
Epigenetic Modifications of Major Depressive Disorder



Abstract
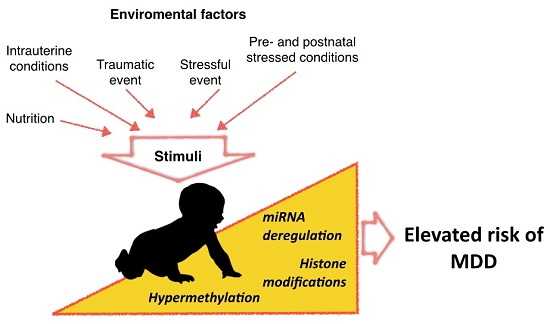
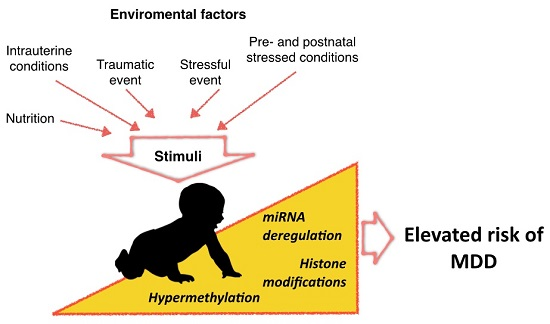
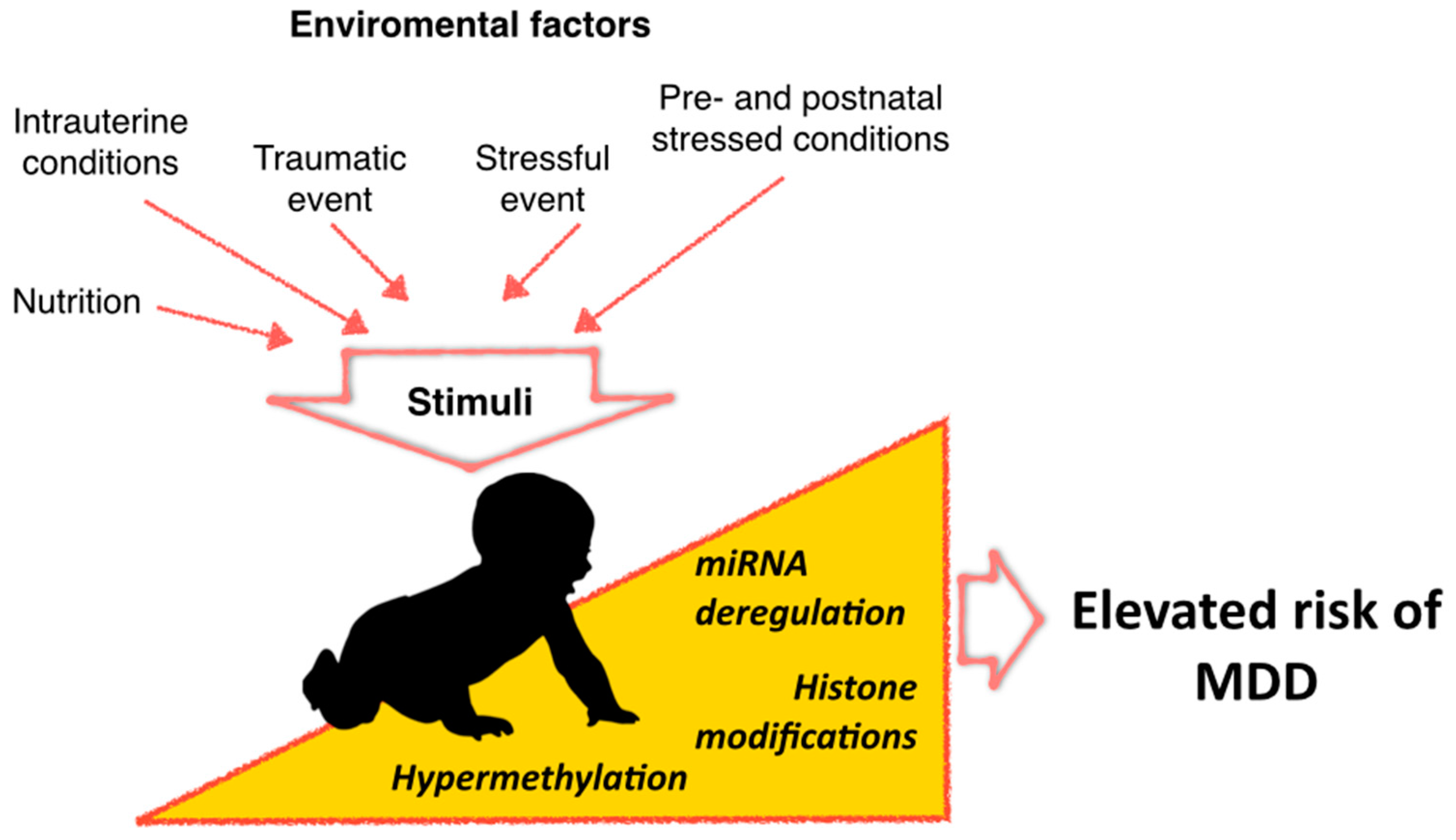
:
1. Introduction
2. Genetics of Major Depression
3. Epigenetic Modifications and Depressive Disorders
3.1. DNA Methylation
3.2. Histone Modification
3.3. Non-Coding RNAs
4. Epigenetics Modifications in MDD Therapy
5. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smith, K. Mental health: A world of depression. Nature 2014, 515, 181. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N.; et al. Global burden of disease attributable to mental and substance use disorders: Findings from the global burden of disease study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- WHO. Mental Health and Older Adults; WHO: Ginebra, Switzerland, 2013. [Google Scholar]
- Sun, H.; Kennedy, P.J.; Nestler, E.J. Epigenetics of the depressed brain: Role of histone acetylation and methylation. Neuropsychopharmacology 2013, 38, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Uher, R.; Payne, J.L.; Pavlova, B.; Perlis, R.H. Major depressive disorder in dsm-5: Implications for clinical practice and research of changes from dsm-iv. Depress Anxiety 2014, 31, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Nemeroff, C.B.; Weinberger, D.; Rutter, M.; MacMillan, H.L.; Bryant, R.A.; Wessely, S.; Stein, D.J.; Pariante, C.M.; Seemuller, F.; Berk, M.; et al. Dsm-5: A collection of psychiatrist views on the changes, controversies, and future directions. BMC Med. 2013, 11, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic epidemiology of major depression: Review and meta-analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, P.; Cohen, S.; Knight, J. Homing in on depression genes. Am. J. Psychiatry 2007, 164, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Klengel, T.; Binder, E.B. Epigenetics, depression and antidepressant treatment. Curr. Pharm. Des. 2012, 18, 5879–5889. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Perlis, R.H.; Jung, J.Y.; Byrne, E.M.; Rueckert, E.; Siburian, R.; Haddad, S.; Mayerfeld, C.E.; Heath, A.C.; Pergadia, M.L.; et al. Multi-locus genome-wide association analysis supports the role of glutamatergic synaptic transmission in the etiology of major depressive disorder. Transl. Psychiatry 2012, 2, e184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Pergadia, M.L.; Blackwood, D.H.; Penninx, B.W.; Gordon, S.D.; Nyholt, D.R.; Ripke, S.; MacIntyre, D.J.; McGhee, K.A.; Maclean, A.W.; et al. Genome-wide association study of major depressive disorder: New results, meta-analysis, and lessons learned. Mol. Psychiatry 2012, 17, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium; Ripke, S.; Wray, N.R.; Lewis, C.M.; Hamilton, S.P.; Weissman, M.M.; Breen, G.; Byrne, E.M.; Blackwood, D.H.; Boomsma, D.I.; et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 2013, 18, 497–511. [Google Scholar]
- Mak, K.K.; Kong, W.Y.; Mak, A.; Sharma, V.K.; Ho, R.C. Polymorphisms of the serotonin transporter gene and post-stroke depression: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.L.; Batten, L.A.; Tremblay, P.; Aldosary, F.; Du, L.; Blier, P. Impact of monoamine-related gene polymorphisms on hippocampal volume in treatment-resistant depression. Acta Neuropsychiatr. 2015, 27, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Brummett, B.H.; Krystal, A.D.; Siegler, I.C.; Kuhn, C.; Surwit, R.S.; Zuchner, S.; Ashley-Koch, A.; Barefoot, J.C.; Williams, R.B. Associations of a regulatory polymorphism of monoamine oxidase—A gene promoter (maoa-uvntr) with symptoms of depression and sleep quality. Psychosom. Med. 2007, 69, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Tadic, A.; Rujescu, D.; Szegedi, A.; Giegling, I.; Singer, P.; Moller, H.J.; Dahmen, N. Association of a maoa gene variant with generalized anxiety disorder, but not with panic disorder or major depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 117B, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tadic, A.; Muller, M.J.; Rujescu, D.; Kohnen, R.; Stassen, H.H.; Dahmen, N.; Szegedi, A. The maoa t941g polymorphism and short-term treatment response to mirtazapine and paroxetine in major depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Dannlowski, U.; Ohrmann, P.; Konrad, C.; Domschke, K.; Bauer, J.; Kugel, H.; Hohoff, C.; Schoning, S.; Kersting, A.; Baune, B.T.; et al. Reduced amygdala-prefrontal coupling in major depression: Association with maoa genotype and illness severity. Int. J. Neuropsychopharmacol. 2009, 12, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Goenjian, A.K.; Noble, E.P.; Steinberg, A.M.; Walling, D.P.; Stepanyan, S.T.; Dandekar, S.; Bailey, J.N. Association of comt and tph-2 genes with dsm-5 based ptsd symptoms. J. Affect. Disord. 2014, 172C, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Nielsen, D.A.; Rosenthal, N.E.; Jefferson, K.; Kaye, W.; Murphy, D.; Altemus, M.; Humphries, J.; Cassano, G.; Rotondo, A.; et al. No coding variant of the tryptophan hydroxylase gene detected in seasonal affective disorder, obsessive-compulsive disorder, anorexia nervosa, and alcoholism. Biol. Psychiatry 1999, 45, 615–619. [Google Scholar] [CrossRef]
