
Identifying Virulence-Associated Genes Using Transcriptomic and Proteomic Association Analyses of the Plant Parasitic Nematode Bursaphelenchus mucronatus

Abstract
:
1. Introduction
2. Results
2.1. Identification of the Re-Isolated Nematodes
2.2. Quantitative Transcript and Protein Profiling
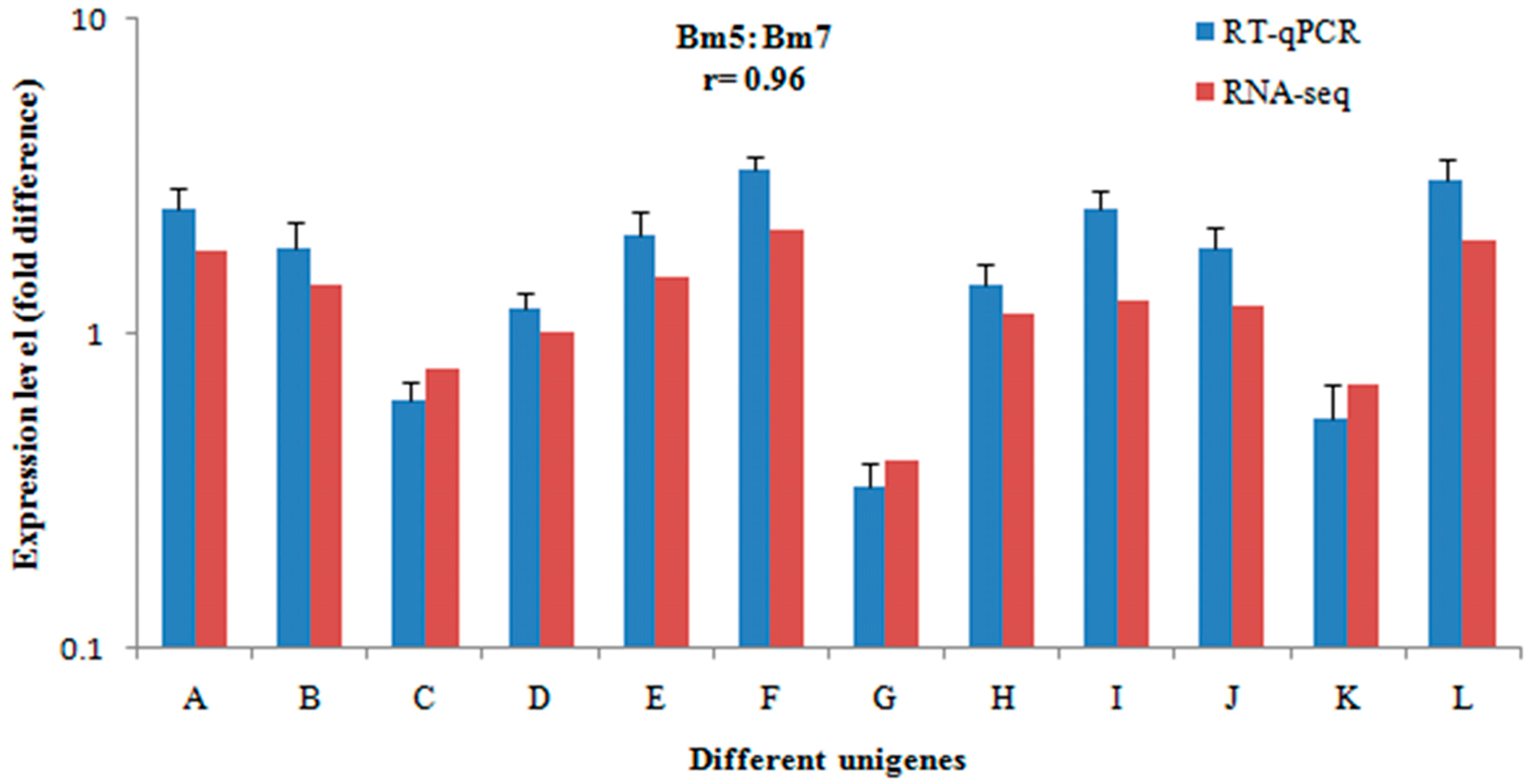
2.3. Validation of Gene Expression Data Using Real-Time Quantitative PCR (qRT-PCR)
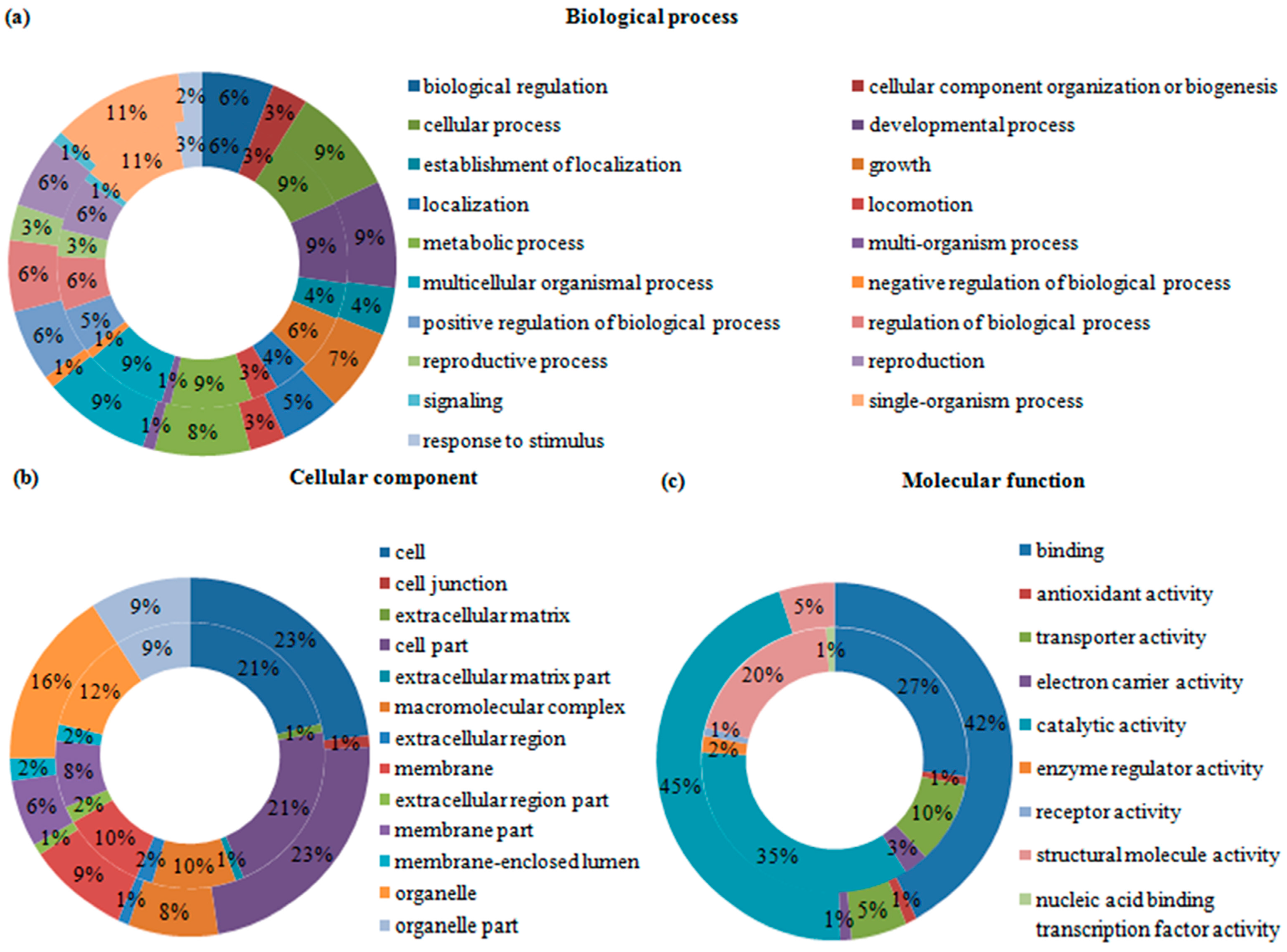
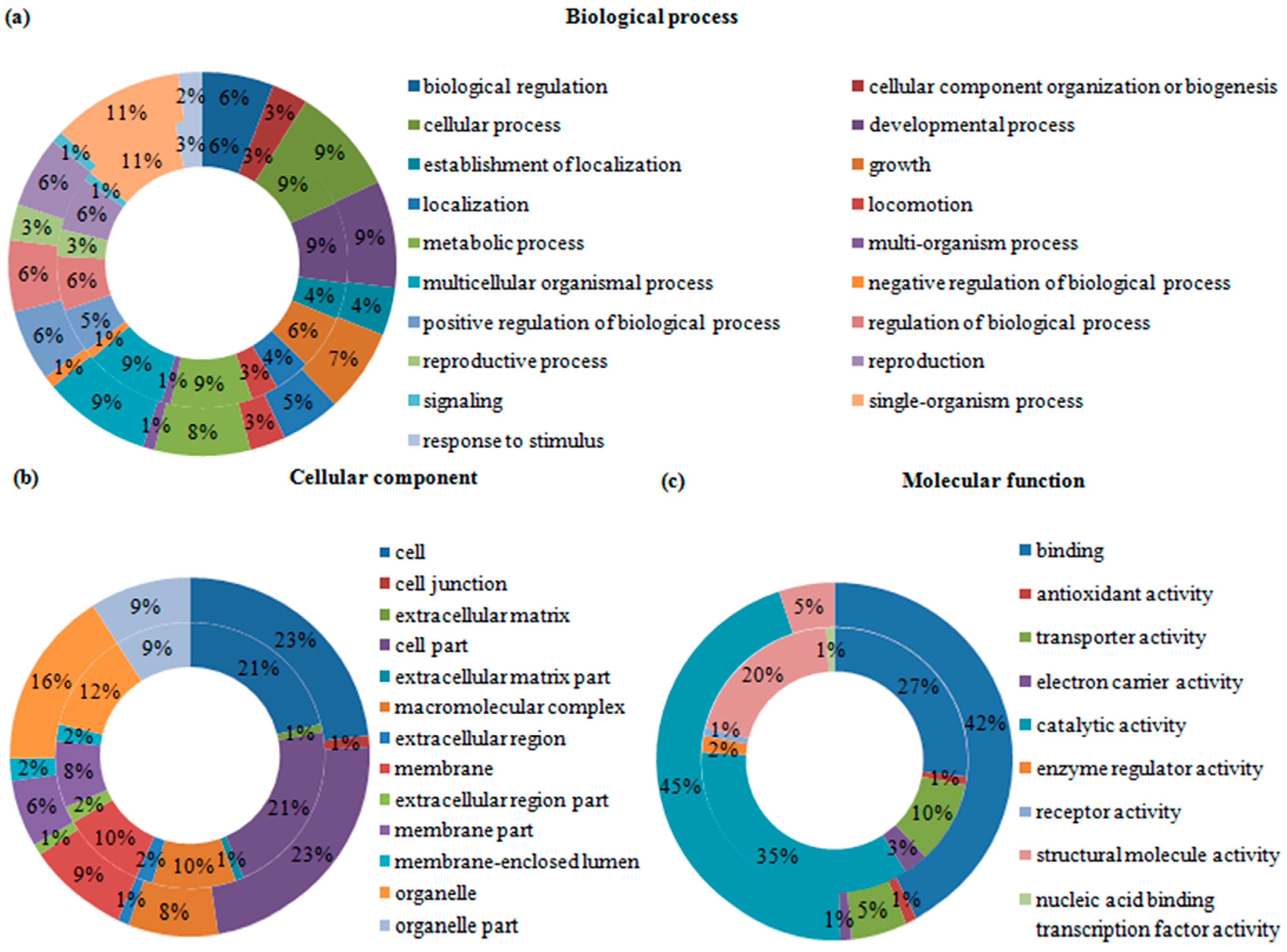
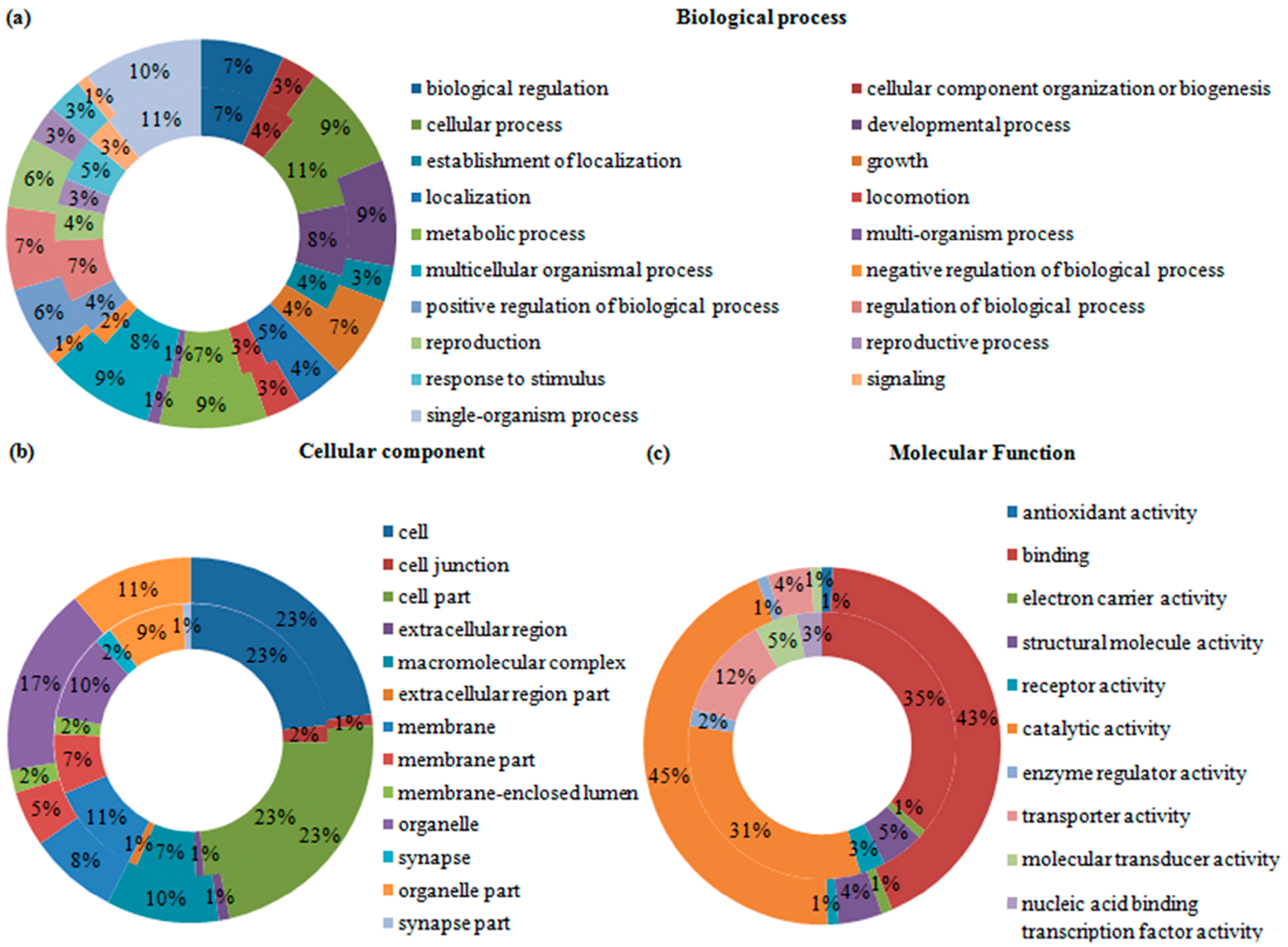
2.4. Gene Ontology (GO) Analysis of Differentially Expressed Transcripts and Proteins
2.5. KEGG Pathway Enrichment Analysis of Differentially Expressed Genes and Proteins
2.6. Correlation Analysis of Protein and RNA Expression
2.7. Analysis of Pathogenesis-Related Genes
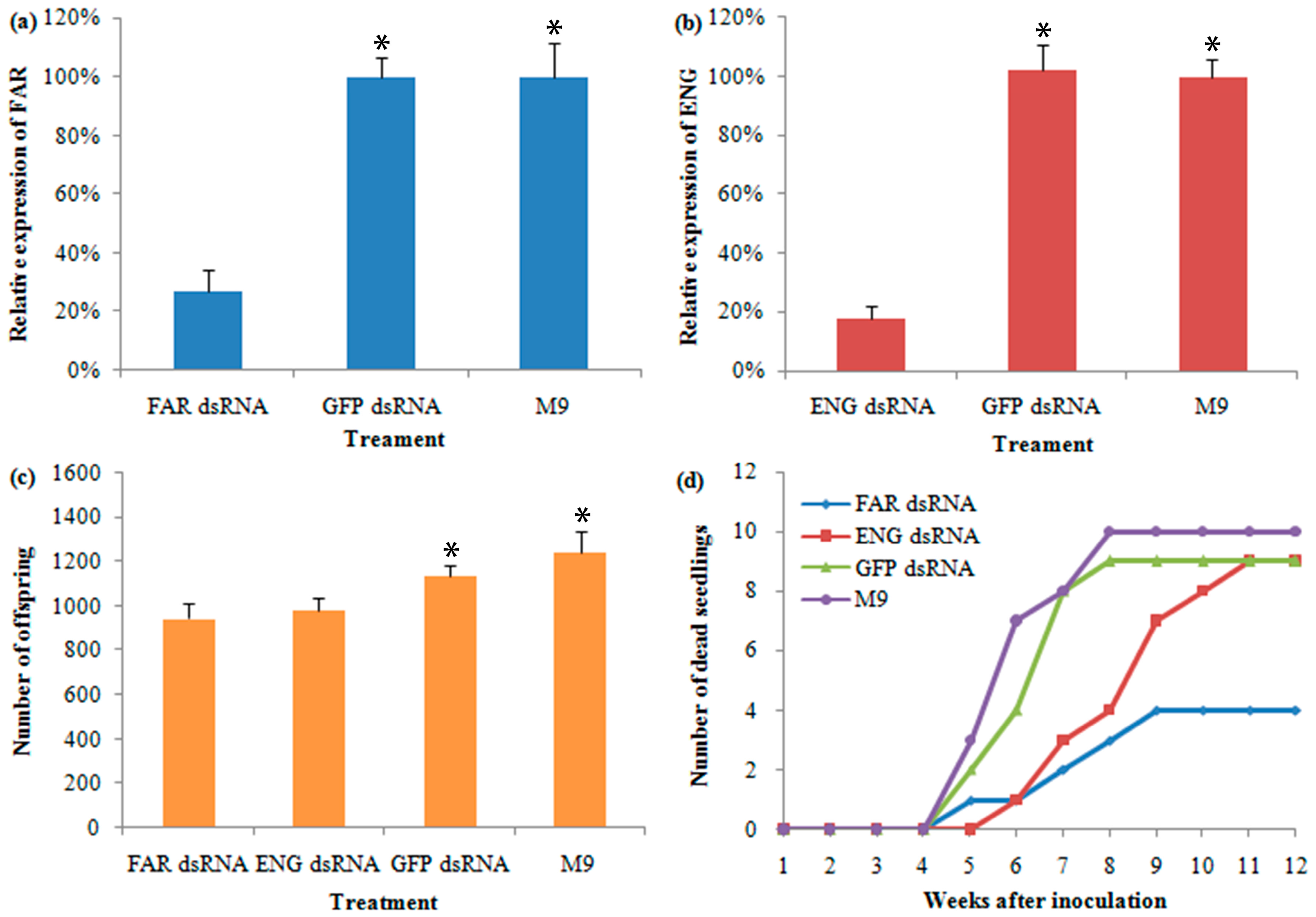
2.8. Characterization and Analysis of RNAi in B. mucronatu
3. Discussion
4. Materials and Methods
4.1. Nematode Sample Preparation
4.2. RNA Preparation and Transcriptome Sequencing
4.3. Redundant Data Filtering and de Novo Assembly
4.4. Unigene Annotation and Classification
4.5. qRT-PCR
4.6. Protein Preparation and iTRAQ Labelling
4.7. Strong Cation Exchange (SCX) Fractionation and LC-ESI-MS/MS Analysis
4.8. Proteomic Data Analysis
4.9. Construction and Analysis of RNAi
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Braasch, H. Bursaphelenchus species in conifers in Europe: Distribution and morphological relationships. Bull. OEPP/EPPO Bull. 2001, 31, 127–142. [Google Scholar] [CrossRef]
- Ryss, A.; Polyanina, K.S.; Popovichev, B.G.; Subbotin, S.A. Description of Bursaphelenchus ulmophilus sp. n. (Nematoda: Parasitaphelenchinae) associated with Dutch elm disease of Ulmus glabra Huds. in the Russian North West. Nematology 2015, 17, 685–703. [Google Scholar] [CrossRef]
- Webster, J.M.; Anderson, R.V.; Baillie, D.L.; Beckenbach, K.; Curran, J.; Rutherford, T.A. DNA probes for differentiating isolates of the pinewood nematode species complex. Rev. Nématol. 1990, 13, 255–263. [Google Scholar]
- Bakke, A.; Anderson, R.V.; Kvamme, T. Pathogenicity of the nematodes Bursaphelenchus xylophilus and B. mucronatus to Pinus sylvestris seedlings: A greenhouse test. Scand. J. For. Res. 1991, 6, 407–412. [Google Scholar] [CrossRef]
- Skarmoutsos, G.; Michalopoulos-Skarmoutsos, H. Pathogenicity of Bursaphelenchus sexdentati, Bursaphelenchus leoni and Bursaphelenchus hellenicus on European pine seedlings. For. Pathol. 2000, 30, 149–156. [Google Scholar]
- Kanzaki, N.; Aikawa, T.; Maehara, N.; Ichihara, Y. An inoculation experiment of Japanese Bursaphelenchus nematodes on Japanese black and red pine, Pinus thunbergii and P. densiflora. J. For. Res. 2010, 16, 325–330. [Google Scholar] [CrossRef]
