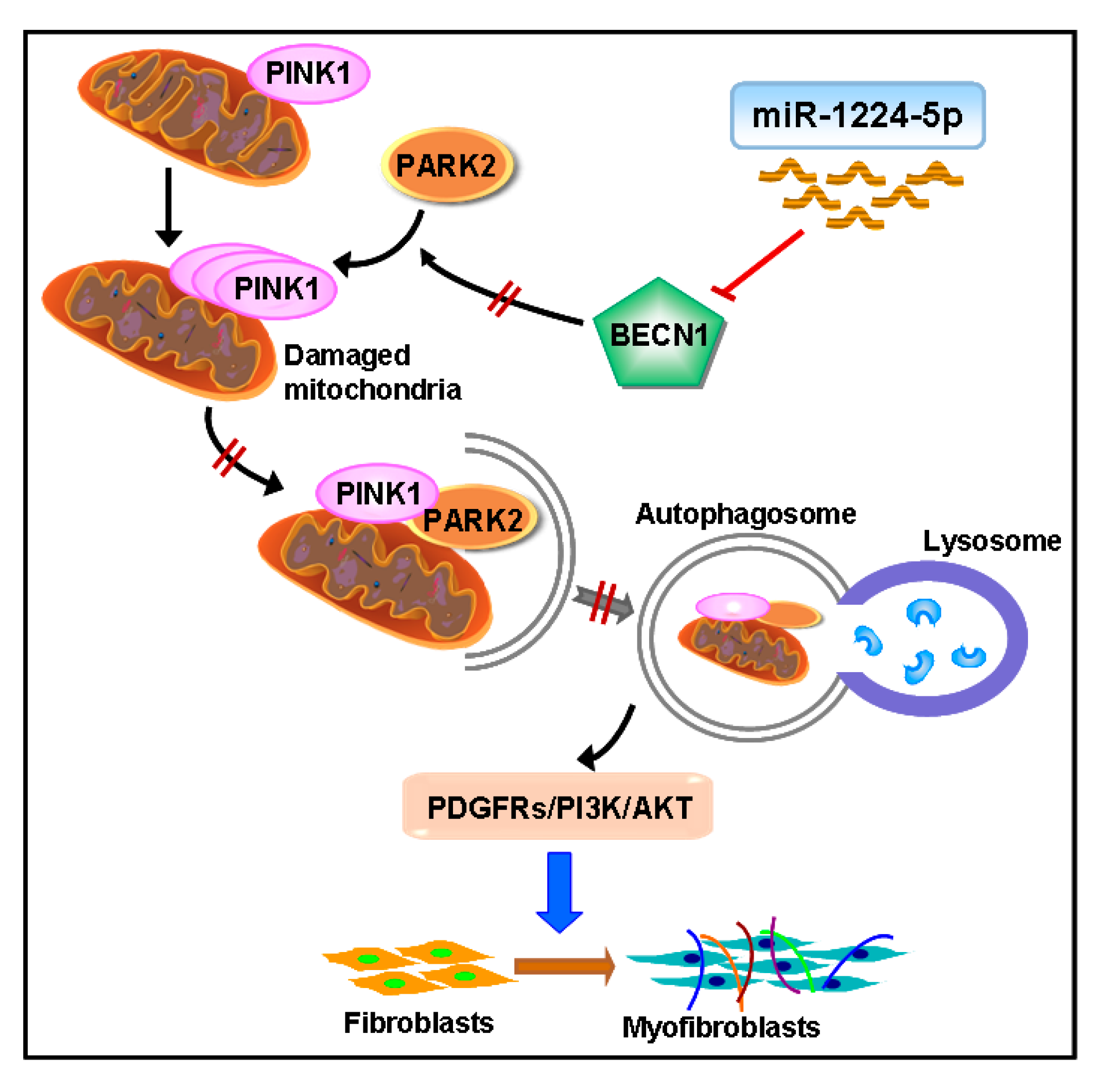
3. Discussion
4. Materials and Methods
4.1. Animals and Grouping
4.2. Fibroblast Culture and Treatment
4.3. Histological Analysis
4.4. Quantitative Real Time PCR (qRT-PCR)
4.5. Western Blot
4.6. Luciferase Assays
4.7. miRNA Inhibitor and siRNA Transfection
4.8. Immunofluorescence Staining
4.9. Co-Immunoprecipitation Assay
4.10. Transmission Electron Microscopy
4.11. Mitochondria Isolation
4.12. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TGF-β1 | Transforming growth factor beta 1 |
| BECN1 | Beclin 1, autophagy-related |
| PARK2 | Parkinson disease (autosomal recessive, juvenile) 2 |
| PDGFR | Platelet-derived growth factor receptor |
| TGF-βRII | Transforming growth factor beta receptor 2 |
| TNF-α | Tumor necrosis factor alpha |
| LPS | Lipopolysaccharide |
| α-SMA | Alpha-smooth muscle actin |
| PINK1 | PTEN induced putative kinase 1 |
References
- Lee, S.; Hayashi, H.; Mastuzaki, H.; Kumagai-Takei, N.; Otsuki, T. Silicosis and autoimmunity. Curr. Opin. Allergy Clin. Immunol. 2017, 17, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.; Yu, I.T.; Chen, W. Silicosis. Lancet 2012, 379, 2008–2018. [Google Scholar] [CrossRef]
- Aggarwal, S.; Mannam, P.; Zhang, J. Differential regulation of autophagy and mitophagy in pulmonary diseases. American journal of physiology. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L433–L452. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Araya, J.; Minagawa, S.; Hara, H.; Saito, N.; Kadota, T. Involvement of park2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J. Immunol. 2016, 197, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. Park2-mediated mitophagy is involved in regulation of hbec senescence in copd pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.; Choi, M.E.; Hashimoto, S.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Autophagy: Friend or foe in lung disease? Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S1), S40–S47. [Google Scholar] [PubMed]
- Mischler, S.E.; Cauda, E.G.; Di Giuseppe, M.; McWilliams, L.J.; St Croix, C.; Sun, M.; Franks, J.; Ortiz, L.A. Differential activation of raw 264.7 macrophages by size-segregated crystalline silica. J. Occup. Med. Toxicol. 2016, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, T.; Li, Y.; Yao, W.; Ji, X.; Wu, Q.; Han, L.; Han, R.; Yan, W.; Yuan, J.; et al. Earthworm extract attenuates silica-induced pulmonary fibrosis through nrf2-dependent mechanisms. Lab. Investig. 2016, 96, 1279–1300. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle micrornas. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ye, H.; Xiang, F.; Song, L.J.; Zhou, L.L.; Cai, P.C.; Zhang, J.C.; Yu, F.; Shi, H.Z.; Su, Y.; et al. Mir-18a-5p inhibits sub-pleural pulmonary fibrosis by targeting tgf-beta receptor ii. Mol. Ther. 2017, 25, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wu, B.; Fan, J.; Han, R.; Luo, C.; Wang, T.; Yang, J.; Han, L.; Zhu, B.; Wei, D.; et al. The anti-fibrotic effects and mechanisms of microrna-486-5p in pulmonary fibrosis. Sci. Rep. 2015, 5, 14131. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Mo, D.; Qin, L.; Wang, C.; Li, A.; Zhao, X.; Wang, X.; Xiao, S.; Wang, Q.; Xie, Y.; et al. Lipopolysaccharide-induced mir-1224 negatively regulates tumour necrosis factor-alpha gene expression by modulating sp1. Immunology 2011, 133, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Seow, Y.; Curtis, H.; Weinberg, M.S.; Wood, M.J. Silencing of parkinson’s disease-associated genes with artificial mirtron mimics of mir-1224. Nucleic Acids Res. 2012, 40, 9863–9875. [Google Scholar] [CrossRef] [PubMed]