- Goenjian, A.K.; Bailey, J.N.; Walling, D.P.; Steinberg, A.M.; Schmidt, D.; Dandekar, U.; Noble, E.P. Association of tph1, tph2, and 5httlpr with ptsd and depressive symptoms. J. Affect. Disord. 2012, 140, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kendler, K.S.; Gardner, C.O.; Prescott, C.A. Toward a comprehensive developmental model for major depression in men. Am. J. Psychiatry 2006, 163, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kendler, K.S.; Gardner, C.O.; Prescott, C.A. Toward a comprehensive developmental model for major depression in women. Am. J. Psychiatry 2002, 159, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Dalton, V.S.; Kolshus, E.; McLoughlin, D.M. Epigenetics and depression: Return of the repressed. J. Affect. Disord. 2014, 155, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lolak, S.; Suwannarat, P.; Lipsky, R.H. Epigenetics of depression. Prog. Mol. Biol. Transl. Sci. 2014, 128, 103–137. [Google Scholar] [PubMed]
- Mateus-Pinheiro, A.; Pinto, L.; Sousa, N. Epigenetic (de)regulation of adult hippocampal neurogenesis: Implications for depression. Clin. Epigenet. 2011, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [CrossRef]
- Wu, C.; Morris, J.R. Genes, genetics, and epigenetics: A correspondence. Science 2001, 293, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Pena, C.J.; Kundakovic, M.; Mitchell, A.; Akbarian, S. Epigenetic basis of mental illness. Neuroscientist 2015. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J. Epigenetic mechanisms of depression. JAMA Psychiatry 2014, 71, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Ward, J.; Taylor, I.A.; Waters, M.; Shi, Y.; Cannon, B.; Kelnar, K.; Kemppainen, J.; Brown, D.; Chen, C.; et al. Identification of mirna changes in alzheimer‘s disease brain and csf yields putative biomarkers and insights into disease pathways. J. Alzheimers Dis. 2008, 14, 27–41. [Google Scholar] [PubMed]
- Li, Y.J.; Xu, M.; Gao, Z.H.; Wang, Y.Q.; Yue, Z.; Zhang, Y.X.; Li, X.X.; Zhang, C.; Xie, S.Y.; Wang, P.Y. Alterations of serum levels of bdnf-related mirnas in patients with depression. PLoS ONE 2013, 8, e63648. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schubeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Junaid, M.A. Lifestyle, pregnancy and epigenetic effects. Epigenomics 2015, 7, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Dulawa, S.C. Epigenetic programing of depression during gestation. Bioessays 2014, 36, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav. Rev. 2015, 48, 70–91. [Google Scholar] [CrossRef] [PubMed]
- Nieratschker, V.; Massart, R.; Gilles, M.; Luoni, A.; Suderman, M.J.; Krumm, B.; Meier, S.; Witt, S.H.; Nothen, M.M.; Suomi, S.J.; et al. Morc1 exhibits cross-species differential methylation in association with early life stress as well as genome-wide association with mdd. Transl. Psychiatry 2014, 4, e429. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Brandwein, C.; Luoni, A.; Sandrini, P.; Calzoni, T.; Deuschle, M.; Cirulli, F.; Riva, M.A.; Gass, P. Morc1 knockout evokes a depression-like phenotype in mice. Behav. Brain Res. 2015, 296, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Frank, M.B.; Victor, T.; Bebak, M.; Marino, J.H.; Bellgowan, P.S.; McKinney, B.A.; Bodurka, J.; Kent Teague, T.; Drevets, W.C. Inflammation and neurological disease-related genes are differentially expressed in depressed patients with mood disorders and correlate with morphometric and functional imaging abnormalities. Brain Behav. Immun. 2013, 31, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.; Turkheimer, E.; Gottesman, I.I.; Bouchard, T.J., Jr. Beyond heritability: Twin studies in behavioral research. Curr. Dir. Psychol. Sci. 2010, 18, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.; Busjahn, A.; Peltonen, L. Classical twin studies and beyond. Nat. Rev. Genet. 2002, 3, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, J.; Slagboom, P.E.; Draisma, H.H.; Martin, N.G.; Boomsma, D.I. The continuing value of twin studies in the omics era. Nat. Rev. Genet. 2012, 13, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Christiansen, L.; von Bornemann Hjelmborg, J.; Christensen, K. Twin methodology in epigenetic studies. J. Exp. Biol. 2015, 218, 134–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Fernandez, J.E.; Spector, T.D.; Bell, J.T. Epigenetics of discordant monozygotic twins: Implications for disease. Genome Med. 2014, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.A.; Tang, T.; Wang, S.C.; Ptak, C.; Oh, G.H.; Wong, A.H.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.N.; Gottesman, I.I.; Wong, A.H. Not really identical: Epigenetic differences in monozygotic twins and implications for twin studies in psychiatry. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151C, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Martins, Y.; Matias, A.; Blickstein, I. Why are monozygotic twins different? J. Perinat. Med. 2011, 39, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Cordova-Palomera, A.; Fatjo-Vilas, M.; Gasto, C.; Navarro, V.; Krebs, M.O.; Fananas, L. Genome-wide methylation study on depression: Differential methylation and variable methylation in monozygotic twins. Transl. Psychiatry 2015, 5, e557. [Google Scholar] [CrossRef] [PubMed]