- Shinya, R.; Ueda, M.; Futai, K.; Takeuchi, Y.; Kuroda, K.; Miura, N. Surface coat proteins of the pine wood nematode, Bursaphelenchus xylophilus: Profiles of stage- and isolate-specific characters. Nematology 2009, 11, 429–438. [Google Scholar] [CrossRef]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.; et al. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Wu, X.; Huang, L.; Tian, M.; Ye, J. Specifically expressed genes of the nematode Bursaphelenchus xylophilus involved with early interactions with pine trees. PLoS ONE 2013, 8, e78063. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Wu, X.Q.; Ye, J.R.; Huang, L. Molecular characterization and functional analysis of three pathogenesis-related cytochrome p450 genes from Bursaphelenchus xylophilus (Tylenchida: Aphelenchoidoidea). Int. J. Mol. Sci. 2015, 16, 5216–5234. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.L.; Ye, J.R.; Wu, X.Q.; Huang, L.; Zhu, L.H.; Lin, S.X. Deep sequencing analyses of pine wood nematode Bursaphelenchus xylophilus microRNAs reveal distinct miRNA expression patterns during the pathological process of pine wilt disease. Gene 2015, 555, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Palomares-Rius, J.E.; Tsai, I.J.; Karim, N.; Akiba, M.; Kato, T.; Maruyama, H.; Takeuchi, Y.; Kikuchi, T. Genome-wide variation in the pinewood nematode Bursaphelenchus xylophilus and its relationship with pathogenic traits. BMC Genom. 2015, 16, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente, C.S.; Ikuyo, Y.; Shinya, R.; Mota, M.; Hasegawa, K. Catalases induction in high virulence pinewood nematode Bursaphelenchus xylophilus under hydrogen peroxide-induced stress. PLoS ONE 2015, 10, e0123839. [Google Scholar] [CrossRef] [PubMed]
- Kulinich, O.A.; Kruglic, I.; Eroshenko, A.S.; Kolosova, N.V. Occurrence and distribution of the nematode Bursaphelenchus mucronatus in the Russian Far East. Russ. J. Nematol. 1994, 2, 113–120. [Google Scholar]
- Pereira, F.; Moreira, C.; Fonseca, L.; van Asch, B.; Mota, M.; Abrantes, I.; Amorim, A. New insights into the phylogeny and worldwide dispersion of two closely related nematode species, Bursaphelenchus xylophilus and Bursaphelenchus mucronatus. PLoS ONE 2013, 8, e56288. [Google Scholar] [CrossRef] [PubMed]
- Mamiya, Y.; Enda, N. Bursaphelenchus mucronatus n. sp. (Nematoda: Aphelenchoididae) from pine wood and its biology and pathogenicity to pine trees. Nematologica 1979, 3, 353–361. [Google Scholar] [CrossRef]
- Mamiya, Y.; Kiyohara, T. Description of Bursaphelenchus lignicolus n. sp. (Nematoda: Aphelenchoididae) from pine wood and histopathology of nematode-infested trees. Nematologica 1971, 18, 120–124. [Google Scholar] [CrossRef]
- Wingfield, M.J.; Blanchette, R.A. The pine-wood nematode, Bursaphelenchus xylophilus, in Minnesota and Wisconsin: Insect associates and transmission studies. Can. J. For. Res. 1983, 13, 1068–1076. [Google Scholar] [CrossRef]
- Schroeder, L.M.; Magnusson, C. Transmission of Bursaphelenchus mucronatus (Nematoda) to branches and bolts of Pinus sylvestris and Picea abies by the cerambycid beetle Monochamus sutor. Scand. J. For. Res. 1992, 7, 107–112. [Google Scholar] [CrossRef]
- Kobayashi, F.; Yamane, A.; Ikeda, T. The Japanese pine sawyer beetle as the vector of pine wilt disease. Annu. Rev. Entomol. 2003, 29, 115–135. [Google Scholar] [CrossRef]
- Wang, Y.; Yamada, T.; Sakaue, D.; Suzuki, K. Variations in life history parameters and their influence on rate of population increase of different pathogenic isolates of the pine wood nematode, Bursaphelenchus xylophilus. Nematology 2005, 7, 459–467. [Google Scholar] [CrossRef]
