
Postnatal High-Fat Diet Increases Liver Steatosis and Apoptosis Threatened by Prenatal Dexamethasone through the Oxidative Effect

 , ,
, ,  and
and
Abstract
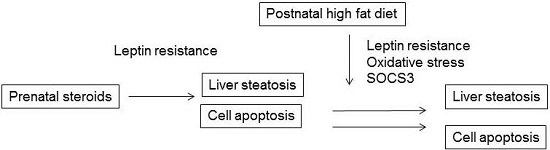
:
1. Introduction
2. Results and Discussion
2.1. Higher AST and Cholesterol in DEX Treated Group and DHF Treated Group
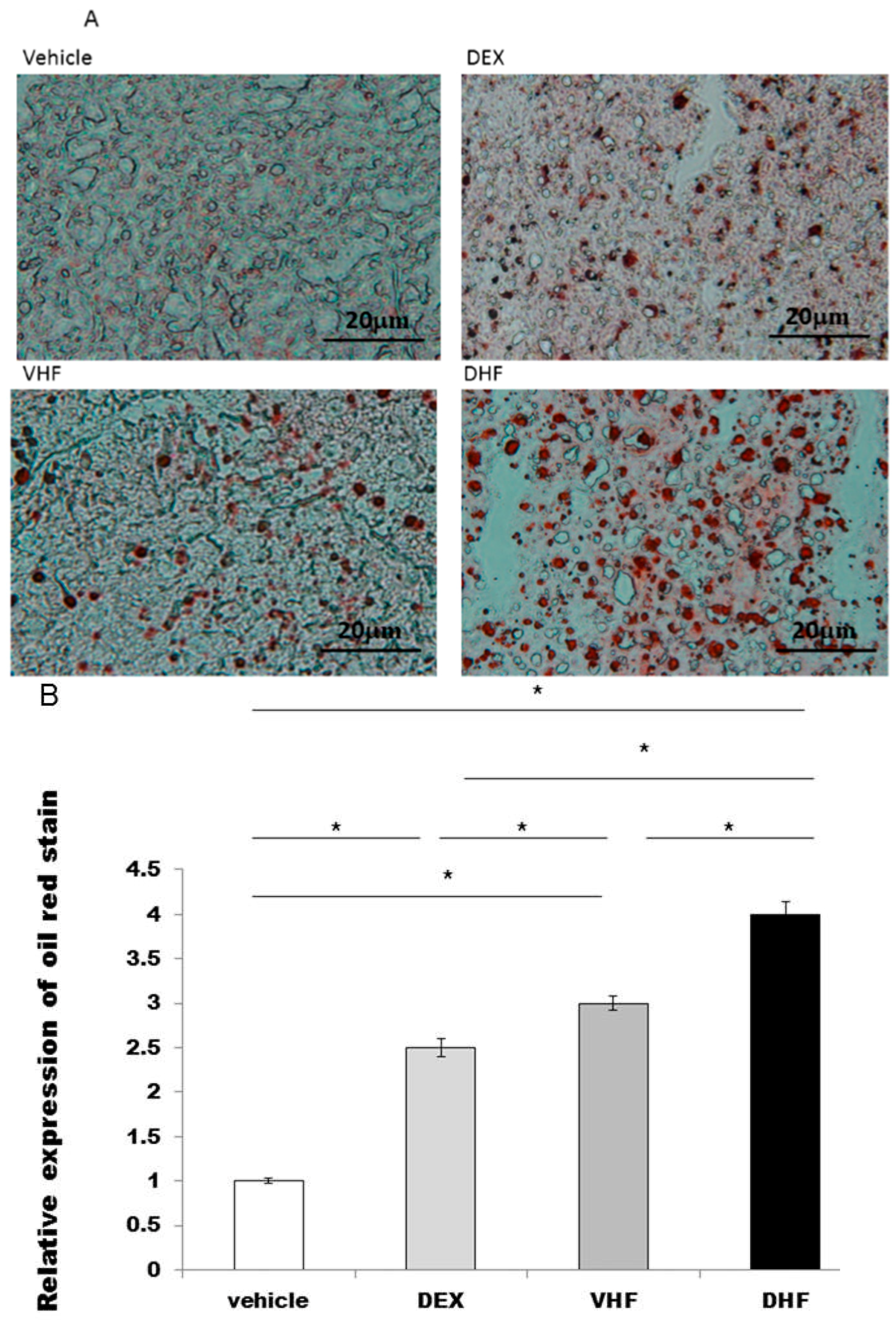
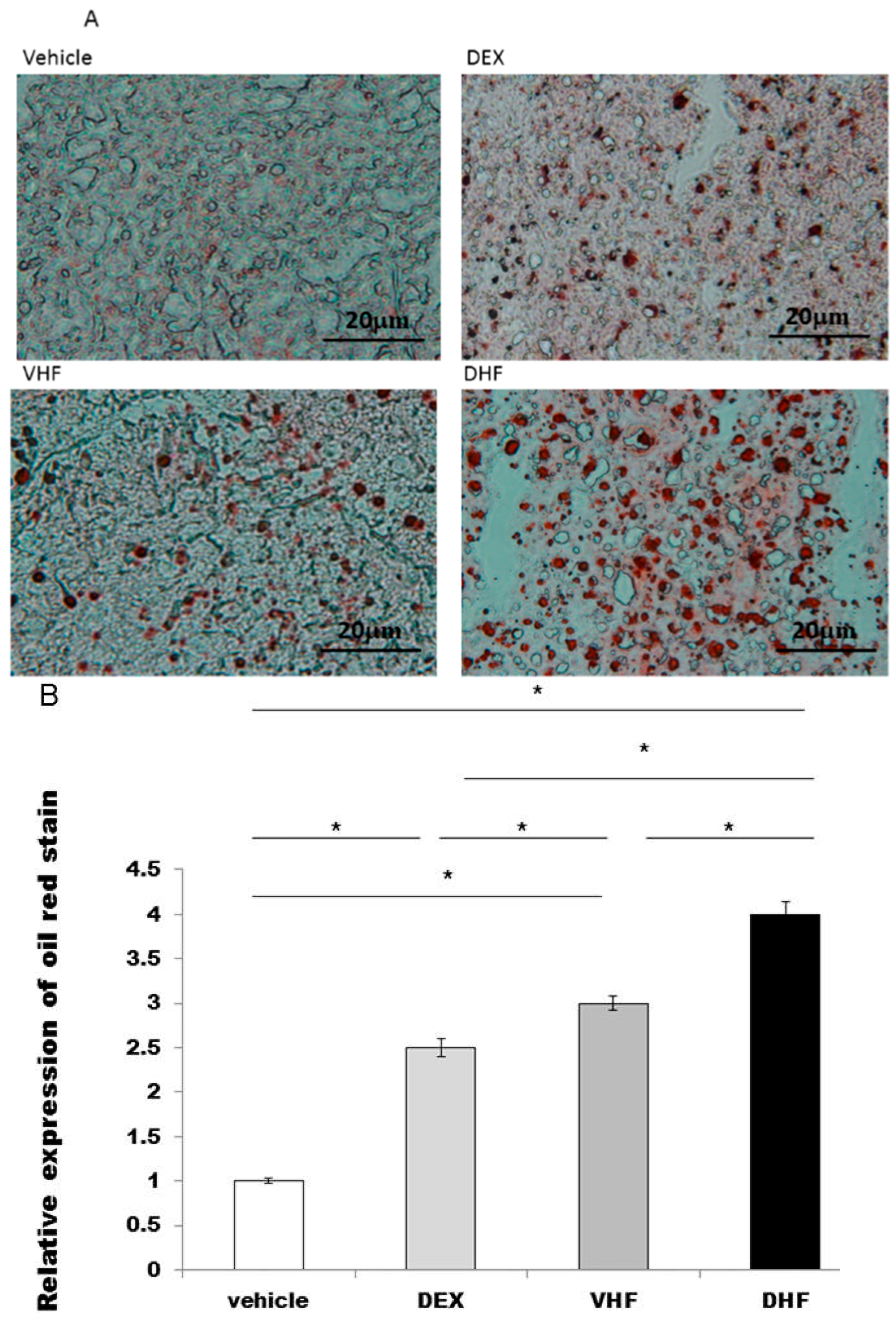
2.2. Liver Steatosis Was More Severe in the DHF Treated Group
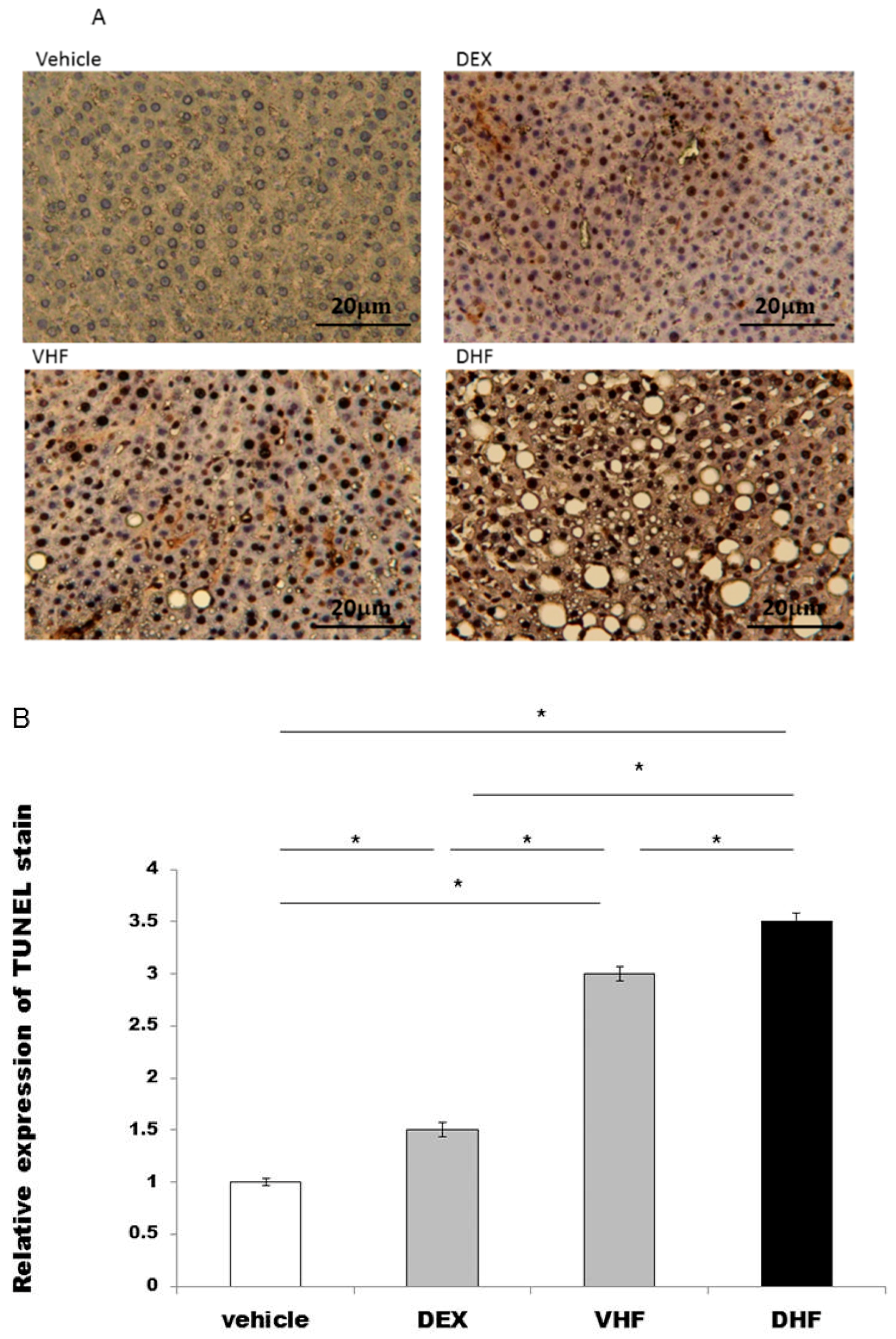
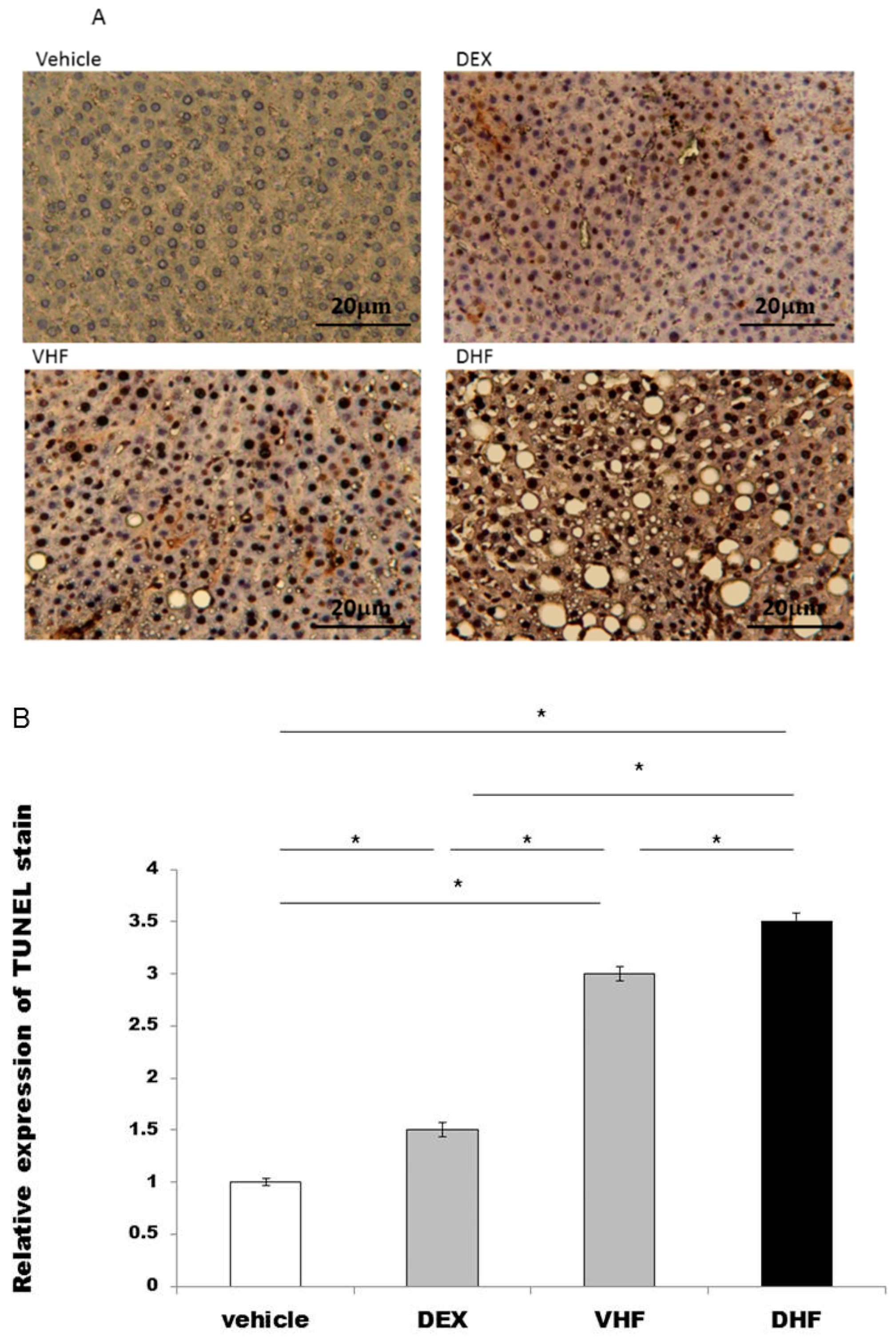
2.3. Apoptosis Was Higher in the DHF Treated Group
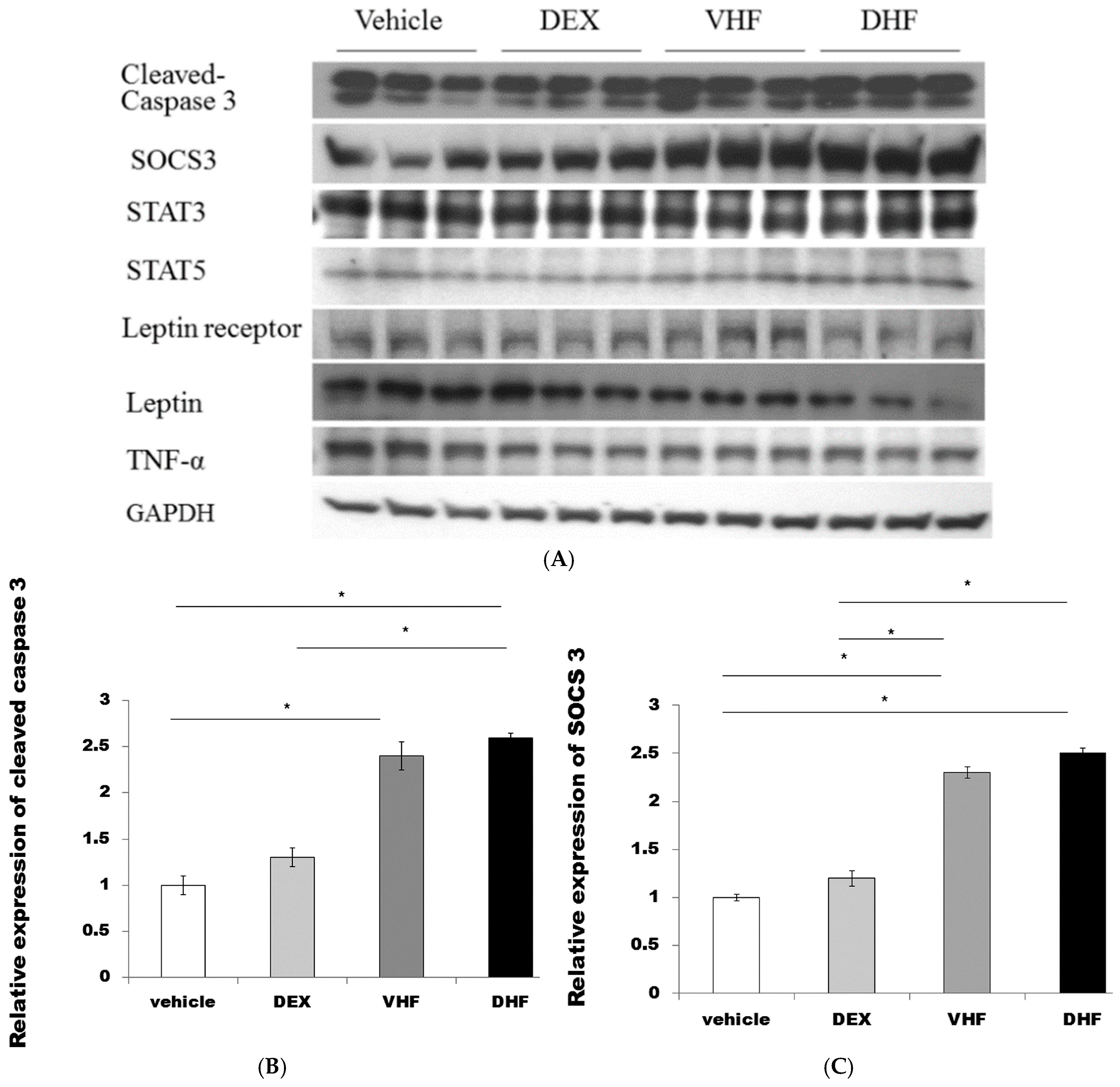
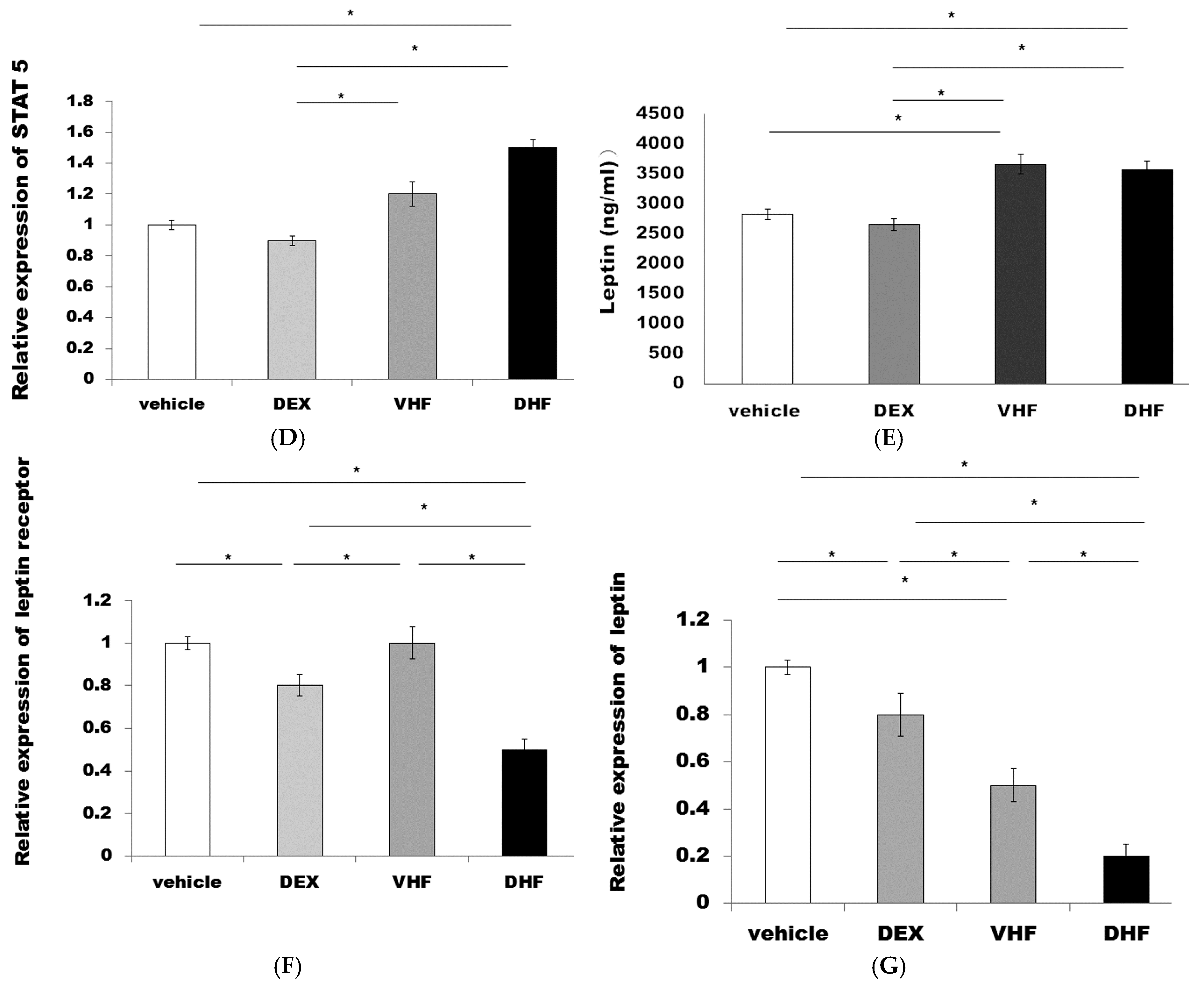
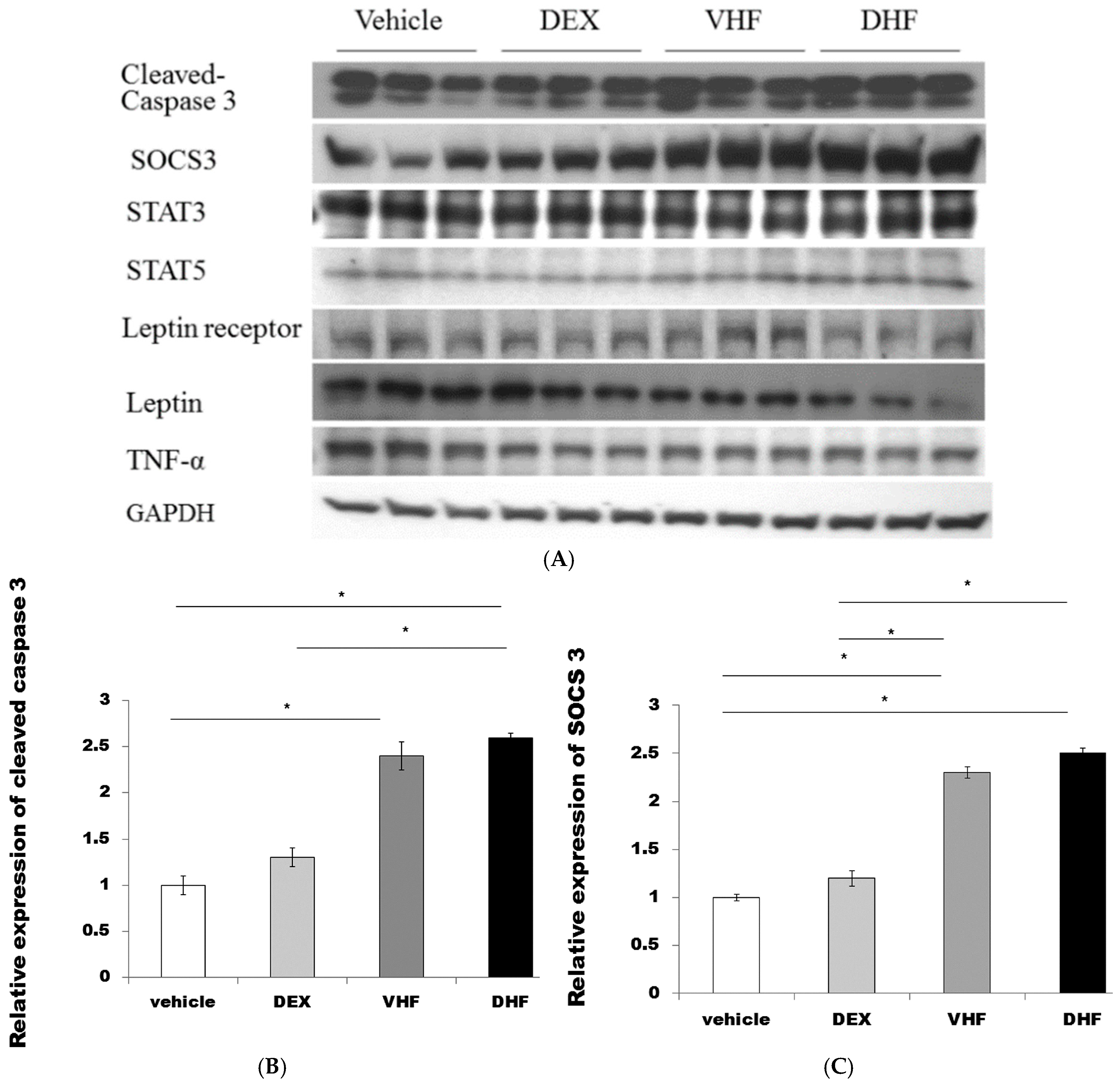
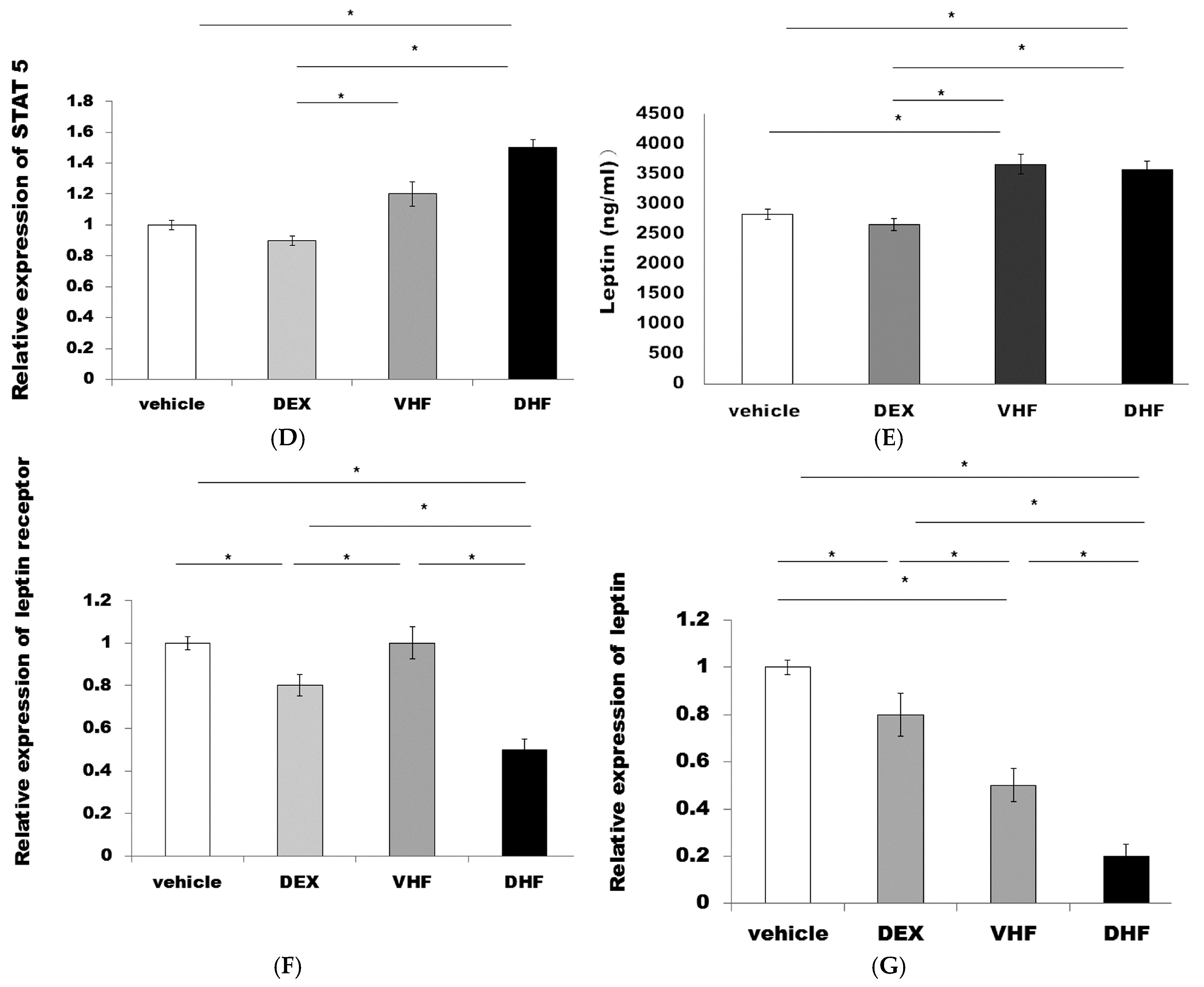
2.4. The Higher Activated Caspase 3 (Apoptosis) and Leptin Resistance in the DHF Treated Group
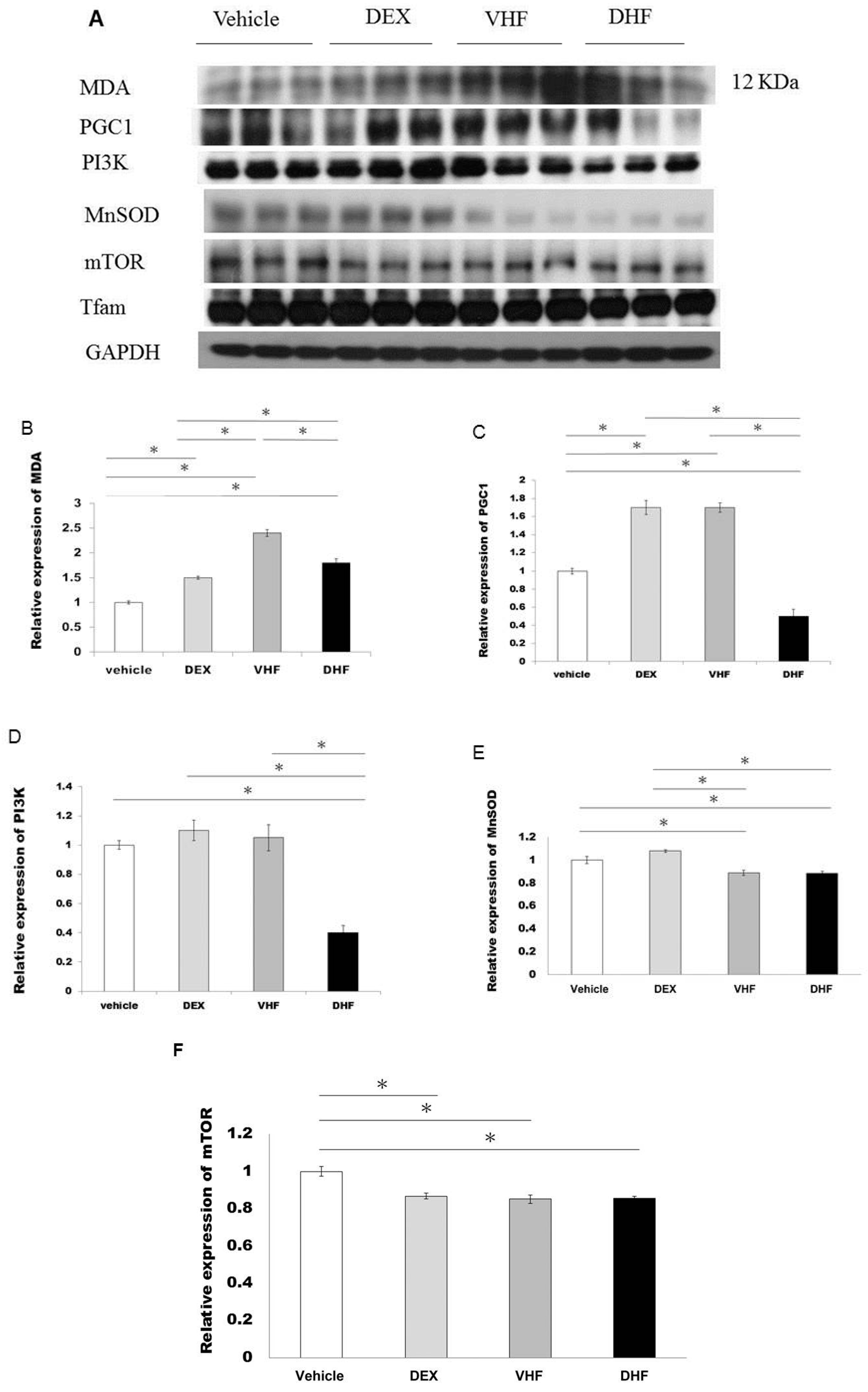
2.5. Increased Oxidative Stress Studies in DHF Treated Group
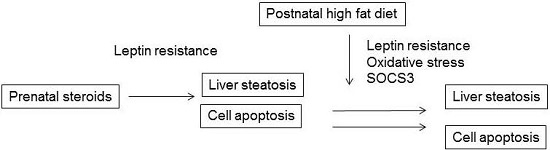
2.6. Discussion
3. Experimental Section
3.1. Animals
3.2. Experimental Procedures and Specimen Collection
3.3. Localization of Oil Red Stain Targets Fat Deposits and Analysis
3.4. TdT-Mediated dUTP Biotin Nick End Labeling (TUNEL)
3.5. Western Blotting Analysis
3.6. Cytokine Secretion with Enzyme-Linked Immunosorbent Assay (ELISA)
3.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kapoor, A.; Petropoulos, S.; Matthews, S.G. Fetal programming of hypothalamic-pituitary-adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Res. Rev. 2008, 57, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Seckl, J. Glucocorticoids, prenatal stress and the programming of disease. Horm. Behav. 