- Pajer, K.; Andrus, B.M.; Gardner, W.; Lourie, A.; Strange, B.; Campo, J.; Bridge, J.; Blizinsky, K.; Dennis, K.; Vedell, P.; et al. Discovery of blood transcriptomic markers for depression in animal models and pilot validation in subjects with early-onset major depression. Transl. Psychiatry 2012, 2, e101. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Chambwe, N.; Klein, S.; Gal, J.; Andrews, S.; Gleason, G.; Shaknovich, R.; Melnick, A.; Campagne, F.; Toth, M. Differential gene body methylation and reduced expression of cell adhesion and neurotransmitter receptor genes in adverse maternal environment. Transl. Psychiatry 2013, 3, e218. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Bruchas, M.R.; Schindler, A.G.; Shankar, H.; Messinger, D.I.; Miyatake, M.; Land, B.B.; Lemos, J.C.; Hagan, C.E.; Neumaier, J.F.; Quintana, A.; et al. Selective p38alpha mapk deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 2011, 71, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Sabunciyan, S.; Aryee, M.J.; Irizarry, R.A.; Rongione, M.; Webster, M.J.; Kaufman, W.E.; Murakami, P.; Lessard, A.; Yolken, R.H.; Feinberg, A.P.; et al. Genome-wide DNA methylation scan in major depressive disorder. PLoS ONE 2012, 7, e34451. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.Q.; Siow, N.L.; Peng, H.B.; Massoulie, J.; Tsim, K.W. Regulation of prima: Membrane anchor of acetylcholinesterase (ache) in neuron and muscle. Chem. Biol. Interact. 2005, 432, 157–158. [Google Scholar] [CrossRef]
- Xie, H.Q.; Choi, R.C.; Leung, K.W.; Chen, V.P.; Chu, G.K.; Tsim, K.W. Transcriptional regulation of proline-rich membrane anchor (prima) of globular form acetylcholinesterase in neuron: An inductive effect of neuron differentiation. Brain Res. 2009, 1265, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Numata, S.; Ishii, K.; Tajima, A.; Iga, J.; Kinoshita, M.; Watanabe, S.; Umehara, H.; Fuchikami, M.; Okada, S.; Boku, S.; et al. Blood diagnostic biomarkers for major depressive disorder using multiplex DNA methylation profiles: Discovery and validation. Epigenetics 2015, 10, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Spijker, S.; van Zanten, J.S.; de Jong, S.; Penninx, B.W.; van Dyck, R.; Zitman, F.G.; Smit, J.H.; Ylstra, B.; Smit, A.B.; Hoogendijk, W.J. Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol. Psychiatry 2010, 68, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Kittel-Schneider, S.; Gessner, A.; Domschke, K.; Neuner, M.; Jacob, C.P.; Buttenschon, H.N.; Boreatti-Hummer, A.; Volkert, J.; Herterich, S.; et al. Cross-disorder analysis of bipolar risk genes: Further evidence of dgkh as a risk gene for bipolar disorder, but also unipolar depression and adult adhd. Neuropsychopharmacology 2011, 36, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Ronai, Z.; Kovacs-Nagy, R.; Szantai, E.; Elek, Z.; Sasvari-Szekely, M.; Faludi, G.; Benkovits, J.; Rethelyi, J.M.; Szekely, A. Glycogen synthase kinase 3 beta gene structural variants as possible risk factors of bipolar depression. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xu, Y.; Sun, N.; Ren, Y.; Liu, Z.; Cao, X.; Zhang, K. The combined effects of the bdnf and gsk3b genes modulate the relationship between negative life events and major depressive disorder. Brain Res. 2010, 1355, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Anacker, C.; Cattaneo, A.; Musaelyan, K.; Zunszain, P.A.; Horowitz, M.; Molteni, R.; Luoni, A.; Calabrese, F.; Tansey, K.; Gennarelli, M.; et al. Role for the kinase sgk1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 8708–8713. [Google Scholar] [CrossRef] [PubMed]
- Stenz, L.; Zewdie, S.; Laforge-Escarra, T.; Prados, J.; La Harpe, R.; Dayer, A.; Paoloni-Giacobino, A.; Perroud, N.; Aubry, J.M. Bdnf promoter i methylation correlates between post-mortem human peripheral and brain tissues. Neurosci. Res. 2015, 91, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed]
- Januar, V.; Ancelin, M.L.; Ritchie, K.; Saffery, R.; Ryan, J. Bdnf promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.H.; Smoller, J.W. The genetics of bipolar disorder. Neuroscience 2009, 164, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Krause, L.; Bell, J.T.; Gao, F.; Ward, K.J.; Wu, H.; Lu, H.; Liu, Y.; Tsai, P.C.; Collier, D.A.; et al. Hypermethylation in the zbtb20 gene is associated with major depressive disorder. Genome Biol. 2014, 15, R56. [Google Scholar] [CrossRef] [PubMed]