- Futai, K. Population dynamics of Bursaphelenchus lignicolus (Nematoda: Aphelenchoididae) and B. mucronatus in pine seedlings. Appl. Entomol. Zool. 1980, 15, 458–464. [Google Scholar]
- Tomminen, J. Pathogenicity studies with Bursaphelenchus mucronatus in Scots pine in Finland. Eur. J. For. Pathol. 1993, 23, 236–243. [Google Scholar] [CrossRef]
- Braasch, H. Pathogenitäts tests mit Bursaphelenchus mucronatus an Kiefern und Fichtensämlingen in Deutschland. Eur. J. For. Pathol. 1996, 26, 205–216. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Lin, M.; Yu, B.Y. Pathogenicity of Bursaphelenchus mucronatus on the seedlings of black pine. J. Nanjing Agric. Univ. 2004, 27, 46–50. [Google Scholar]
- Akbulut, S.; Yuksel, B.; Serin, M.; Baysal, I.; Erdem, M. Pathogenicity of Bursaphelenchus mucronatus in pine seedlings under greenhouse conditions. Turk. J. Agric. For. 2007, 31, 169–173. [Google Scholar]
- Dayi, M.; Akbulut, S. Pathogenicity testing of four Bursaphelenchus species on conifer seedlings under greenhouse conditions. For. Pathol. 2012, 42, 213–219. [Google Scholar] [CrossRef]
- Akbulut, S.; Yüksel, B.; Serin, M.; Erdem, M. Comparison of pathogenic potential of Bursaphelenchus species on conifer seedlings between greenhouse and outdoor conditions. Phytoparasitica 2014, 43, 209–214. [Google Scholar] [CrossRef]
- Zhou, L.F.; Chen, F.M.; Wang, J.C.; Pan, H.Y.; Ye, J.R. Virulence of Bursaphelenchus mucronatus to pine seedlings and trees under field conditions. For. Pathol. 2016. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Reidegeld, K.A.; Meyer, H.E.; Warscheid, B. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. Proteomics 2007, 7, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Zieske, L.R. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.A.; Palanisamy, N.; Brenner, J.C.; Cao, X.; Kalyana-Sundaram, S.; Luo, S.; Khrebtukova, I.; Barrette, T.R.; Grasso, C.; Yu, J.; et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 12353–12358. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Cheng, X.Y.; Wang, Y.S.; Luo, J.; Mao, Z.C.; Ferris, V.R.; Xie, B.Y. Comparative transcriptomics of two pathogenic pinewood nematodes yields insights into parasitic adaptation to life on pine hosts. Gene 2012, 505, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Trampczynska, A.; Clemens, S. Comparative transcriptome analysis of toxic metal responses in Arabidopsis thaliana and the Cd2+-hypertolerant facultative metallophyte Arabidopsis halleri. Plant Cell Environ. 2006, 29, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.U.; Chen, G.Q. Identification of hydroxy fatty acid and triacylglycerol metabolism-related genes in lesquerella through seed transcriptome analysis. BMC Genom. 2015, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hu, H.; Yu, D.; Sun, Z.; He, X.; Zhang, J.; Chen, Q.; Tian, R.; Fan, J. Candidate resistant genes of sand pear (Pyrus pyrifolia Nakai) to Alternaria alternata revealed by transcriptome sequencing. PLoS ONE 2015, 10, e0135046. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Mu, H.; Zhang, H.; Chandramouli, K.H.; Qian, P.-Y.; Wong, C.K.C.; Qiu, J.-W. Understanding the regulation of estivation in a freshwater snail through iTRAQ-based comparative proteomics. J. Proteome Res. 2013, 12, 5271–5280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.Y.; Yan, H.; Wang, L.L.; Yan, W.J.; Shui, Y.B.; Beebe, D.C. Quantitative proteomics analysis by iTRAQ in human nuclear cataracts of different ages and normal lens nuclei. Proteom. Clin. Appl. 2015, 9, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Chen, F.; Duan, S.; Gao, X.; Liu, G.; Chen, Y.; Dixon, W.; Xiao, H.; Cao, Y. iTRAQ-based quantitative proteomic analysis of the antimicrobial mechanism of peptide F1 against Escherichia coli. J. Agric. Food Chem. 2015, 63, 7190–7197. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.M. National Eradication Programme for the Pinewood Nematode. In Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Mota, M.M., Vieira, P., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 5–14. [Google Scholar]
- Ding, X.; Ye, J.; Lin, S.; Wu, X.; Li, D.; Nian, B. Deciphering the molecular variations of pine wood nematode Bursaphelenchus xylophilus with different virulence. PLoS ONE 2016, 11, e0156040. [Google Scholar] [CrossRef] [PubMed]
- Futai, K. Developmental rate and population growth of Bursaphelenchus lignicolus (Nematoda: Aphelenchoididae) and B. mucronatus. Appl. Entomol. Zool. 1980, 15, 115–122. [Google Scholar]
- Iwahori, H.; Futai, K. Comparative movement speed of pathogenic and nonpathogenic isolates of Bursaphelenchus nematodes. Appl. Entomol. Zool. 1995, 30, 159–167. [Google Scholar]
- De Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Silva, G.M.; Marcotte, E.M. Protein expression regulation under oxidative stress. Mol. Cell. Proteom. 2011, 10, M111-009217. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, K.C.; Vermeire, A.C.J.J.; Harrison, A.L.M.; Bungiro, A.R.D.; Grant, D.W.; Husain, B.S.Z.; Cappello, M. Characterization of a fatty acid and retinol binding protein orthologue from the hookworm Ancylostoma ceylanicum. Int. J. Parasitol. 2009, 39, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Iberkleid, I.; Vieira, P.; de Almeida Engler, J.; Firester, K.; Spiegel, Y.; Horowitz, S.B. Fatty acid- and retinol-binding protein, Mj-FAR-1 induces tomato host susceptibility to root-knot nematodes. PLoS ONE 2013, 8, e64586. [Google Scholar] [CrossRef] [PubMed]
- Tripp, C.; Frank, R.S.; Selkirk, M.E.; Liang, T.; Grieve, M.M.; Frank, G.R.; Grieve, R.B. Dirofilaria immitis: molecular cloning and expression of a cDNA encoding a selenium-independent secreted glutathione peroxidase. Exp. Parasitol. 1998, 88, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Reavy, B.; Smant, G.; Prior, A.E. Glutathione peroxidases of the potato cyst nematode Globodera rostochiensis. Gene 2004, 324, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.; Robertson, W.; Sobczak, M.; Helder, J.; Tetaud, E.; Ariyanayagam, M.; Ferguson, M.; Fairlamb, A.; Jones, J. Cloning, expression and functional characterisation of a peroxiredoxin from the potato cyst nematode Globodera rostochiensis. Mol. Biochem. Parasitol. 2000, 111, 41–49. [Google Scholar] [CrossRef]