- Divya, T.; Sureshkumar, A.; Sudhandiran, G. Autophagy induction by celastrol augments protection against bleomycin-induced experimental pulmonary fibrosis in rats: Role of adaptor protein p62/sqstm1. Pulm. Pharmacol. Ther. 2017, 45, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.; Wollin, L.; Wirth, J.; Gantner, F.; Lämmle, B.; Wex, E. Olodaterol shows anti-fibrotic efficacy in vitro and in vivo models of pulmonary fibrosis. Br. J. Pharmacol. 2017, 74, 3848–3864. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Zhou, J. CXCR7 attenuates the TGF-β-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. Biosyst. 2017, 13, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. Becn1 is involved in the initiation of mitophagy: It facilitates park2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Tsai, C.W. Carnosic acid attenuates 6-hydroxydopamine-induced neurotoxicity in sh-sy5y cells by inducing autophagy through an enhanced interaction of parkin and beclin1. Mol. Neurobiol. 2017, 54, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, S.; Sun, Y.; Li, S.; Ning, Y.; Dong, Y.; Shang, Y.; Bai, C. Microrna-34/449 targets igfbp-3 and attenuates airway remodeling by suppressing nur77-mediated autophagy. Cell Death Dis. 2017, 8, e2998. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Ji, X.; Rong, R.; Li, Y.; Yao, W.; Yuan, J.; Wu, Q.; Yang, J.; Yan, W.; Han, L.; et al. Mir-449a regulates autophagy to inhibit silica-induced pulmonary fibrosis through targeting bcl2. J. Mol. Med. (Berl.) 2016, 94, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Cheng, Y.; Yang, J.; Wang, W.; Fang, S.; Zhang, W.; Han, B.; Zhou, Z.; Yao, H.; Chao, J.; et al. Bbc3 in macrophages promoted pulmonary fibrosis development through inducing autophagy during silicosis. Cell Death Dis. 2017, 8, e2657. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. Usp8 and park2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase pink1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage akt1 kinase-mediated mitophagy modulates apoptosis resistance and pulmonary fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Lai, Y.C.; Romero, Y.; Brands, J.; St Croix, C.M.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.S.; et al. Pink1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. Microrna-137 is a novel hypoxia-responsive microrna that inhibits mitophagy via regulation of two mitophagy receptors fundc1 and nix. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.Q.; Zhang, L.; Wan, H.Y.; Liu, M.; Li, X.; Tang, H. Creb1-driven expression of mir-320a promotes mitophagy by down-regulating vdac1 expression during serum starvation in cervical cancer cells. Oncotarget 2015, 6, 34924–34940. [Google Scholar] [PubMed]
- Ricci, A.; Cherubini, E.; Scozzi, D.; Pietrangeli, V.; Tabbi, L.; Raffa, S.; Leone, L.; Visco, V.; Torrisi, M.R.; Bruno, P.; et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J. Cell. Physiol. 2013, 228, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. Pink1 and becn1 relocalize at mitochondria-associated membranes during mitophagy and promote er-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. Pink1- and park2-mediated local mitophagy in distal neuronal axons. Autophagy 2015, 11, 187–189. [Google Scholar] [PubMed]