2011, 59, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Drake, A.J.; Raubenheimer, P.J.; Kerrigan, D.; McInnes, K.J.; Seckl, J.R.; Walker, B.R. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology 2010, 151, 1581–1587. [Google Scholar] [CrossRef] [PubMed]
- Tamashiro, K.L.; Terrillion, C.E.; Hyun, J.; Koenig, J.I.; Moran, T.H. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes 2009, 58, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Parente, L.B.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Deleterious effects of high-fat diet on perinatal and postweaning periods in adult rat offspring. Clin. Nutr. 2008, 27, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B.; Schaart, G.; Mensink, R.P.; Schrauwen, P. Three weeks on a high-fat diet increases intrahepatic lipid accumulation and decreases metabolic flexibility in healthy overweight men. J. Clin. Endocrinol. Metab. 2011, 96, E691–E695. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Yu, H.R.; Chen, C.C.; Tiao, M.M.; Hsu, C.N.; Lin, Y.J.; Kuo, K.C.; Huang, L.T. Maternal melatonin therapy rescues prenatal dexamethasone and postnatal high-fat diet induced programmed hypertension in male rat offspring. Front. Physiol. 2015, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- Yida, Z.; Imam, M.U.; Ismail, M.; Ismail, N.; Ideris, A.; Abdullah, M.A. High fat diet-induced inflammation and oxidative stress are attenuated by N-acetylneuraminic acid in rats. J. Biomed. Sci. 2015, 22, 96. [Google Scholar] [CrossRef] [PubMed]
