
Roles of RNA-Binding Proteins in DNA Damage Response

Department of Radiation Oncology, Johns Hopkins University, School of Medicine, Baltimore, MD 21231, USA
Int. J. Mol. Sci. 2016, 17(3), 310; https://doi.org/10.3390/ijms17030310
Submission received: 4 February 2016
/
Revised: 18 February 2016
/
Accepted: 22 February 2016
/
Published: 27 February 2016
(This article belongs to the Special Issue DNA Damage and Repair in Degenerative Diseases 2016)
{kind=link}
Abstract
:Living cells experience DNA damage as a result of replication errors and oxidative metabolism, exposure to environmental agents (e.g., ultraviolet light, ionizing radiation (IR)), and radiation therapies and chemotherapies for cancer treatments. Accumulation of DNA damage can lead to multiple diseases such as neurodegenerative disorders, cancers, immune deficiencies, infertility, and also aging. Cells have evolved elaborate mechanisms to deal with DNA damage. Networks of DNA damage response (DDR) pathways are coordinated to detect and repair DNA damage, regulate cell cycle and transcription, and determine the cell fate. Upstream factors of DNA damage checkpoints and repair, “sensor” proteins, detect DNA damage and send the signals to downstream factors in order to maintain genomic integrity. Unexpectedly, we have discovered that an RNA-processing factor is involved in DNA repair processes. We have identified a gene that contributes to glioblastoma multiforme (GBM)’s treatment resistance and recurrence. This gene, RBM14, is known to function in transcription and RNA splicing. RBM14 is also required for maintaining the stem-like state of GBM spheres, and it controls the DNA-PK-dependent non-homologous end-joining (NHEJ) pathway by interacting with KU80. RBM14 is a RNA-binding protein (RBP) with low complexity domains, called intrinsically disordered proteins (IDPs), and it also physically interacts with PARP1. Furthermore, RBM14 is recruited to DNA double-strand breaks (DSBs) in a poly(ADP-ribose) (PAR)-dependent manner (unpublished data). DNA-dependent PARP1 (poly-(ADP) ribose polymerase 1) makes key contributions in the DNA damage response (DDR) network. RBM14 therefore plays an important role in a PARP-dependent DSB repair process. Most recently, it was shown that the other RBPs with intrinsically disordered domains are recruited to DNA damage sites in a PAR-dependent manner, and that these RBPs form liquid compartments (also known as “liquid-demixing”). Among the PAR-associated IDPs are FUS/TLS (fused in sarcoma/translocated in sarcoma), EWS (Ewing sarcoma), TARF15 (TATA box-binding protein-associated factor 68 kDa) (also called FET proteins), a number of heterogeneous nuclear ribonucleoproteins (hnRNPs), and RBM14. Importantly, various point mutations within the FET genes have been implicated in pathological protein aggregation in neurodegenerative diseases, specifically with amyotrophic lateral sclerosis (ALS), and frontotemporal lobe degeneration (FTLD). The FET proteins also frequently exhibit gene translocation in human cancers, and emerging evidence shows their physical interactions with DDR proteins and thus implies their involvement in the maintenance of genome stability.
1. Introduction
Involvements of RNA-binding proteins (RBPs) in DNA damage response (DDR) have been described, and growing evidence suggests their roles in DNA repair. Large-scale proteomic and genetic screenings have identified RNA processing factors including RBPs in major functional categories of ATM (ataxia-telangiectasia mutated)/ATR (ataxia telangiectagia and Rad3 related) substrates [1] and in the prevention of genomic instability [2,3]. However, mechanisms of their involvement in DDR regulation remain elusive. Recent findings on liquid demixing produced by poly(ADP-ribose) (PAR)-dependent recruitments of RBPs shed new light on how DDR might be initiated.