- Mitchelmore, C.; Kjaerulff, K.M.; Pedersen, H.C.; Nielsen, J.V.; Rasmussen, T.E.; Fisker, M.F.; Finsen, B.; Pedersen, K.M.; Jensen, N.A. Characterization of two novel nuclear btb/poz domain zinc finger isoforms: Association with differentiation of hippocampal neurons, cerebellar granule cells, and macroglia. J. Biol. Chem. 2002, 277, 7598–7609. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, E.H.; Tonchev, A.B.; Stoykova, A.; Chowdhury, K. Regulation of archicortical arealization by the transcription factor zbtb20. Hippocampus 2012, 22, 2144–2156. [Google Scholar] [CrossRef] [PubMed]
- Bagot, R.C.; Labonte, B.; Pena, C.J.; Nestler, E.J. Epigenetic signaling in psychiatric disorders: Stress and depression. Dialogues Clin. Neurosci. 2014, 16, 281–295. [Google Scholar] [PubMed]
- Covington, H.E., 3rd; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J., 3rd; Wu, E.Y.; et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.A.; Lin, C.L.; Crusio, W.E.; Akbarian, S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 2007, 62, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Arent, C.O.; Valvassori, S.S.; Fries, G.R.; Stertz, L.; Ferreira, C.L.; Lopes-Borges, J.; Mariot, E.; Varela, R.B.; Ornell, F.; Kapczinski, F.; et al. Neuroanatomical profile of antimaniac effects of histone deacetylases inhibitors. Mol. Neurobiol. 2011, 43, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R.; Ibrahim, L.; Zarate, C.A., Jr. Histone deacetylases and mood disorders: Epigenetic programming in gene-environment interactions. CNS Neurosci. Ther. 2011, 17, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Renthal, W.; Maze, I.; Krishnan, V.; Covington, H.E., 3rd; Xiao, G.; Kumar, A.; Russo, S.J.; Graham, A.; Tsankova, N.; Kippin, T.E.; et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 2007, 56, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Hobara, T.; Uchida, S.; Otsuki, K.; Matsubara, T.; Funato, H.; Matsuo, K.; Suetsugi, M.; Watanabe, Y. Altered gene expression of histone deacetylases in mood disorder patients. J. Psychiatr. Res. 2010, 44, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Iga, J.; Ueno, S.; Yamauchi, K.; Numata, S.; Kinouchi, S.; Tayoshi-Shibuya, S.; Song, H.; Ohmori, T. Altered hdac5 and creb mrna expressions in the peripheral leukocytes of major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Cruceanu, C.; Alda, M.; Nagy, C.; Freemantle, E.; Rouleau, G.A.; Turecki, G. H3k4 tri-methylation in synapsin genes leads to different expression patterns in bipolar disorder and major depression. Int. J. Neuropsychopharmacol. 2013, 16, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Huang, H.S. Epigenetic regulation in human brain-focus on histone lysine methylation. Biol. Psychiatry 2009, 65, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Porton, B.; Kao, H.T.; Greengard, P. Characterization of transcripts from the synapsin iii gene locus. J. Neurochem. 1999, 73, 2266–2271. [Google Scholar] [CrossRef] [PubMed]
- Cesca, F.; Baldelli, P.; Valtorta, F.; Benfenati, F. The synapsins: Key actors of synapse function and plasticity. Prog. Neurobiol. 2010, 91, 313–348. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Raimondi, A.; Lignani, G.; Benfenati, F.; Baldelli, P. Synapsins: From synapse to network hyperexcitability and epilepsy. Semin. Cell Dev. Biol. 2011, 22, 408–415. [Google Scholar] [PubMed]
- He, L.; Hannon, G.J. Micrornas: Small rnas with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [PubMed]
- Mendell, J.T. Micrornas: Critical regulators of development, cellular physiology and malignancy. Cell Cycle 2005, 4, 1179–1184. [Google Scholar] [PubMed]
- Esteller, M. Non-coding rnas in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of mirnas and sirnas. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microrna biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fan, J.; Belasco, J.G. Micrornas direct rapid deadenylation of mrna. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Nishida, A.; Hara, K.; Kamemoto, T.; Suetsugi, M.; Fujimoto, M.; Watanuki, T.; Wakabayashi, Y.; Otsuki, K.; McEwen, B.S.; et al. Characterization of the vulnerability to repeated stress in fischer 344 rats: Possible involvement of microrna-mediated down-regulation of the glucocorticoid receptor. Eur. J. Neurosci. 2008, 27, 2250–2261. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Fitzsimons, C.P.; Datson, N.A.; Meijer, O.C.; Vreugdenhil, E. Glucocorticoid signaling and stress-related limbic susceptibility pathway: About receptors, transcription machinery and microrna. Brain Res. 2009, 1293, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Belzeaux, R.; Bergon, A.; Jeanjean, V.; Loriod, B.; Formisano-Treziny, C.; Verrier, L.; Loundou, A.; Baumstarck-Barrau, K.; Boyer, L.; Gall, V.; et al. Responder and nonresponder patients exhibit different peripheral transcriptional signatures during major depressive episode. Transl. Psychiatry 2012, 2, e185. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y. Evidence demonstrating role of micrornas in the etiopathology of major depression. J. Chem. Neuroanat. 2011, 42, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Lugli, G.; Rizavi, H.S.; Torvik, V.I.; Turecki, G.; Dwivedi, Y. Microrna expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS ONE 2012, 7, e33201. [Google Scholar] [CrossRef] [PubMed]