- Nichols, T.W., Jr. Mitochondria of mice and men: Moderate magnetic fields in obesity and fatty liver. Med. Hypotheses 2012, 79, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [PubMed]
- Carabelli, J.; Burgueno, A.L.; Rosselli, M.S.; Gianotti, T.F.; Lago, N.R.; Pirola, C.J.; Sookoian, S. High fat diet-induced liver steatosis promotes an increase in liver mitochondrial biogenesis in response to hypoxia. J. Cell. Mol. Med. 2011, 15, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Khaoustov, V.I.; Krishnan, B.; Cai, W.; Stoll, B.; Burrin, D.G.; Yoffe, B. Total parenteral nutrition induces liver steatosis and apoptosis in neonatal piglets. J. Nutr. 2006, 136, 2547–2552. [Google Scholar] [PubMed]
- Ducroc, R.; Sakar, Y.; Fanjul, C.; Barber, A.; Bado, A.; Lostao, M.P. Luminal leptin inhibits l-glutamine transport in rat small intestine: Involvement of ASCT2 and B0AT1. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G179–G185. [Google Scholar] [CrossRef] [PubMed]
- Tiao, M.M.; Huang, L.T.; Chen, C.J.; Sheen, J.M.; Tain, Y.L.; Chen, C.C.; Kuo, H.C.; Huang, Y.H.; Tang, K.S.; Chu, E.W.; et al. Melatonin in the regulation of liver steatosis following prenatal glucocorticoid exposure. Biomed. Res. Int. 2014, 2014, 942172. [Google Scholar] [CrossRef] [PubMed]
- Ahima, R.S.; Osei, S.Y. Leptin signaling. Physiol. Behav. 2004, 81, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Bjorbaek, C.; El-Haschimi, K.; Frantz, J.D.; Flier, J.S. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999, 274, 30059–30065. [Google Scholar] [CrossRef] [PubMed]