2. Control of DNA Damage Response (DDR)-Gene Expression by RNA-Binding Protein (RBP)
A large body of evidence suggests that RNA-processing factors regulate the expression of several genes involved in DDR. For example, one of the FET proteins in Ewing sarcoma (EWS) depletion results in alternative splicing (AS) changes of genes involved in DNA repair and genotoxic stress signaling, including ABL1, CHK2, and MAP4K2. Chromatin and RNA crosslinking immunoprecipitation experiments show that EWS co-transcriptionally binds to its target RNAs. This association is decreased after UV irradiation of cells, concomitant with transient enrichment of EWS in nucleoli and with AS changes that parallel those induced by EWS depletion that lead to reduced c-ABL expression [4].
BRCA1 (breast cancer 1), one of the key players in the cell cycle checkpoint and homologous recombination (HR), interacts with the mRNA splicing factor BCLAF1. This BRCA1-BCLAF1 complex regulates mRNA splicing of DDR genes ATRIP, BACH1, and EXO1 in response to DNA damage, although AS of these genes were not detected in this study. Importantly, ATM/ATR-dependent phosphorylation of BRCA1 at Ser1423 is required for its interaction with BCLAF1 [5].
Levels of the tumor suppressor and the DNA damage checkpoint protein p53 are increased in response to DNA damage [6]. The p53 protein level is primarily regulated through ubiquitin-mediated proteolysis, and DNA damage-induced phosphorylation inhibits this degradation process [7]. However, p53 mRNA stability and translation are shown as strong regulators of p53 expression [8]. The RNA-binding protein HuR (Hu antigen R) binds to p53 mRNA and enhances its translation [8]. In response to ionizing radiation (IR)-induced damage, HuR targets mRNAs that encode DDR-, apoptosis-, and proliferation-related proteins, such as 53BP1, MDM2, BAX, K-Ras, and p21 [9,10,11]. Importantly, one of the DNA damage checkpoint kinases, CHK2, phosphorylates HuR in response to DNA damage, and the CHK2-dependent phosphorylation induces HuR’s association and dissociation from its target mRNAs. CHK2 phosphorylation-induced dissociation of HuR is import for cell survival after IR exposure [10]. It is also required for regulation of the mRNA expression encoding the longevity and stress-response protein SIRT1 [12]. HuR binds to SIRT1 mRNA and stabilizes it, leading to increased SIRT1 expression levels. Oxidative stress triggers the dissociation of HuR from SIRT1, promoting SIRT1 mRNA decay. The influence of HuR function by ATM-CHK2-dependent phosphorylation underscores the intricate connections between the RBP and DNA damage and stress responsiveness [12].
The RNA-binding protein RBMX has been identified as a protein involved in DDR by a genome-wide siRNA screen to find components of the mammalian HR machinery using a well-characterized GFP (green fluorescent protein)-based HR reporter [13]. RBMX is a heterogeneous nuclear ribonucleoprotein that has a role in alternative splicing [14]. Knockdown of RBMX inhibits HR, and RBMX accumulates at DNA lesions through multiple domains in a PARP1-dependent manner. However, PARP1 inhibition does not cause HR defects, and inhibition of HR by RBMX knockdown seems to be caused by the reduced expression of BRCA2. Similar effects have been observed with knockdown of some of the other pre-mRNA-processing genes that have been identified by the screening [13].