- Smant, G.; Stokkermans, J.P.; Yan, Y.; de Boer, J.M.; Baum, T.J.; Wang, X.; Hussey, R.S.; Gommers, F.J.; Henrissat, B.; Davis, E.L. Endogenous cellulases in animals: isolation of β-1,4-endoglucanase genes from two species of plant-parasitic cyst nematodes. Proc. Natl. Acad. Sci. USA 1998, 95, 4906–4911. [Google Scholar] [CrossRef] [PubMed]
- Rosso, M.N.; Favery, B.; Piotte, C.; Arthaud, L.; Boer, J.M.D.; Hussey, R.S.; Bakker, J.; Baum, T.J.; Abad, P. Isolation of a cDNA encoding a β-1,4-endoglucanase in the root-knot nematode Meloidogyne incognita and expression analysis during plant parasitism. Mol. Plant-Microbe Interact. 1999, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Loosening of plant cell walls by expansins. Nature 2000, 407, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Ling, Q.; Urszula, K.; Roze, E.H.A.; Aska, G.; Herman, P.; Jeroen, N.; Hein, O.; Jones, J.T.; Arjen, S.; Geert, S. Plant degradation: A nematode expansin acting on plants. Nature 2004, 427, 30. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.B.; Lu, Q.; Liang, J.; Zhang, X.Y. Functional analysis of the cellulose gene of the pine wood nematode, Bursaphelenchus xylophilus, using RNA interference. Genet. Mol. Res. 2011, 10, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Rehman, S.G.; Jones, J.T. Functional analysis of pathogenicity proteins of the potato cyst nematode Globodera rostochiensis using RNAi. Mol. Plant-Microbe Interact. 2005, 18, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Bakhetia, M.; Urwin, P.E.; Atkinson, H.J. qPCR analysis and RNAi define pharyngeal gland cell-expressed genes of Heterodera glycines required for initial interactions with the host. Mol. Plant-Microbe Interact. 2007, 20, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Prior, A.; Jones, J.T.; Blok, V.C.; Beauchamp, J.; Mcdermott, L.; Cooper, A.; Kennedy, M.W. A surface-associated retinol- and fatty acid-binding protein (Gp-FAR-1) from the potato cyst nematode Globodera pallida: Lipid binding activities, structural analysis and expression pattern. Biochem. J. 2001, 356, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Viglierchio, D.R.; Schmitt, R.V. On the methodology of nematode extraction from field samples: Baermann funnel modifications. J. Nematol. 1983, 15, 438–444. [Google Scholar] [PubMed]
- Lewis, J.A.; Fleming, J.T. Basic culture methods. Methods Cell Biol. 1995, 48, 3–29. [Google Scholar] [PubMed]
- Kanzaki, N. Taxonomy and systematics of Bursaphelenchus nematodes (pine wilt disease). J. Jpn. For. Soc. 2006, 88, 392–406. [Google Scholar] [CrossRef]