- Maier, I.B.; Stricker, L.; Ozel, Y.; Wagnerberger, S.; Bischoff, S.C.; Bergheim, I. A low fructose diet in the treatment of pediatric obesity: A pilot study. Pediatr. Int. 2011, 53, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Volynets, V.; Machann, J.; Kuper, M.A.; Maier, I.B.; Spruss, A.; Konigsrainer, A.; Bischoff, S.C.; Bergheim, I. A moderate weight reduction through dietary intervention decreases hepatic fat content in patients with non-alcoholic fatty liver disease (NAFLD): A pilot study. Eur. J. Nutr. 2013, 52, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Coon, A.B.; Robinson, S.M.; Moinuddin, A.; Shultz, J.M.; Nakaoke, R.; Morley, J.E. Triglycerides induce leptin resistance at the blood-brain barrier. Diabetes 2004, 53, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.L.; Zuloaga, D.G.; Hiroi, R.; Foradori, C.D.; Legare, M.E.; Handa, R.J. Prenatal dexamethasone exposure potentiates diet-induced hepatosteatosis and decreases plasma IGF-I in a sex-specific fashion. Endocrinology 2012, 153, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Lehavi, O.; Aizenstein, O.; Evans, M.I.; Yaron, Y. 2nd-trimester maternal serum human chorionic gonadotropin and α-fetoprotein levels in male and female fetuses with Down syndrome. Fetal Diagn. Therapy 2005, 20, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Gallou-Kabani, C.; Gabory, A.; Tost, J.; Karimi, M.; Mayeur, S.; Lesage, J.; Boudadi, E.; Gross, M.S.; Taurelle, J.; Vige, A.; et al. Sex- and diet-specific changes of imprinted gene expression and DNA methylation in mouse placenta under a high-fat diet. PLoS ONE 2010, 5, e14398. [Google Scholar] [CrossRef] [PubMed]