3. Direct Roles of RBPs in DDR
In addition to their roles in AS and the expression of DDR genes, direct involvement of RBPs in DDR has been shown. PRP19 is an ubiquitin ligase and an important regulator of pre-mRNA splicing [15,16,17]. During splicing, PRP19 ubiquitylates the U4 small nuclear ribonucleic particles (snRNP) component PRP3, leading to the stabilization of the U4/U6.U5 snRNP [18]. PRP19 interacts with RNA polymerase II, and couples RNA processing and transcription [19,20]. Interestingly, yeast PRP19 (also called PSO4) has been identified by independent genetic screenings for splicing as well as DDR mutants [21,22,23]. It has also been shown in human cells that PRP19 interacts with WRN (Werner syndrome, RecQ helicase-like), which has DNA helicase and nuclease activities, for the processing of DNA interstrand cross-links and with terminal deoxynucleotidyl transferase for DNA repair [24,25]. A core component of the putative E3 ubiquitin ligase complex of PRP19, CDC5L, interacts with ATR. CDC5L-depleted cells are sensitive to replication blocking agents, and are defective in S-phase checkpoint. Furthermore, CDC5L is required for the activation of downstream effectors or mediators of ATR checkpoint function, such as CHK1, RAD17, and FANCD2 proteins [26]. Recent proteomic screenings have identified PRP19 as a protein that interacts with RPA (replication protein A)-coated single-stranded DNA (ssDNA) [27,28]. PRP19 binds to RPA directly in vitro, and localizes to sites of DNA damage via RPA in cells. PRP19 promotes ubiquitylation of RPA in a DNA damage-induced manner, and facilitates accumulation of ATRIP (ATR interacting protein) at the site of DNA damage. Depletion of PRP19 compromises phosphorylation of RPA32 and CHK1, leading to defective recovery of stalled replication forks and impaired fork progression on damaged DNA. Importantly, PRP19 mutants that are unable to bind to RPA or function as an E3 ligase failed to support the ATR response, suggesting that full activation of ATR is driven by ubiquitylation-mediated circuitry orchestrated by RPA-ssDNA and PRP19. These results imply that the RNA-processing protein in undamaged cells transforms into a DNA damage sensor during DDR, revealing an unexpected interplay between these two fundamental processes [27].
Heterogeneous nuclear ribonucleoprotein U-like (hnRNPUL) proteins 1 and 2 have been identified as binding partners for the DNA double-strand break (DSB) sensor complex MRE11-RAD50-NBS1 (MRN). The hnRNPUL 1 and 2 proteins are recruited to DNA damage in an MRE11-dependent manner. They stimulate DNA-end resection and promote ATR-dependent signaling and DSB repair by HR. Furthermore, hnRNPUL 1 and 2 function downstream of MRN and CtIP to promote recruitment of BLM (Bloom syndrome, RecQ helicase-like) helicase to DSBs [29,30]
We originally identified RBM14 as a gene that radio-sensitizes glioblastoma multiforme (GBM) spheres when knocked down. RBM14 is highly expressed in embryonic tissues and in stem cells, and has been implicated in RNA splicing as well as in transcription. It is thought that RBM14 controls transcription-coupled alternative splicing in a manner that depends on the promoter [31,32]. RBM14 contains two N-terminal RNA recognition motifs (RRMs) and a prion-like domain (PLD). This PLD is found in oncoproteins EWS and TLS/FUS family proteins that are mutated both in human cancers and neurodegenerative diseases [33,34]. Amplification of RBM14 has been observed in human cancers [35]. The RRM domains enable it to act as a regulator of mRNA splicing. The PLD is required for interaction with TRBP (thyroid-hormone-receptor-binding protein), p300, SYT (synovial sarcoma translocation protein), and RUNX2, and exhibits transcription regulation activity [36,37,38,39]. RBM14 (also called CoAA) is alternatively spliced to produce a smaller protein called CoAM that lacks the PLD, which acts in a dominant negative fashion to regulate transcription [37,40]. Our results indicate that RBM14 controls the non-homologous end-joining (NHEJ) pathway of DSB repair, and is involved in maintaining the stem-like state of GBM spheres. RBM14 knockdown cells are sensitive to IR. RBM14 interacts with KU80, and autophosphorylation of DNA-PK is compromised in RBM14 knockdown cells. These results indicate that RBM14 is required for the NHEJ pathway [41]. Most importantly, knockdown of RBM14 reduces tumorigenicity and also radio-sensitizes GBM stem-like cells in vivo [41]. RBM14 is a component of paraspeckles that are nuclear bodies composed of RBPs and the long noncoding RNA NEAT1 [42,43]. Interestingly, the other paraspeckle proteins, FUS and NONO, have also been implicated in DDR. NONO is recruited to laser-induced DSB sites in a PAR-dependent manner, and stimulates NHEJ and represses HR [44]. FUS is also recruited to laser-induced DSB sites in a PAR-dependent manner. However, knockdown of FUS inhibits both NHEJ and HR [45,46,47,48,49]. The function of FUS in DDR involves its direct interaction with histone deacetylase 1 (HDAC1). Knockdown of FUS abolishes γH2AX formation in response to DSBs [45], whereas RBM14 knockdown induces prolonged γH2AX foci [41]. These results indicate that there might be several layers of controls to initiate a particular repair process by different PAR-recruited RBPs.