- Lugli, G.; Torvik, V.I.; Larson, J.; Smalheiser, N.R. Expression of micrornas and their precursors in synaptic fractions of adult mouse forebrain. J. Neurochem. 2008, 106, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Bocchio-Chiavetto, L.; Maffioletti, E.; Bettinsoli, P.; Giovannini, C.; Bignotti, S.; Tardito, D.; Corrada, D.; Milanesi, L.; Gennarelli, M. Blood microrna changes in depressed patients during antidepressant treatment. Eur. Neuropsychopharmacol. 2013, 23, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Caputo, V.; Sinibaldi, L.; Fiorentino, A.; Parisi, C.; Catalanotto, C.; Pasini, A.; Cogoni, C.; Pizzuti, A. Brain derived neurotrophic factor (bdnf) expression is regulated by micrornas mir-26a and mir-26b allele-specific binding. PLoS ONE 2011, 6, e28656. [Google Scholar] [CrossRef] [PubMed]
- Baraniskin, A.; Kuhnhenn, J.; Schlegel, U.; Maghnouj, A.; Zollner, H.; Schmiegel, W.; Hahn, S.; Schroers, R. Identification of micrornas in the cerebrospinal fluid as biomarker for the diagnosis of glioma. Neuro Oncol. 2012, 14, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Baraniskin, A.; Kuhnhenn, J.; Schlegel, U.; Chan, A.; Deckert, M.; Gold, R.; Maghnouj, A.; Zollner, H.; Reinacher-Schick, A.; Schmiegel, W.; et al. Identification of micrornas in the cerebrospinal fluid as marker for primary diffuse large b-cell lymphoma of the central nervous system. Blood 2011, 117, 3140–3146. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of micrornas in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microrna spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating micrornas as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.F.; Wu, N.; Wang, L.; Li, J. Circulating micrornas: A novel class of potential biomarkers for diagnosing and prognosing central nervous system diseases. Cell. Mol. Neurobiol. 2013, 33, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Bjorkqvist, M. Hsa-mir-34b is a plasma-stable microrna that is elevated in pre-manifest huntington's disease. Hum. Mol. Genet. 2011, 20, 2225–2237. [Google Scholar] [CrossRef] [PubMed]
- Redell, J.B.; Moore, A.N.; Ward, N.H.; Hergenroeder, G.W.; Dash, P.K. Human traumatic brain injury alters plasma microrna levels. J. Neurotrauma 2010, 27, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Tian, Y.; Ander, B.P.; Xu, H.; Stamova, B.S.; Zhan, X.; Turner, R.J.; Jickling, G.; Sharp, F.R. Brain and blood microrna expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J. Cereb. Blood Flow Metab. 2010, 30, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Vreugdenhil, E.; Verissimo, C.S.; Mariman, R.; Kamphorst, J.T.; Barbosa, J.S.; Zweers, T.; Champagne, D.L.; Schouten, T.; Meijer, O.C.; de Kloet, E.R.; et al. Microrna 18 and 124a down-regulate the glucocorticoid receptor: Implications for glucocorticoid responsiveness in the brain. Endocrinology 2009, 150, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.M.; Sun, X.Y.; Guo, W.; Zhong, A.F.; Niu, W.; Zhao, L.; Dai, Y.H.; Guo, Z.M.; Zhang, L.Y.; Lu, J. Differential expression of microrna in peripheral blood mononuclear cells as specific biomarker for major depressive disorder patients. J. Psychiatr. Res. 2014, 59, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Liu, Y.; Wang, X.; Wu, J.; Liu, K.; Zhou, J.; Liu, L.; Zhang, C. Identification of differential micrornas in cerebrospinal fluid and serum of patients with major depressive disorder. PLoS ONE 2015, 10, e0121975. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sundquist, K.; Hedelius, A.; Palmer, K.; Memon, A.A.; Sundquist, J. Circulating microRNA-144–5p is associated with depressive disorders. Clin. Epigenet. 2015, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Vialou, V.; Feng, J.; Robison, A.J.; Nestler, E.J. Epigenetic mechanisms of depression and antidepressant action. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 59–87. [Google Scholar] [CrossRef] [PubMed]
- Perisic, T.; Zimmermann, N.; Kirmeier, T.; Asmus, M.; Tuorto, F.; Uhr, M.; Holsboer, F.; Rein, T.; Zschocke, J. Valproate and amitriptyline exert common and divergent influences on global and gene promoter-specific chromatin modifications in rat primary astrocytes. Neuropsychopharmacology 2010, 35, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, N.; Zschocke, J.; Perisic, T.; Yu, S.; Holsboer, F.; Rein, T. Antidepressants inhibit DNA methyltransferase 1 through reducing g9a levels. Biochem. J. 2012, 448, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Group, U.E.R. Efficacy and safety of electroconvulsive therapy in depressive disorders: A systematic review and meta-analysis. Lancet 2003, 361, 799–808. [Google Scholar]