- Burgermeister, W.; Kai, M.; Braasch, H.; Buchbach, E. ITS-RFLP patterns for differentiation of 26 Bursaphelenchus species (Nematoda: Parasitaphelenchidae) and observations on their distribution. Russ. J. Nematol. 2005, 29–42. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Z.; Dong, W.; Sun, C.; Wang, H.; Song, A.; He, L.; Fang, W.; Chen, F.; Teng, N. Transcriptomic and proteomic analysis reveals mechanisms of embryo abortion during chrysanthemum cross breeding. Sci. Rep. 2014, 4, 6536. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Hou, L.; Zhao, Y.; Huang, X.; Huang, Y.; Xia, S.; Gu, W.; Wang, W. iTRAQ-based proteomic study of the effects of Spiroplasma eriocheiris on Chinese mitten crab Eriocheir sinensis Hemocytes. Fish Shellfish Immunol. 2014, 40, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Hooiveld, G.J.; Zhang, W.; Li, M.; Zhao, F.; Zhu, J.; Xu, X.; Muller, M.; Li, C.; Zhou, G. Comparative proteomics provides insights into metabolic responses in rat liver to isolated soy and meat proteins. J. Proteome Res. 2016, 15, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Zhang, H.; Guo, X.; Cui, Q.; Wang, J.; Dai, J. Proteomic analysis of cell proliferation in a human hepatic cell line (HL-7702) induced by perfluorooctane sulfonate using iTRAQ. J. Hazard. Mater. 2015, 299, 361–370. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptome Data | Proteome Data | ||
|---|---|---|---|
| Raw reads (pair) | 114,154,604 | Total spectra | 329,868 |
| Clean reads (pair) | 107,097,176 | Peptide number | 16,205 |
| Total nucleotide length (bp) | 9,638,745,840 | Unique number | 12,452 |
| Unigene number | 40,355 | Protein number | 5092 |
| Average length (bp) | 1220 | - | - |
| N50 length (bp) | 2007 | - | - |
| Pathway | DEGs 1 | Genes 2 | p-Value | Pathway ID |
|---|---|---|---|---|
| Ribosome | 358 | 520 | 4.35 × 10−30 | ko03010 |
| Amoebiasis | 362 | 576 | 1.50 × 10−19 | ko05146 |
| Pathogenic Escherichia coli infection | 177 | 280 | 1.50 × 10−10 | ko05130 |
| Dilated cardiomyopathy | 213 | 356 | 2.76 × 10−9 | ko05414 |
| Focal adhesion | 452 | 837 | 8.36 × 10−9 | ko04510 |
| Alzheimer’s disease | 295 | 521 | 9.96 × 10−9 | ko05010 |
| Huntington’s disease | 288 | 515 | 7.34 × 10−8 | ko05016 |
| Cardiac muscle contraction | 222 | 387 | 1.67 × 10−7 | ko04260 |
| Hypertrophic cardiomyopathy | 203 | 352 | 3.27 × 10−7 | ko05410 |
| Viral myocarditis | 153 | 255 | 3.55 × 10−7 | ko05416 |
| Extracellular matrix-receptor interaction | 255 | 458 | 6.56 × 10−7 | ko04512 |
| Tight junction | 262 | 475 | 1.29 × 10−6 | ko04530 |
| Oxidative phosphorylation | 242 | 436 | 1.72 × 10−6 | Ko00190 |
| Salmonella infection | 204 | 362 | 2.81 × 10−6 | Ko05132 |
| Melanogenesis | 124 | 208 | 6.87 × 10−6 | Ko04916 |
| Parkinson’s disease | 238 | 435 | 8.45 × 10−6 | Ko05012 |
| Salivary secretion | 162 | 284 | 1.14 × 10−5 | Ko04970 |
| Gastric acid secretion | 187 | 338 | 3.19 × 10−5 | Ko04971 |
| Endocytosis | 223 | 411 | 3.30 × 10−5 | Ko04144 |
| Phagosome | 181 | 328 | 5.14 × 10−5 | Ko04145 |
| Endocrine and other factor-regulated calcium reabsorption | 99 | 166 | 5.43 × 10−5 | Ko04961 |
| Calcium signalling pathway | 260 | 494 | 1.26 × 10−4 | Ko04020 |
| Vibrio cholerae infection | 188 | 349 | 2.19 × 10−4 | Ko05110 |
| Ribosome biogenesis in eukaryotes | 141 | 256 | 3.66 × 10−4 | Ko03008 |
| Vascular smooth muscle contraction | 332 | 654 | 5.38 × 10−4 | Ko04270 |
| African trypanosomiasis | 22 | 29 | 5.83 × 10−4 | Ko05143 |
| GnRH signalling pathway | 133 | 242 | 5.94 × 10−4 | Ko04912 |