Like FUS and NONO, several other RBPs are recruited to laser-induced DSBs in a PAR- or PARP-dependent manner. These RBPs are intrinsically disordered proteins (IDPs) that contain an unstructured “prion”-like domain (PLD). PLDs are a subset of low complexity regions, enriched in unchanged polar amino acids and glycines, with similarities to the yeast prion protein [43,50]. Importantly, PLDs are often found in RBPs that drive protein aggregation in neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS) [51]. These RBPs are detected on laser tracks within one minute after laser irradiation, and are excluded from the laser tracks shortly (within 10–15 min, depending on conditions of laser irradiation) [29,30,44,45,50,52]. It was shown recently that some of these RBPs, FUS, EWS, TAF15, and RBM14, form a PAR-dependent liquid-like compartment by phase separation at the laser-induced DSB sites [43,50]. This phase separation, also referred to as liquid demixing, requires the PLDs [43,50,51]. PLD-containing proteins/IDPs can phase-separate from the soluble intracellular space, and are disposed to aggregating under pathological conditions, forming “functional aggregates”. The nucleic acid–mimicking biopolymer PAR nucleates intracellular liquid demixing. PAR-levels are significantly increased at sites of DNA damage. PAR seeds RBPs/IDPs for liquid demixing, resulting in the rapid yet transient and fully reversible association of various RBPs/IDPs at DSB sites. Liquid-demixing relies on electrostatic interactions between positively charged RGG repeats (found in the PLDs) and negatively charged PAR, and is amplified by aggregation of PLDs. It has been proposed that PAR-seeded liquid demixing is a general mechanism to dynamically reorganize the soluble nuclear space. Deflected phase separation is implicated in pathological protein aggregation that is believed to cause neurological diseases [51]. However, it is not understood how these RBPs are involved in DNA repair pathways. This phase separation by PAR-seeded RBPs/IDPs is likely to provide orchestration of the earliest cellular DDR prior to the recruitment of classical “sensor” proteins (see Figure 1 for a model).
Immediate recruitment of prion-like RBPs to sites of DNA damage followed by phase separation requires PARP1. PARP1, which catalyzes the attachment of ADP-ribose units to target proteins, plays wide-ranging roles in cellular processes including DNA repair and transcription. PARP1 is rapidly recruited to DNA damage sites including single-strand nicks and DSBs, where its catalytic activity is enhanced by up to orders of magnitude, resulting in the synthesis of protein-conjugated long branches of ADP-ribose chains [53,54]. The best-known roles of PARP1 are associated with DNA damage and genomic maintenance, with specific roles in base-excision/single-strand break repair. However, roles of PARP1 in DSB repair remain controversial. PARP1 is activated by DSBs, and is implicated in classical and alternative (c/alt) NHEJ and HR pathways [54]. PARP1 is involved in the early recruitment of the MRN complex to DSBs [55], and interacts with cNHEJ components such as KU and DNA-PK [56,57]. However, PARP1-deficient cells do not exhibit cNHEJ defects [58]. Instead, biochemical studies suggest that PARP1 facilitates altNHEJ and inhibits cNHEJ [59,60,61]. PARP-depleted cells show little effect on HR efficiency by homology-directed repair [62]. However, PARP1 has been strongly implicated in recovery from stalled replication forks that is mediated by HR [58,63,64,65]. In fact, PARP1 promotes recruitment of MRE11 and RAD51 specifically in response to stalled replication forks [55,66]. Understanding of PARylation/PARP1’s exact role in response to DNA damage requires further investigation. The PAR-dependent liquid-demixing seems to provide a platform for RBPs which contain PLDs (IDPs). This discovery is a new clue for exploring mechanisms of PAR-dependent initiation/selection of DDR pathways.
