
Pseudogenes and Their Genome-Wide Prediction in Plants

Abstract
:1. Introduction
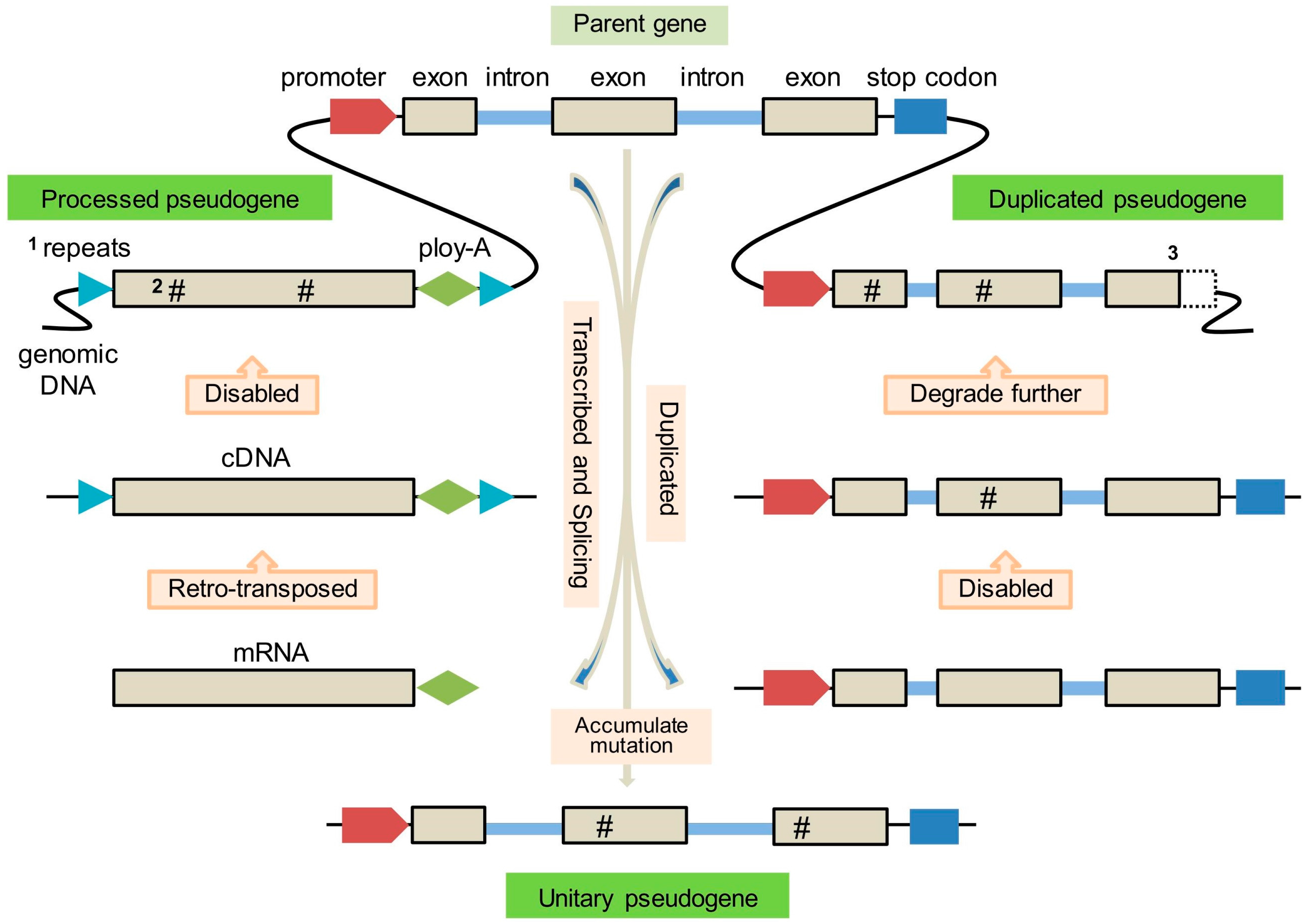
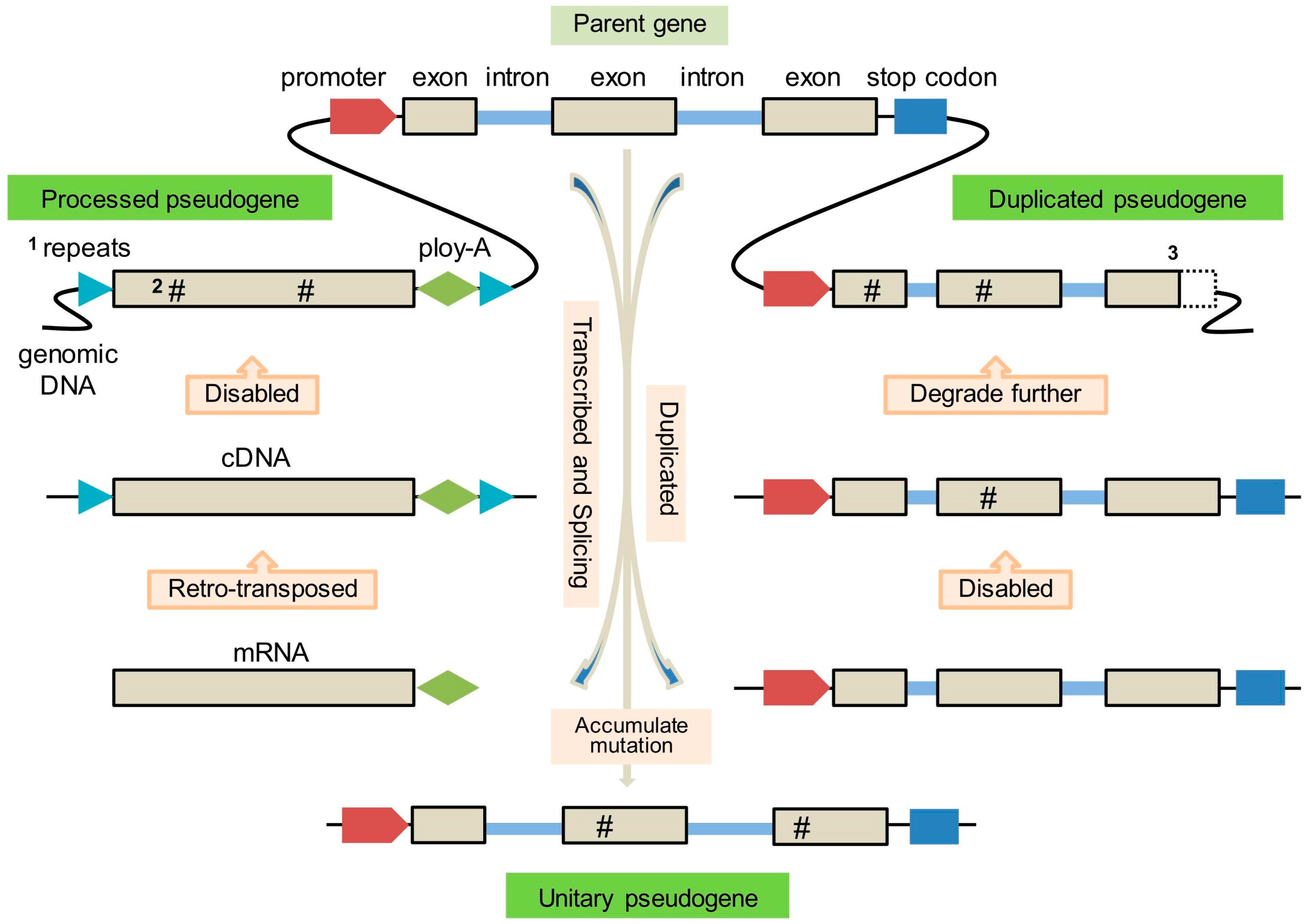
2. Origin and Formation of Pseudogenes
2.1. Processed Pseudogenes
2.2. Duplicated Pseudogenes
2.3. Unitary Pseudogenes
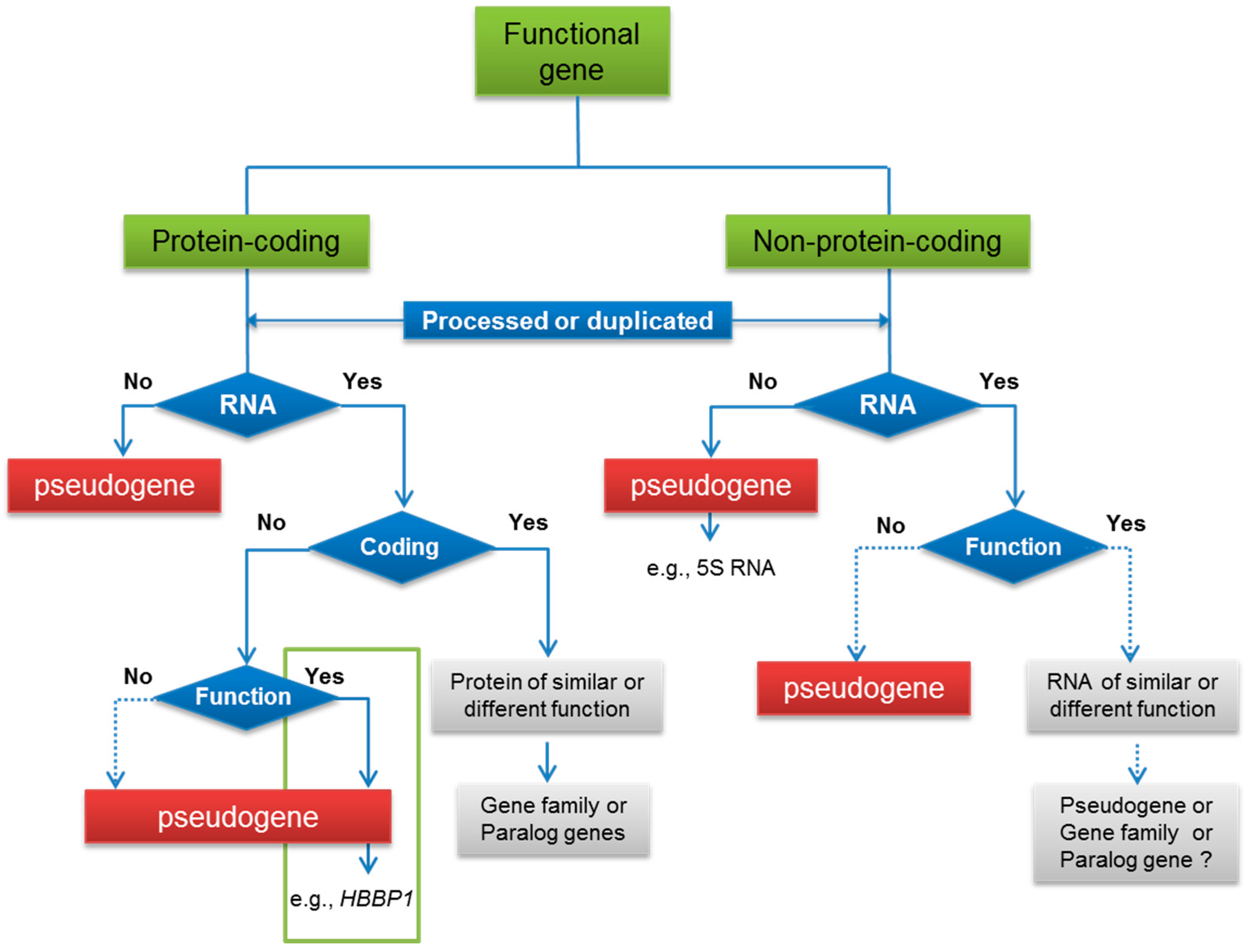
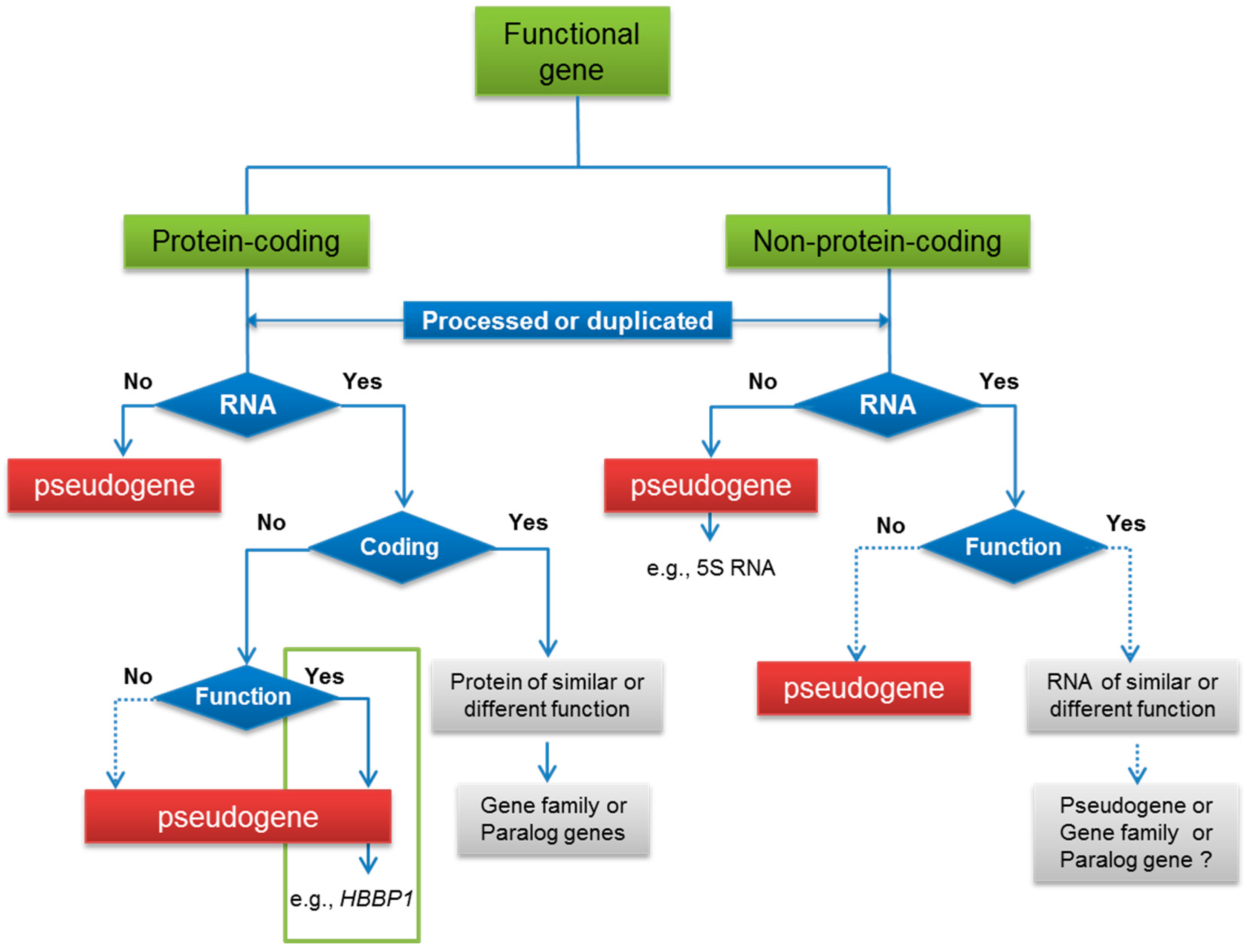
3. Functional Pseudogenes
4. Pseudogenes for Evolutionary Study
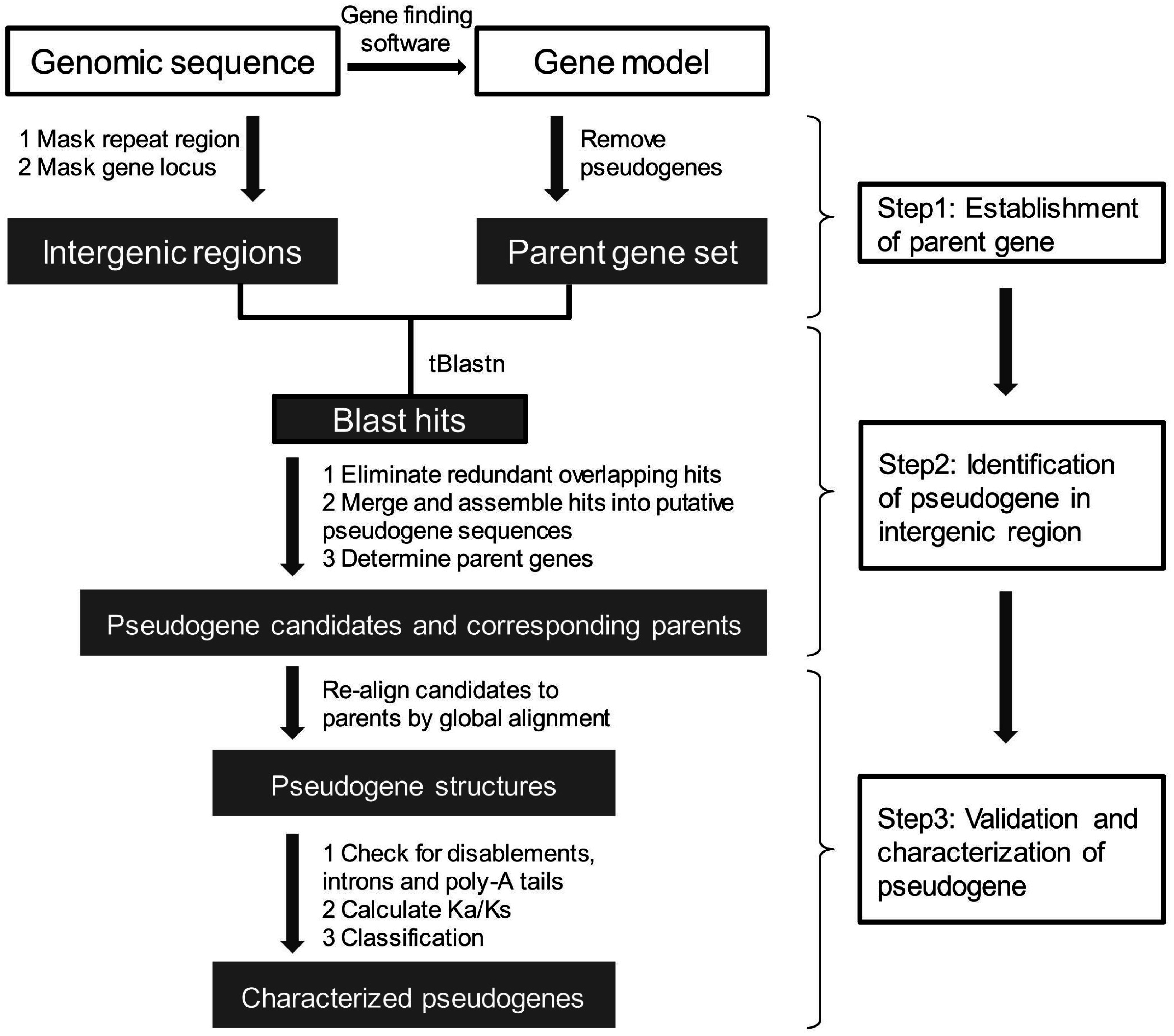
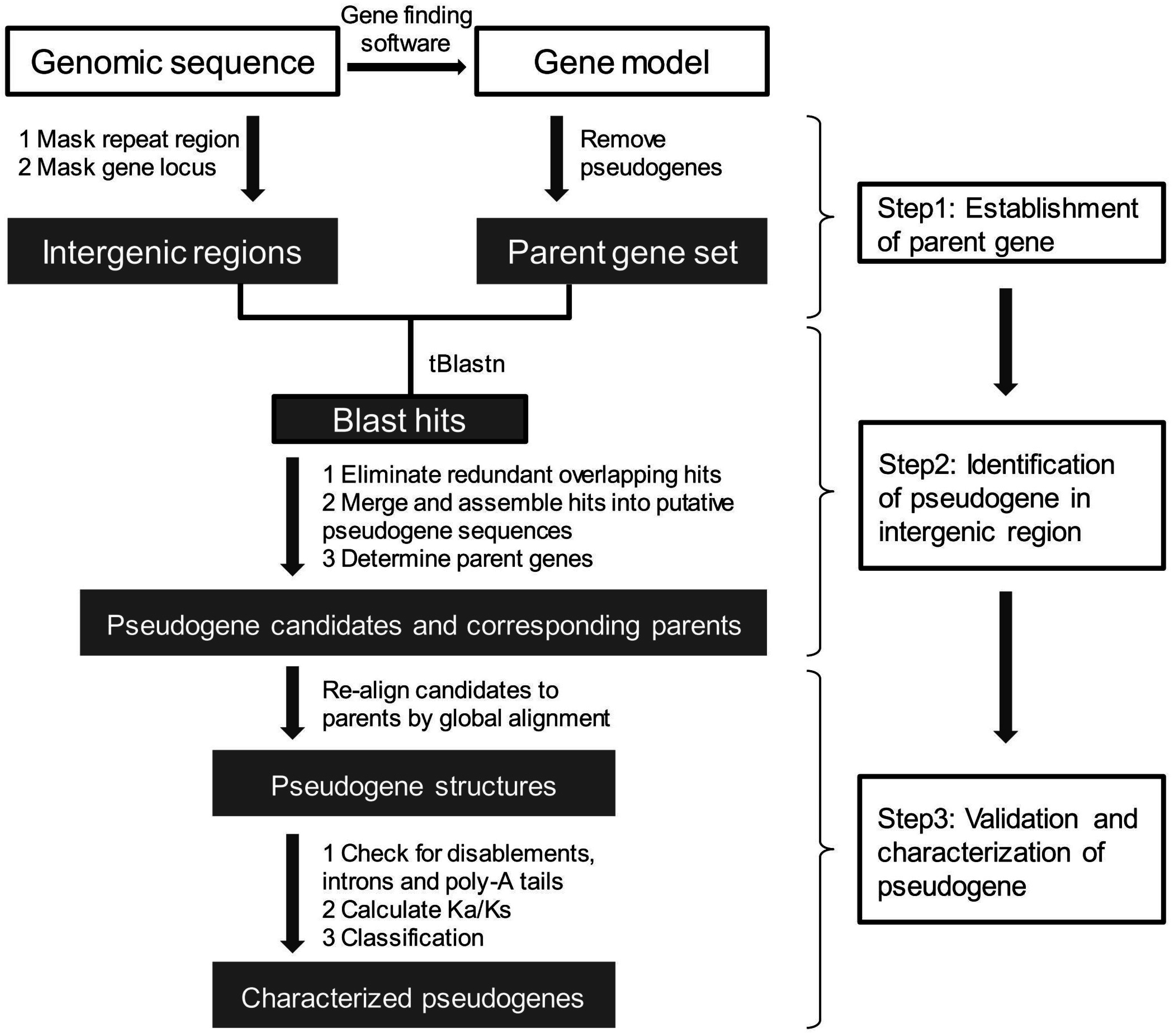
5. Pseudogene Prediction