- Tsankova, N.M.; Kumar, A.; Nestler, E.J. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J. Neurosci. 2004, 24, 5603–5610. [Google Scholar] [CrossRef] [PubMed]
- Heyer, M.P.; Kenny, P.J. Microrna-mediated repression combats depression. Neuron 2014, 83, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Baudry, A.; Mouillet-Richard, S.; Schneider, B.; Launay, J.M.; Kellermann, O. Mir-16 targets the serotonin transporter: A new facet for adaptive responses to antidepressants. Science 2010, 329, 1537–1541. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S. Small RNAs: The enigma of prozac resolved. Nat. Rev. Neurosci. 2010, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.M.; Mouillet-Richard, S.; Baudry, A.; Pietri, M.; Kellermann, O. Raphe-mediated signals control the hippocampal response to sri antidepressants via mir-16. Transl. Psychiatry 2011, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.P.; Lim, R.; Cruceanu, C.; Crapper, L.; Fasano, C.; Labonte, B.; Maussion, G.; Yang, J.P.; Yerko, V.; Vigneault, E.; et al. Mir-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat. Med. 2014, 20, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Issler, O.; Haramati, S.; Paul, E.D.; Maeno, H.; Navon, I.; Zwang, R.; Gil, S.; Mayberg, H.S.; Dunlop, B.W.; Menke, A.; et al. MicroRNA 135 is essential for chronic stress resiliency, antidepressant efficacy, and intact serotonergic activity. Neuron 2014, 83, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Heller, E.A.; Cates, H.M.; Pena, C.J.; Sun, H.; Shao, N.; Feng, J.; Golden, S.A.; Herman, J.P.; Walsh, J.J.; Mazei-Robison, M.; et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat. Neurosci. 2014, 17, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Damez-Werno, D.M.; Scobie, K.N.; Shao, N.Y.; Dias, C.; Rabkin, J.; Koo, J.W.; Korb, E.; Bagot, R.C.; Ahn, F.H.; et al. Acf chromatin-remodeling complex mediates stress-induced depressive-like behavior. Nat. Med. 2015, 21, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Xu, J.; Yu, H.; Nie, B.; Li, N.; Luo, C.; Li, H.; Liu, F.; Bai, Y.; Shan, B.; et al. Delineation of early and later adult onset depression by diffusion tensor imaging. PLoS ONE 2014, 9, e112307. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Cheng, Y.; Chai, P.; Lu, Z.; Li, H.; Luo, C.; Li, X.; Li, L.; Zhou, Q.; Chen, B.; et al. White-matter volume reduction and the protective effect of immunosuppressive therapy in systemic lupus erythematosus patients with normal appearance by conventional magnetic resonance imaging. J. Rheumatol. 2010, 37, 974–986. [Google Scholar] [CrossRef] [PubMed]

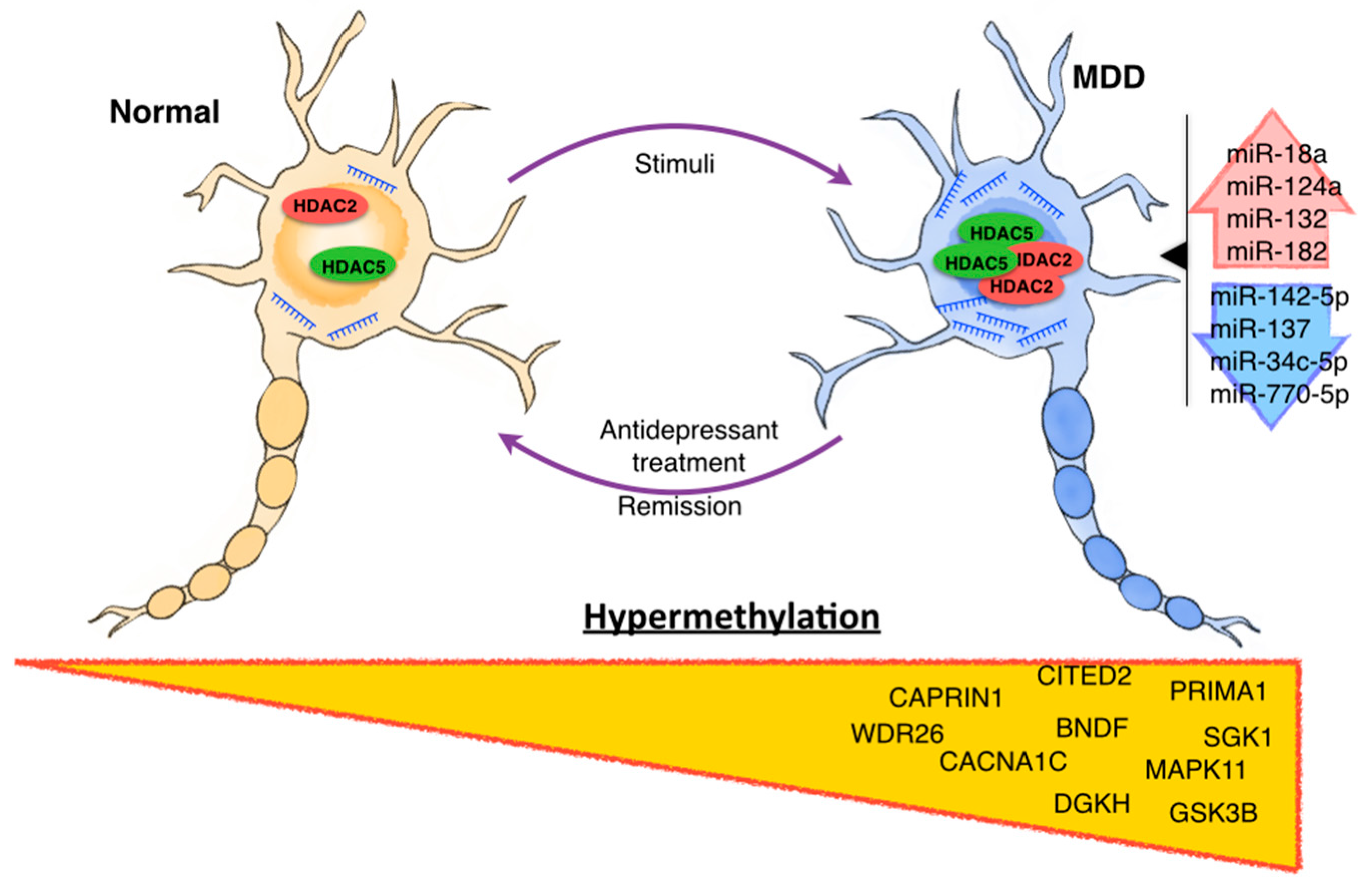

{kind=link}
{kind=link}
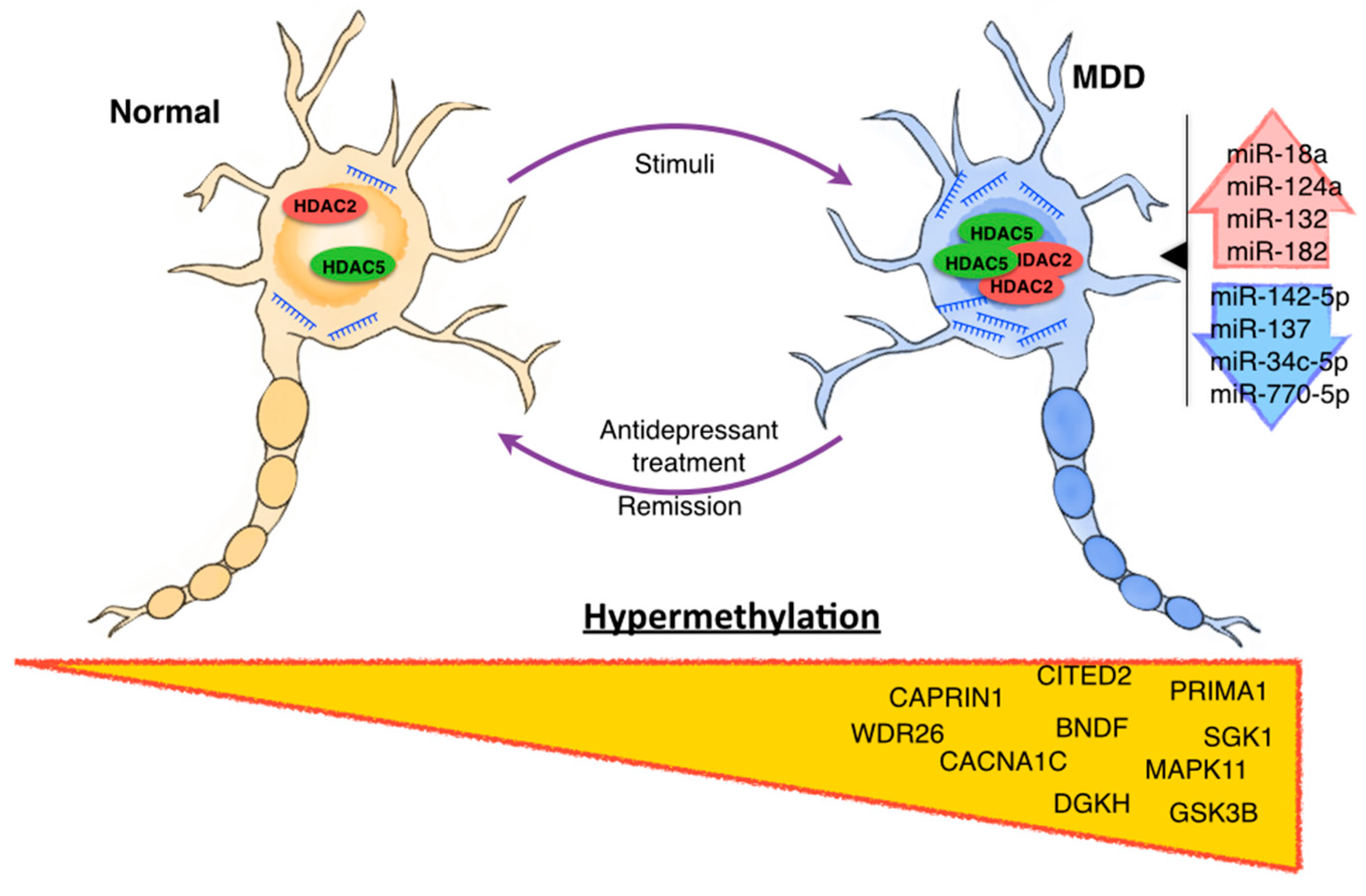{kind=link}
| Reference | Sample Characteristics | Study | Tissue | Diagnosis | Platform | Gene Associated | Potential Relevance of Gene in Depressive Disorders |
|---|---|---|---|---|---|---|---|
| Córdoba-Palomera et al. [49] 2015 | 17 MZ pairs Caucasian Spanish adult twins | Genome-wide DNA methylation | Peripheral blood | Anxious or depressive disorder | Illumina Infinium HumanMethylation450 Beadchip | WDR26 | Prospective blood transcriptomic marker for depression [11,50]. |
| CACNA1C | Susceptibility factor for depressive psychopathology [11]. Methylation changes have been associated with risk factors for depressive disorders [51,52]. | ||||||
| MAPK11 | Associated with depression phenotypes [53]. | ||||||
| Sabunciyan et al. [54] 2012 | 39 individuals with MDD from Stanley Medical Research Institute | Genome-wide DNA methylation | Post-mortem frontal cortex | MDD | Comprehensive High-throughput Arrays for Relative Methylation (CHARM) | PRIMA1 | Encodes a protein that functions to organize AChE into tetramers, and to anchor AChE to neural cell membranes [55,56]. |
| Numata et al. [57] 2015 | 29 Medication-free patients with MDD | Genome-wide DNA methylation | Peripheral leukocytes | MDD | Infinium HumanMethylation450 BeadChips | CAPRIN1 | Potential blood marker of major depressive disorder [58]. |
| CITED2 | Differentially expressed in the mood disorder, associated with neurological or psychiatric diseases [40]. | ||||||
| DGKH | Risk gene for bipolar disorder [59,66]. | ||||||
| Januar et al. [65] 2015 | 183 patients with MDD >65 years-old | High-throughput DNA methylation profiling | Buccal tissue | MDD | Sequenom MassARRAY | BDNF | Promotes the proliferation, differentiation and survival of neurons, crucial for neural plasticity and cognitive function [64]. Potential biomarker of depression [65]. |