Mutations in proteins that contain the PLDs of low sequence complexity are associated with neurodegenerative and aging-associated diseases because the disordered sequences are prone to aggregation. FUS is a prion-like protein containing intrinsically disordered domains, and mutations in the FUS gene are implicated in causes of the neurodegenerative disease ALS. An in vitro “aging” experiment demonstrated that liquid droplets of FUS protein convert with time from a liquid to an aggregated state, and that this conversion is accelerated by patient-derived mutations. Therefore, the physiological role of FUS and presumably the other prion-like RBPs, the formation of dynamic liquid-like compartments through intrinsically disordered regions, faces trade-off between functionality and risk of pathological aggregations, causing ALS and, presumably, other age-related diseases [51].
4. Conclusions
Recent discovery of the “liquid-demixing” state created by PAR and IDPs at damaged DNA areas opened up a new paradigm in the field. Further investigation on how these prion-like domain-containing proteins orchestrate DNA repair pathways will deepen our understanding of neurological diseases and cancers caused by mutations in these proteins.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Lackner, D.H.; Durocher, D.; Karlseder, J. A siRNA-based screen for genes involved in chromosome end protection. PLoS ONE 2011, 6, e21407. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Minana, B.; Valcarcel, J. The ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Furneaux, H.; Cheng, H.; Caldwell, M.C.; Hutter, D.; Liu, Y.; Holbrook, N.; Gorospe, M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000, 20, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Hagner, P.R.; Zhang, Y.; Dai, B.; Lehrmann, E.; Becker, K.G.; Keene, J.D.; Gorospe, M.; Liu, Z.; Gartenhaus, R.B. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011, 117, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Zhang, Z.; Raitskin, O.; Hiller, M.; Benderska, N.; Hartmann, A.M.; Bracco, L.; Elliott, D.; Ben-Ari, S.; Soreq, H.; et al. Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J. Biol. Chem. 2009, 284, 14303–14315. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.P.; Cheng, S.C. The PRP19-associated complex is required for specifying interactions of U5 and U6 with pre-mRNA during spliceosome activation. J. Biol. Chem. 2005, 280, 31190–31199. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.P.; Kao, D.I.; Tsai, W.Y.; Cheng, S.C. The PRP19p-associated complex in spliceosome activation. Science 2003, 302, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Kao, D.I.; Chan, S.P.; Kao, T.C.; Lin, J.Y.; Cheng, S.C. Functional links between the PRP19-associated complex, U4/U6 biogenesis, and spliceosome recycling. RNA 2006, 12, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Werner, S.L.; Neubauer, J.; Stegmeier, F.; Aspden, J.; Rio, D.; Harper, J.W.; Elledge, S.J.; Kirschner, M.W.; Rape, M. The