- Penaloza, C.; Estevez, B.; Orlanski, S.; Sikorska, M.; Walker, R.; Smith, C.; Smith, B.; Lockshin, R.A.; Zakeri, Z. Sex of the cell dictates its response: Differential gene expression and sensitivity to cell death inducing stress in male and female cells. FASEB J. 2009, 23, 1869–1879. [Google Scholar] [CrossRef] [PubMed]
- Ivey, R.; Desai, M.; Green, K.; Sinha-Hikim, I.; Friedman, T.C.; Sinha-Hikim, A.P. Additive effects of nicotine and high-fat diet on hepatocellular apoptosis in mice: Involvement of caspase 2 and inducible nitric oxide synthase-mediated intrinsic pathway signaling. Horm. Metab. Res. 2014, 46, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Sugden, M.C.; Langdown, M.L.; Munns, M.J.; Holness, M.J. Maternal glucocorticoid treatment modulates placental leptin and leptin receptor expression and materno-fetal leptin physiology during late pregnancy, and elicits hypertension associated with hyperleptinaemia in the early-growth-retarded adult offspring. Eur. J. Endocrinol. 2001, 145, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Matsuzaki, T.; Munkhzaya, M.; Tungalagsuvd, A.; Kawami, T.; Murakami, M.; Yamasaki, M.; Kato, T.; Kuwahara, A.; Yasui, T.; et al. Prenatal exposure to glucocorticoids affects body weight, serum leptin levels, and hypothalamic neuropeptide-Y expression in pre-pubertal female rat offspring. Int. J. Dev. Neurosci. 