| Nieratschker et al. [38] 2014 | 8 mothers and their infants with prenatal stressed conditions. 9 pregnant rats with prenatal stressed conditions | Genome-wide association | Peripheral leukocytes and refrontal cortex of adult rats | MDD | Methylated DNA immunoprecipitation (MeDIP) and pyrosequencing | MORC1 | Candidate gene for major depressive disorder related to early life stress in rodents, primates and humans [38]. Evokes a depression-like phenotype in mice [39]. |
| Davies et al. [67] 2014 | 50 monozygotic twin pairs from the UK and Australia discordant for depression | Genome-wide DNA methylation | Whole blood and brain tissue samples | MDD | MeDIP-Sequencing | ZBTB20 | Important for the hormonal hippocampal function, crucial for the regionalization and volume of archicortex, playing a role in depression [68,69]. |
| Reference | Sample Characteristics | Tissue | Diagnosis | Platform | Epigenetic Modification Evaluated | Gene and Histone Modification Associated | Main Findings |
|---|---|---|---|---|---|---|---|
| Cruceanu et al. [80] 2013 | Individuals with bipolar disorder type I (n = 13) or MDD (n = 18) and controls (n = 14) with no psychiatric history | Post-mortem prefrontal cortex (PCF) from Broadman Area (BA) 10 | BD or MDD | Chromatin immunoprecipitation (ChIP) and Quantitative real-time PCR | Histone modification | SYN2 H3K4me3 | H3K4me3 increase in MDD patients and correlated with gene expression of SYN2 [80]. |
| Covington et al. [71] 2009 | C57BL/6J male mice with chronic social defeat stress (n = 6) and control mice (n = 10). Patients depress postmortem (n = 8) | Brain tissue | Depression | Immunohistochemistry, Western blot and Illumina MouseWG-8 V2.0 array | Histone modification | H3K14ac | Transiently decreased and then stably increased of H3K14ac in the NAc of mice after chronic social defeat stress, correlated with a reduction in HDAC2 levels [71]. |
| Hobara et al. [78] 2010 | Mood disorder patients in a depressive and remissive state | Peripheral white cells | MDD and BD | Quantitative real-time PCR | Expression of HDACs | HDAC2 and HDAC5 | Gene expression of HDAC2 and HDAC5 were significantly increased in MDD patients in depressive state compare to controls subjects, while during remissive state, HDACs expression was comparable to controls subjects, suggesting a state-dependent alteration [78]. |
| Iga et al. [79] 2007 | Patients diagnosed with MDD according to DSM-IV (n = 25) and controls (n = 25) | Peripheral leucocytes | MDD | Quantitative real-time PCR | Expression of HDACs | HDAC5 | HDAC5 mRNA levels were significantly higher in drug-free depressive patients than controls [79]. |
| Renthal et al. [77] 2007 | Mice with chronic social defeat stress | Brain tissue | Depression | Immnunohistochemistry, ChIP and microarray | Histone modification and expression of HDACs | HDAC5 | HDAC5 expression was decrease in a model with social defeat stress, imipramine treatment increased HDAC5 expression [77]. |
| Reference | Sample Characteristics | Tissue | Diagnosis | Platform | miRNAs Associated | Main Findings |
|---|---|---|---|---|---|---|
| Uchida et al. [94] 2008 | SH-SY5Y cells and Male rats Fisher 344 (F344) and Sprague-Dawley (SD) control with repeated restraint stress | Neuron cell lines Hypothalamic paraventricular nucleus | ------ | ----- | miR-18a | Overexpressed in repeated restraint stress model. Its expression inhibits translation of the glucocorticoid receptor in neuron cell culture. |
| Vreugdenhil et al. [97,111] 2009 | NS1 cells | Neuron cell lines | ------ | Luciferase reporter assay | miR-18a and miR-124a | miR-18a and miR-124a decrease protein expression of glucocorticoid receptor by luciferase reporter assay in NS1 cells. |
| Caputo et al. [101] 2011 | HeLa cells | Cervix epithelial cell line | Schizophrenia | Luciferase reporter assay | miR-132 and miR-182 | These miRNAs regulate the expression of BDNF by Allele-Specific Binding [101]. |
| Smalheiser et al. [98] 2012 | Antidepressant-free depressed suicide (n = 18) and well-matched non-psychiatric control subjects (n = 17) | Tissue, prefrontal cortex (Brodmann Area 9) | Depression | PCR miltiplex | miR-142-5p, miR-137, miR-489, miR-148b, miR-101, miR-324-5p, miR-301a, miR-146a, miR-335, miR-494, miR-20b, miR-376a*, miR-190, miR-155, miR-660, miR-130a, miR-27a, miR-497, miR-10a, miR-20a, miR-142-3p | miRs significantly downregulated in the prefrontal cortex of depressed patients compared with normal controls, many of them implicated in cellular growth and differentiation and some of them showed high synaptic enrichment [98,99]. |
| Belzeaux et al. [96] 2012 | 16 severe MDE patients and 13 matched controls | Peripheral blood mononuclear cells | Major depressive episode | Microarray SurePrint G3 Human GE 8 x 60 K | miR-107, miR-133a, miR-148a, miR-200c, miR-381, miR-425-3p, miR-494, miR-517b, miR-579, miR-589, miR-636, miR-652, miR-941, miR-1243 | miRs significantly deregulated between MDE patients and controls. These miRs help finding a gene combination useful to predict treatment response [96]. |