PRP19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010, 24, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Chanarat, S.; Seizl, M.; Strasser, K. The PRP19 complex is a novel transcription elongation factor required for trex occupancy at transcribed genes. Genes Dev. 2011, 25, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Boyne, A.R.; Millhouse, S.R.; Manley, J.L. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-PRP19 complex. Genes Dev. 2011, 25, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Tarn, W.Y.; Tsao, T.Y.; Abelson, J. PRP19: A novel spliceosomal component. Mol. Cell. Biol. 1993, 13, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Tarn, W.Y.; Lee, K.R.; Cheng, S.C. The yeast PRP19 protein is not tightly associated with small nuclear RNAs, but appears to associate with the spliceosome after binding of U2 to the pre-mRNA and prior to formation of the functional spliceosome. Mol. Cell. Biol. 1993, 13, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dusterhoft, A.; Henriques, J.A.; Brendel, M. Allelism of PSO4 and PRP19 links pre-mRNA processing with recombination and error-prone DNA repair in saccharomyces cerevisiae. Nucleic Acids Res. 1996, 24, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.N.; Mitchell, B.S. Role of human PSO4 in mammalian DNA repair and association with terminal deoxynucleotidyl transferase. Proc. Natl. Acad. Sci. USA 2003, 100, 10746–10751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kaur, R.; Lu, X.; Shen, X.; Li, L.; Legerski, R.J. The PSO4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 2005, 280, 40559–40567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kaur, R.; Akhter, S.; Legerski, R.J. CDC5L interacts with atr and is required for the S-phase cell-cycle checkpoint. EMBO Rep. 2009, 10, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Huang, J. The pso4 protein complex associates with replication protein a (RPA) and modulates the activation of ataxia telangiectasia-mutated and RAD3-related (ATR). J. Biol. Chem. 2014, 289, 6619–6626. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Jiang, J.; Ma, J.; Dai, S.; Xu, T.; Li, H.; Yasui, A. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS ONE 2013, 8, e60208. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhang, Z.J.; Oldenburg, M.; Ayala, M.; Zhang, S.C. Human embryonic stem cell-derived dopaminergic neurons reverse functional deficit in parkinsonian rats. Stem Cells 2008, 26, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Dowhan, D.H.; Li, X.; Larkin, K.; Ko, L.; Berget, S.M.; O’Malley, B.W. CoAA, a nuclear receptor coactivator protein at the interface of transcriptional coactivation and RNA splicing. Mol. Cell. Biol. 2004, 24, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Law, W.J.; Cann, K.L.; Hicks, G.G. TLS, EWS and TAF15: A model for transcriptional integration of gene expression. Brief. Funct. Genom. Proteom. 