2014, 36, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bispham, J.; Budge, H.; Mostyn, A.; Dandrea, J.; Clarke, L.; Keisler, D.H.; Symonds, M.E.; Stephenson, T. Ambient temperature, maternal dexamethasone, and postnatal ontogeny of leptin in the neonatal lamb. Pediatr. Res. 2002, 52, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Gautron, L.; Elmquist, J.K. Sixteen years and counting: An update on leptin in energy balance. J. Clin. Investig. 2011, 121, 2087–2093. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, E.H.; Martinsen, M.; Strom, V.; Hansen, K.E.; Ravuri, C.S.; Gong, N.; Jobling, M. Long-term fasting in the anadromous Arctic charr is associated with downregulation of metabolic enzyme activity and upregulation of leptin A1 and SOCS expression in the liver. J. Exp. Biol. 2013, 216, 3222–3230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, Y.T.; Kakuma, T.; Lee, Y.; Kalra, S.P.; Kalra, P.S.; Pan, W.; Unger, R.H. Leptin resistance of adipocytes in obesity: Role of suppressors of cytokine signaling. Biochem. Biophys. Res. Commun. 2000, 277, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Bjorbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef]
- Heeba, G.H.; Morsy, M.A. Fucoidan ameliorates steatohepatitis and insulin resistance by suppressing oxidative stress and inflammatory cytokines in experimental non-alcoholic fatty liver disease. Environ. Toxicol. Pharmacol. 2015, 40, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, A.; Zang, S.; Wang, C.; Song, G.; Li, X.; Zhu, Y.; Yu, X.; Li, L.; Wang, Y.; Duan, L. Jinlida reduces insulin resistance and ameliorates liver oxidative stress in high-fat fed rats. J. Ethnopharmacol. 