- McWilliams, T.G.; Muqit, M.M. Pink1 and parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and pink1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Jacoupy, M.; Corvol, J.C.; Corti, O. Pink1/parkin-dependent mitochondrial surveillance: From pleiotropy to parkinson’s disease. Front. Mol. Neurosci. 2017, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Suliman, H.B. Mitochondrial dysfunction in lung pathogenesis. Annu. Rev. Physiol. 2017, 79, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. Pdgf-a and pdgf-b induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Dadrich, M.; Nicolay, N.H.; Flechsig, P.; Bickelhaupt, S.; Hoeltgen, L.; Roeder, F.; Hauser, K.; Tietz, A.; Jenne, J.; Lopez, R.; et al. Combined inhibition of tgfbeta and pdgf signaling attenuates radiation-induced pulmonary fibrosis. Oncoimmunology 2016, 5, e1123366. [Google Scholar] [CrossRef] [PubMed]
- Borkham-Kamphorst, E.; Weiskirchen, R. The pdgf system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Xing, R.G.; Chen, L.; Liu, C.R.; Miao, Z.G. Pi3k/akt signaling is involved in the pathogenesis of bleomycininduced pulmonary fibrosis via regulation of epithelialmesenchymal transition. Mol. Med. Rep. 2016, 14, 5699–5706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, Y.; Liang, C.; Yang, W. Ccn5 overexpression inhibits profibrotic phenotypes via the pi3k/akt signaling pathway in lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis and in an in vivo model of lung fibrosis. Int. J. Mol. Med. 2014, 33, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Sharawy, M.H.; El-Agamy, D.S.; Shalaby, A.A.; Ammar el, S.M. Protective effects of methyl palmitate against silica-induced pulmonary fibrosis in rats. Int. Immunopharmacol. 2013, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Lesion Severity Grade | Average Severity Grade | Lesion Distribution Grade | Average Distribution Grade | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 0 | 1 | 2 | 3 | 4 | 5 | |||
| Saline | 6 | 0 | 6 | 0 | ||||||||||
| Silica | 1 | 1 | 1 | 3 | 3.5 ± 1.8 ** | 1 | 3 | 1 | 1 | 2.5 ± 1.4 ** | ||||
| Silica + anta-NC | 1 | 2 | 1 | 2 | 3.2 ± 1.7 ** | 2 | 1 | 2 | 1 | 2.5 ± 1.5 ** | ||||
| Silica + anta-1224-5p | 3 | 2 | 1 | 1.0 ± 1.5 ## | 3 | 2 | 1 | 0.8 ± 1.2 ## | ||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Xu, T.; Liu, Y.; Li, Y.; Yuan, J.; Yao, W.; Xu, Q.; Yan, W.; Ni, C. miR-1224-5p Mediates Mitochondrial Damage to Affect Silica-Induced Pulmonary Fibrosis by Targeting BECN1. Int. J. Mol. Sci. 2017, 18, 2357. https://doi.org/10.3390/ijms18112357
Wu Q, Xu T, Liu Y, Li Y, Yuan J, Yao W, Xu Q, Yan W, Ni C. miR-1224-5p Mediates Mitochondrial Damage to Affect Silica-Induced Pulmonary Fibrosis by Targeting BECN1. International Journal of Molecular Sciences. 2017; 18(11):2357. https://doi.org/10.3390/ijms18112357
Chicago/Turabian StyleWu, Qiuyun, Tiantian Xu, Yi Liu, Yan Li, Jiali Yuan, Wenxi Yao, Qi Xu, Weiwen Yan, and Chunhui Ni. 2017. "miR-1224-5p Mediates Mitochondrial Damage to Affect Silica-Induced Pulmonary Fibrosis by Targeting BECN1" International Journal of Molecular Sciences 18, no. 11: 2357. https://doi.org/10.3390/ijms18112357