2006, 5, 8–14. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, L.; McCall, P.; Hatziieremia, S.; Catlow, J.; Adams, C.; McArdle, P.; Seywright, M.; Tanahill, C.; Paul, A.; Underwood, M.; et al. Nuclear factor κB predicts poor outcome in patients with hormone-naive prostate cancer with high nuclear androgen receptor. Hum. Pathol. 2012, 43, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Yang, Z.; Xiong, S.; Zhang, L.; Blanchard, K.L.; Peiper, S.C.; Dynan, W.S.; Tuan, D.; Ko, L. Gene amplification and associated loss of 5′ regulatory sequences of CoAA in human cancers. Oncogene 2007, 26, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Perani, M.; Antonson, P.; Hamoudi, R.; Ingram, C.J.; Cooper, C.S.; Garrett, M.D.; Goodwin, G.H. The proto-oncoprotein SYT interacts with SYT-interacting protein/co-activator activator (SIP/CoAA), a human nuclear receptor co-activator with similarity to ews and TLS/FUS family of proteins. J. Biol. Chem. 2005, 280, 42863–42876. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Chin, W.W.; Ko, L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM). J. Biol. Chem. 2001, 276, 33375–33383. [Google Scholar] [CrossRef] [PubMed]
- Verreman, K.; Baert, J.L.; Verger, A.; Drobecq, H.; Ferreira, E.; de Launoit, Y.; Monte, D. The coactivator activator CoAA regulates PEA3 group member transcriptional activity. Biochem. J. 2011, 439, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Koibuchi, N.; Chin, W.W. Synovial sarcoma translocation (SYT) encodes a nuclear receptor coactivator. Endocrinology 2005, 146, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Brooks, Y.S.; Wang, G.; Yang, Z.; Smith, K.K.; Bieberich, E.; Ko, L. Functional pre-mRNA trans-splicing of coactivator coaa and corepressor RBM4 during stem/progenitor cell differentiation. J. Biol. Chem. 2009, 284, 18033–18046. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Eberhart, C.G.; Kai, M. RNA binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget 2014, 5, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Benard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Krietsch, J.; Caron, M.C.; Gagne, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. Parp activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Akhmedov, A.T.; Delacote, F.; Durrbach, A.; Lopez, B.S. Human POMp75 is identified as the pro-oncoprotein TLS/FUS: Both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 1999, 18, 4515–4521. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grofte, M.; Rask, M.B.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the als protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Dernoncourt, E.; Delteil, C.; Froment, C.; Schiltz, O.; Salles, B.; Frit, P.; Calsou, P. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014, 42, 9047–9062. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Pears, C.J.; Couto, C.A.; Wang, H.Y.; Borer, C.; Kiely, R.; Lakin, N.D. The role of ADP-ribosylation in regulating DNA double-strand break repair. Cell Cycle 2012, 11, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef] [PubMed]