| Aminoacyl-tRNA biosynthesis | 73 | 123 | 5.97 × 10−4 | Ko00970 |
| Cholinergic synapse | 109 | 194 | 6.21 × 10−4 | Ko04725 |
| Chemokine signalling pathway | 159 | 296 | 7.57 × 10−4 | Ko04062 |
| Antigen processing and presentation | 98 | 175 | 1.31 × 10−3 | Ko04612 |
| RNA transport | 238 | 463 | 1.34 × 10−3 | Ko03013 |
| Herpes simplex infection | 160 | 303 | 1.92 × 10−3 | Ko05168 |
| Hedgehog signalling pathway | 61 | 104 | 2.40 × 10−3 | Ko04340 |
| Staphylococcus aureus infection | 26 | 38 | 2.42 × 10−3 | Ko05150 |
| Transcriptional misregulation in cancer | 165 | 317 | 3.57 × 10−3 | Ko05202 |
| Phototransduction | 71 | 126 | 4.61 × 10−3 | Ko04744 |
| Prion diseases | 72 | 128 | 4.62 × 10−3 | Ko05020 |
| Pancreatic secretion | 133 | 255 | 7.53 × 10−3 | Ko04972 |
| Morphine addiction | 98 | 183 | 7.88 × 10−3 | Ko05032 |
| Sulphur relay system | 32 | 53 | 1.40 × 10−2 | Ko04122 |
| Olfactory transduction | 109 | 209 | 1.44 × 10−2 | Ko04740 |
| Bacterial invasion of epithelial cells | 131 | 255 | 1.47 × 10−2 | Ko05100 |
| Spliceosome | 274 | 559 | 1.50 × 10−2 | Ko03040 |
| Long-term depression | 71 | 131 | 1.52 × 10−2 | Ko04730 |
| GABAergic synapse | 110 | 212 | 1.69 × 10−2 | Ko04727 |
| Dopaminergic synapse | 148 | 292 | 1.76 × 10−2 | Ko04728 |
| Legionellosis | 73 | 136 | 1.85 × 10−2 | Ko05134 |
| Protein digestion and absorption | 303 | 624 | 1.93 × 10−2 | Ko04974 |
| Glutamatergic synapse | 112 | 218 | 2.25 × 10−2 | Ko04724 |
| Toxoplasmosis | 100 | 193 | 2.28 × 10−2 | Ko05145 |
| Retrograde endocannabinoid signalling | 111 | 216 | 2.29 × 10−2 | Ko04723 |
| Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 63 | 117 | 2.51 × 10−2 | Ko05412 |
| Type I diabetes mellitus | 8 | 10 | 2.51 × 10−2 | Ko04940 |
| Neuroactive ligand-receptor interaction | 223 | 455 | 2.61 × 10−2 | Ko04080 |
| Influenza A | 155 | 310 | 2.68 × 10−2 | Ko05164 |
| Gap junction | 109 | 214 | 3.19 × 10−2 | Ko04540 |
| Shigellosis | 90 | 176 | 4.27 × 10−2 | Ko05131 |
| ErbB signalling pathway | 99 | 195 | 4.30 × 10−2 | Ko04012 |
| Fc gamma receptor-mediated phagocytosis | 115 | 229 | 4.41 × 10−2 | Ko04666 |
| Synaptic vesicle cycle | 110 | 219 | 4.77 × 10−2 | Ko04721 |
| mRNA surveillance pathway | 169 | 345 | 4.81 × 10−2 | Ko03015 |
| Regulation of actin cytoskeleton | 363 | 766 | 4.98 × 10−2 | Ko04810 |
| Pathway | DEPs 1 | Proteins 2 | p-Value | Pathway ID |
|---|---|---|---|---|
| Metabolic pathways | 444 | 1119 | 2.96 × 10−18 | Ko01100 |
| Valine, leucine and isoleucine degradation | 61 | 111 | 1.39 × 10−8 | Ko00280 |
| Glycolysis/Gluconeogenesis | 67 | 134 | 3.60 × 10−7 | Ko00010 |
| Fatty acid metabolism | 67 | 136 | 7.28 × 10−7 | Ko00071 |
| Pyruvate metabolism | 51 | 98 | 2.05 × 10−6 | Ko00620 |
| Citrate cycle (TCA cycle) | 42 | 76 | 2.17 × 10−7 | Ko00020 |
| β-Alanine metabolism | 38 | 70 | 1.17 × 10−5 | Ko00410 |
| Aminoacyl-tRNA biosynthesis | 26 | 42 | 1.29 × 10−5 | Ko00970 |
| Oxidative phosphorylation | 71 | 160 | 3.72 × 10−5 | Ko00190 |
| Propanoate metabolism | 42 | 84 | 5.74 × 10−5 | Ko00640 |
| Parkinson’s disease | 63 | 150 | 6.58 × 10−4 | Ko05012 |
| Peroxisome | 71 | 174 | 8.22 × 10−4 | Ko04146 |