2015, 162, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, L.; Wang, H.; Narisi, B.; Chen, B. Li-Gan-Shi-Liu-Ba-Wei-San improves non-alcoholic fatty liver disease through enhancing lipid oxidation and alleviating oxidation stress. J. Ethnopharmacol. 2015, 176, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.H. Activation of the PI3K/Akt pathway by oxidative stress mediates high glucose-induced increase of adipogenic differentiation in primary rat osteoblasts. J. Cell. Biochem. 2013, 114, 2595–2602. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Arias, N.; Macarulla, M.T.; Aguirre, L.; Miranda, J.; Portillo, M.P. Liver delipidating effect of a combination of resveratrol and quercetin in rats fed an obesogenic diet. J. Physiol. Biochem. 2015, 71, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Garstka, H.L.; Schmitt, W.E.; Schultz, J.; Sogl, B.; Silakowski, B.; Perez-Martos, A.; Montoya, J.; Wiesner, R.J. Import of mitochondrial transcription factor A (TFAM) into rat liver mitochondria stimulates transcription of mitochondrial DNA. Nucleic Acids Res. 2003, 31, 5039–5047. [Google Scholar] [CrossRef] [PubMed]
- Tiao, M.M.; Wang, F.S.; Huang, L.T.; Chuang, J.H.; Kuo, H.C.; Yang, Y.L.; Huang, Y.H. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis 2014, 19, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Huang, L.T.; Huang, Y.H.; Tiao, M.M.; Tang, K.S.; Liou, C.W. The effect of the red wine polyphenol resveratrol on a rat model of biliary obstructed cholestasis: Involvement of anti-apoptotic signalling, mitochondrial biogenesis and the induction of autophagy. Apoptosis 2012, 17, 871–879. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | DEX | VHF | DHF | |