- Mandraju, R.; Chekuri, A.; Bhaskar, C.; Duning, K.; Kremerskothen, J.; Kondapi, A.K. Topoisomerase iibeta associates with Ku70 and PARP-1 during double strand break repair of DNA in neurons. Arch. Biochem. Biophys. 2011, 516, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Weinfeld, M.; Calsou, P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J. Mol. Biol. 2006, 356, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair 2011, 10, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. PARP-1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate MRE11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
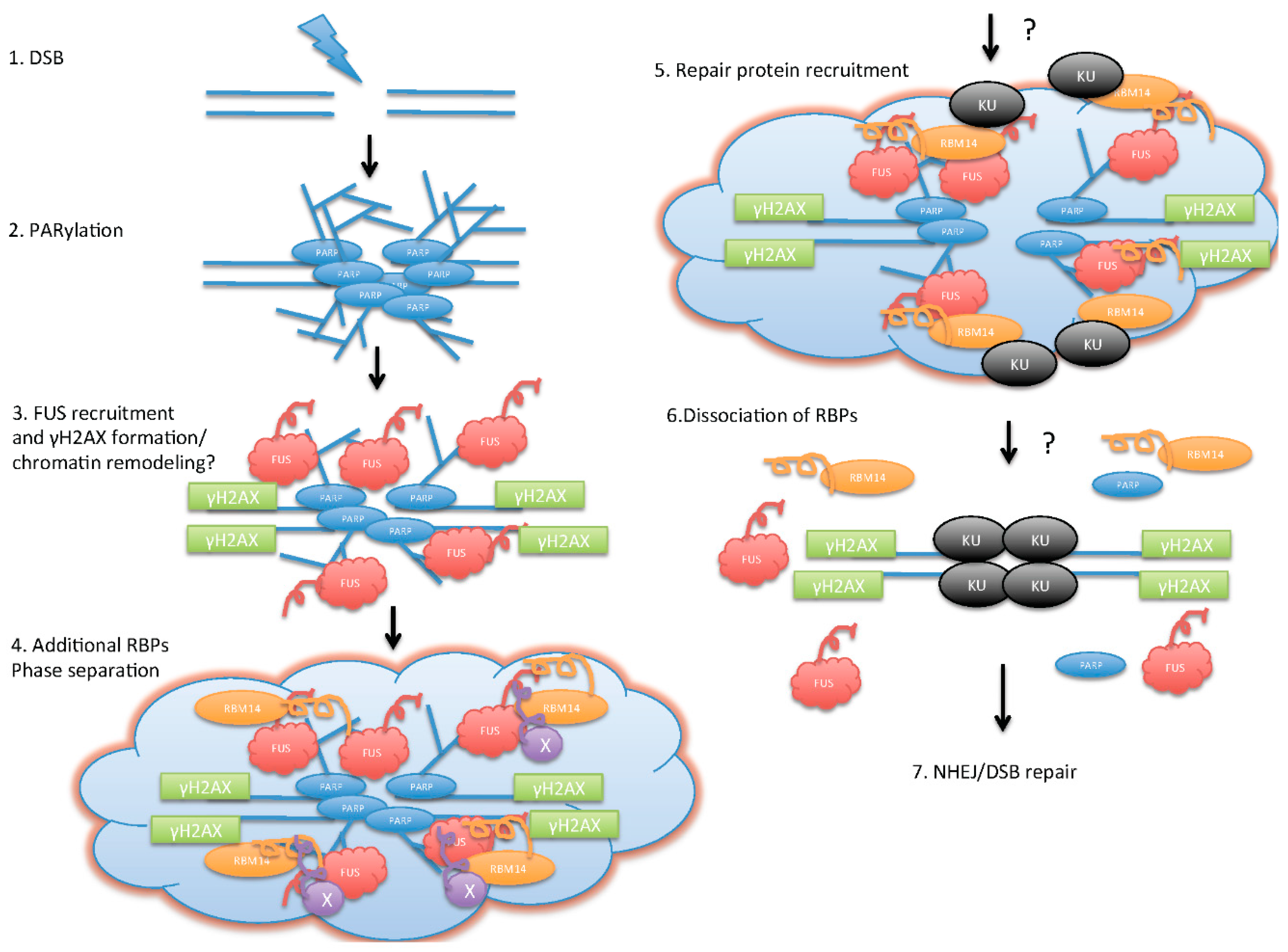
Figure 1.
Illustration of model for poly(ADP-ribose) (PAR)/RNA-binding proteins (RBPs)-mediated initiation of DNA double-strand break (DSB) repair. Upon DSB formation (1); PARylation by PARP occurs around the DSB area, and creates platforms for RBPs (2); FUS (fused in sarcoma)proteins are recruited to the DSB sites in a PAR-dependent manner, and this step allows γH2AX formation (3); The additional RBPs with “prion”-like domains (PLDs) are recruited to the DSB sites, and form “functional” aggregation. This step starts initiation of phase separation/liquid-demixing (4); Phase separation creates an environment for the DSB repair protein recruitment (5); PAR degradation by PARG followed by dissociation of RBPs (6) allows DSB repair; (7) non-homologous end-joining (NHEJ)/DSB repair.
Figure 1.
Illustration of model for poly(ADP-ribose) (PAR)/RNA-binding proteins (RBPs)-mediated initiation of DNA double-strand break (DSB) repair. Upon DSB formation (1); PARylation by PARP occurs around the DSB area, and creates platforms for RBPs (2); FUS (fused in sarcoma)proteins are recruited to the DSB sites in a PAR-dependent manner, and this step allows γH2AX formation (3); The additional RBPs with “prion”-like domains (PLDs) are recruited to the DSB sites, and form “functional” aggregation. This step starts initiation of phase separation/liquid-demixing (4); Phase separation creates an environment for the DSB repair protein recruitment (5); PAR degradation by PARG followed by dissociation of RBPs (6) allows DSB repair; (7) non-homologous end-joining (NHEJ)/DSB repair.

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. https://doi.org/10.3390/ijms17030310
AMA Style
Kai M. Roles of RNA-Binding Proteins in DNA Damage Response. International Journal of Molecular Sciences. 2016; 17(3):310. https://doi.org/10.3390/ijms17030310
Chicago/Turabian StyleKai, Mihoko. 2016. "Roles of RNA-Binding Proteins in DNA Damage Response" International Journal of Molecular Sciences 17, no. 3: 310. https://doi.org/10.3390/ijms17030310
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.