| Alzheimer’s disease | 67 | 167 | 1.93 × 10−3 | Ko05010 |
| Butanoate metabolism | 28 | 58 | 1.95 × 10−3 | Ko00650 |
| Retinol metabolism | 48 | 113 | 2.13 × 10−3 | Ko00830 |
| Tryptophan metabolism | 41 | 94 | 2.44 × 10−3 | Ko00380 |
| Galactose metabolism | 21 | 41 | 2.85 × 10−3 | Ko00052 |
| Huntington’s disease | 72 | 184 | 2.89 × 10−3 | Ko05016 |
| Arginine and proline metabolism | 40 | 92 | 2.94 × 10−3 | Ko00330 |
| Glyoxylate and dicarboxylate metabolism | 23 | 47 | 3.89 × 10−3 | Ko00630 |
| Peroxisome proliferator-activated signalling pathway | 29 | 63 | 3.99 × 10−3 | Ko03320 |
| Metabolism of xenobiotics by cytochrome P450 | 48 | 116 | 4.02 × 10−3 | Ko00980 |
| Pentose phosphate pathway | 23 | 48 | 5.42 × 10−3 | Ko00030 |
| Drug metabolism–cytochrome P450 | 44 | 107 | 6.59 × 10−3 | Ko00982 |
| Lysine degradation | 27 | 60 | 7.82 × 10−3 | Ko00310 |
| Biosynthesis of unsaturated fatty acids | 17 | 34 | 9.56 × 10−3 | Ko01040 |
| Glutathione metabolism | 40 | 98 | 1.08 × 10−2 | Ko00480 |
| Phenylalanine metabolism | 26 | 59 | 1.23 × 10−2 | Ko00360 |
| Glycerolipid metabolism | 26 | 59 | 1.23 × 10−2 | Ko00561 |
| Starch and sucrose metabolism | 22 | 49 | 1.61 × 10−2 | Ko00500 |
| Synthesis and degradation of ketone bodies | 7 | 11 | 1.99 × 10−2 | Ko00072 |
| Alanine, aspartate and glutamate metabolism | 20 | 45 | 2.40 × 10−2 | Ko00250 |
| Tyrosine metabolism | 27 | 65 | 2.58 × 10−2 | Ko00350 |
| Mismatch repair | 4 | 5 | 2.92 × 10−2 | Ko03430 |
| Pentose and glucuronate inter conversions | 33 | 83 | 2.93 × 10−2 | Ko00040 |
| Arachidonic acid metabolism | 30 | 75 | 3.36 × 10−2 | Ko00590 |
| Lysosome | 47 | 128 | 4.70 × 10−2 | Ko04142 |
| α-Linolenic acid metabolism | 11 | 23 | 4.91 × 10−2 | Ko00592 |
| Gene ID | Annotation | Fold Change Bm5:Bm7 | |
|---|---|---|---|
| Protein | RNA | ||
| Uni5244 | Glutathione peroxidase | 3.65 | 1.60 |
| CL3566.Contig2 | Expansin-like protein | 1.79 | 1.45 |
| CL4567.Contig1 | Fatty acid- and retinol-binding protein | 11.66 | 1.94 |
| CL5080.Contig2 | β-1,4-Endoglucanase | 5.34 | 1.51 |
| CL8429.Contig1 | Peroxiredoxin | 4.38 | 1.41 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Chen, F.; Pan, H.; Ye, J.; Dong, X.; Li, C.; Lin, F. Identifying Virulence-Associated Genes Using Transcriptomic and Proteomic Association Analyses of the Plant Parasitic Nematode Bursaphelenchus mucronatus. Int. J. Mol. Sci. 2016, 17, 1492. https://doi.org/10.3390/ijms17091492
Zhou L, Chen F, Pan H, Ye J, Dong X, Li C, Lin F. Identifying Virulence-Associated Genes Using Transcriptomic and Proteomic Association Analyses of the Plant Parasitic Nematode Bursaphelenchus mucronatus. International Journal of Molecular Sciences. 2016; 17(9):1492. https://doi.org/10.3390/ijms17091492
Chicago/Turabian StyleZhou, Lifeng, Fengmao Chen, Hongyang Pan, Jianren Ye, Xuejiao Dong, Chunyan Li, and Fengling Lin. 2016. "Identifying Virulence-Associated Genes Using Transcriptomic and Proteomic Association Analyses of the Plant Parasitic Nematode Bursaphelenchus mucronatus" International Journal of Molecular Sciences 17, no. 9: 1492. https://doi.org/10.3390/ijms17091492