|---|---|---|---|---|
| Weight (gm) | 581.6 ± 29.2 * | 610.3 ± 21.6 † | 679.0 ± 30.7 *,† | 686.1 ± 29.9 *,† |
| AST/GOT (U/L) | 81.8 ± 30.5 * | 107.4 ± 10.4 † | 146.3 ± 11.3 *,† | 313.1 ± 43.3 *,† |
| Cholesterol (mg/dL) | 78.8 ± 6.1 * | 137.0 ± 4.8 † | 81.2 ± 7.3 * | 98.0 ± 2.3 *,† |
| Triglyceride (mg/dL) | 105.2 ± 19.0 * | 137.8 ± 31.9 † | 93.3 ± 13.6 * | 103.1 ± 12.5 * |
| HDL (mg/dL) | 48.8 ± 3.6 * | 38.2 ± 2.9 † | 48.3 ± 4.1 * | 41.9 ± 2.9 † |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.-H.; Chen, C.-J.; Tang, K.-S.; Sheen, J.-M.; Tiao, M.-M.; Tain, Y.-L.; Chen, C.-C.; Chu, E.-W.; Li, S.-W.; Yu, H.-R.; et al. Postnatal High-Fat Diet Increases Liver Steatosis and Apoptosis Threatened by Prenatal Dexamethasone through the Oxidative Effect. Int. J. Mol. Sci. 2016, 17, 369. https://doi.org/10.3390/ijms17030369
Huang Y-H, Chen C-J, Tang K-S, Sheen J-M, Tiao M-M, Tain Y-L, Chen C-C, Chu E-W, Li S-W, Yu H-R, et al. Postnatal High-Fat Diet Increases Liver Steatosis and Apoptosis Threatened by Prenatal Dexamethasone through the Oxidative Effect. International Journal of Molecular Sciences. 2016; 17(3):369. https://doi.org/10.3390/ijms17030369
Chicago/Turabian StyleHuang, Ying-Hsien, Chih-Jen Chen, Kuo-Shu Tang, Jiunn-Ming Sheen, Mao-Meng Tiao, You-Lin Tain, Chih-Cheng Chen, En-Wei Chu, Shih-Wen Li, Hong-Ren Yu, and et al. 2016. "Postnatal High-Fat Diet Increases Liver Steatosis and Apoptosis Threatened by Prenatal Dexamethasone through the Oxidative Effect" International Journal of Molecular Sciences 17, no. 3: 369. https://doi.org/10.3390/ijms17030369