| Bocchio-Chiavetto et al. [100] 2013 | 10 patients with MD, the sample was extracted before and after treatment | Blood | MDD | TaqMan Array Human MicroRNA A + B Cards Set v3.0 | UP: miR-130b*, miR-505*, miR-29b-2*, miR-26b, miR-22*, miR-26a, miR-664, miR-494, let-7d, let-7g, let-7e, let-7f, miR-629, miR-106b*, miR-103, miR-191, miR-128, miR-502-3p, miR-374b, miR-132, miR-30d, miR-500, miR-589, miR-183, miR-574-3p, miR-140-3p, miR-335, miR-361-5p. DOWN: miR-34c-5p and miR-770-5p | Associated with neuronal brain function, such as neuroactive ligand–receptor interaction, axon guidance, long-term potentiation and depression [100]. |
| Li et al. [33] 2013 | 40 patients and 40 healthy controls | Serum | MDD | Real time PCR | miR-132 and miR-182 | The expression of these miRs was negatively correlated with BDNF expression [33]. |
| Fan et al. [112] 2014 | 81 MDD patients and 46 healthy controls | Peripheral blood mononuclear cells | MDD | Affymetrix miRNA 3.0 array | miRNA-26b, miRNA-1972, miRNA-4485, miRNA-4498, and miRNA-4743 | Overexpressed in MDD patients, and would regulate pathways associated with nervous system and brain functions [112]. |
| Wan et al. [113] 2015 | 1° cohort: 6 depressed and 6 control patients. 2° cohort: 32 MDD patients and 21 healthy individuals | Peripheral blood mononuclear cells | MDD | microRNA PCR Panel (V3.M) | let-7d-3p, miR-34a-5p, miR-221-3p, miR-451a | Potential MDD biomarkers [113]. |
| Wang et al. [114] 2015 | 169 patients and 52 controls | Plasma | Depression | Serum/Plasma Focus microRNA PCR Panel | miR-144-5p | miR-144-5p levels are associated with depressive symptoms, and the detection of this miR in plasma could be a potential peripheral biomarker for pathologic processes related to depression [114]. |
| Study | ClinicalTrials.gov Identifier | Status | Phase | Aims | Intervention | Condition | Publications |
|---|---|---|---|---|---|---|---|
| Paliperidone and lithium in the treatment of suicidality—treatment indication and epigenetic regulation (AFSP) | NCT01134731 | Completed | Phase 4 | To identify an efficient pharmacotherapy for the acute management of suicidality and the epigenetic regulation associated with the treatment. | Paliperidone and lithium | MDD Suicidality | Not provided |
| Epigenetic regulation of brain-derived neurotrophic factor (BDNF) in major depression | NCT01182103 | Completed | ----- | To detect the associations between BDNF, DNA methylation, histone modification, depressive symptoms, suicidal behavior and antidepressant responses in MDD patients, check the correlation between blood BDNF protein and RNA and BDNF rs6265 gene, and discuss the possible mechanisms of epigenetic regulation of BDNF in Taiwanese MDD patients. | ----- | MDD | Not provided |
| A neuroimaging and epigenetic investigation of antidepressants in depression | NCT00703742 | Completed | ----- | To find out the structural or functional effects of SSRI in MDD; to explore the DNA methylation status in depression; to find special abnormalities in depression secondary to other disease (autoimmune disease like systemic lupus erythematosus). | Escitalopram | Depression secondary to other disease | [128,129] |
| miRNAs, suicide, and ketamine—plasma exosomal microRNAs as novel biomarkers for suicidality and treatment outcome | NCT02418195 | Recruiting participants | Phase 2 | To examine whether neural-derived exosomal miRNAs are differentially expressed that are specific to suicidal ideation or behavior, and which by affecting specific miRNA targets and pathways, are associated with suicidal behavior and response to ketamine. | ketamine | MDD | Not provided |
| Add-On Study of MSI-195 (S-adenosyl-l-methionine, SAMe) for patients with major depressive disorder (MDD) | NCT01912196 | Ongoing | Phase 2 | To determine the efficacy and safety of 800 mg MSI-195 in reducing symptoms of depression in Major Depressive Disorder (MDD) patients with inadequate response to current antidepressant therapy. | MSI-195 and Placebo | MDD | Not provided |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saavedra, K.; Molina-Márquez, A.M.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Epigenetic Modifications of Major Depressive Disorder. Int. J. Mol. Sci. 2016, 17, 1279. https://doi.org/10.3390/ijms17081279
Saavedra K, Molina-Márquez AM, Saavedra N, Zambrano T, Salazar LA. Epigenetic Modifications of Major Depressive Disorder. International Journal of Molecular Sciences. 2016; 17(8):1279. https://doi.org/10.3390/ijms17081279
Chicago/Turabian StyleSaavedra, Kathleen, Ana María Molina-Márquez, Nicolás Saavedra, Tomás Zambrano, and Luis A. Salazar. 2016. "Epigenetic Modifications of Major Depressive Disorder" International Journal of Molecular Sciences 17, no. 8: 1279. https://doi.org/10.3390/ijms17081279