5.1. Establishment of a Set of Parent Genes
5.2. Pseudogene Identification from Intergenic Regions
5.2.1. Identification of Candidate Pseudogenes and Their Parent Genes
5.2.2. Pseudogenes Validation and Classification
5.3. Comparisons of Bioinformatics Tools for Pseudogene Prediction in Plants
6. Pseudogenes in Plants
7. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PPG | processed pseudogene |
| DPG | duplicated pseudogene |
| UPG | unitary pseudogene |
| WGD | whole genome duplication |
| TE | transposable element |
| ORF | open reading frame |
| RT | reverse transcriptase |
| mRNA | messenger RNA |
| miRNA | microRNA |
| siRNA | short interfering RNA |
| piRNA | piwi-interacting RNA |
| RP | ribosomal protein |
| MYA | million years ago |
| NOS | nitric oxide synthase |
| Ka/Ks | non-synonymous/synonymous substitutions rate |
| CpG | C-phosphate-G |
References
- Jacq, C.; Miller, J.R.; Brownlee, G.G. A pseudogene structure in 5S DNA of Xenopus laevis. Cell 1977, 12, 109–120. [Google Scholar] [CrossRef]
- Torrents, D.; Suyama, M.; Zdobnov, E.; Bork, P. A genome-wide survey of human pseudogenes. Genome Res. 2003, 13, 2559–2567. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Lin, L.; Chen, B.; Dai, J. L1 elements, processed pseudogenes and retrogenes in mammalian genomes. IUBMB Life 2006, 58, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Tutar, Y. Pseudogenes. Comp. Funct. Genom. 2012, 2012, 424526. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Hegyi, H.; Balasubramanian, S.; Luscombe, N.M.; Bertone, P.; Echols, N.; Johnson, T.; Gerstein, M. Molecular fossils in the human genome: Identification and analysis of the pseudogenes in chromosomes 21 and 22. Genome Res. 2002, 12, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, R.; Gerstein, M. Genomics: Protein fossils live on as RNA. Nature 2008, 453, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Lehti-Shiu, M.D.; Thibaud-Nissen, F.; Prakash, T.; Buell, C.R.; Shiu, S.H. Evolutionary and expression signatures of pseudogenes in Arabidopsis and rice. Plant Physiol. 2009, 151, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Carriero, N.; Zheng, D.; Karro, J.; Harrison, P.M.; Gerstein, M. PseudoPipe: An automated pseudogene identification pipeline. Bioinformatics 2006, 22, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Van Baren, M.J.; Brent, M.R. Iterative gene prediction and pseudogene removal improves genome annotation. Genome Res. 2006, 16, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Harrison, P.M.; Liu, Y.; Gerstein, M. Millions of years of evolution preserved: A comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003, 13, 2541–2558. [Google Scholar] [CrossRef] [PubMed]
- Hardison, R.; Miller, W. Use of long sequence alignments to study the evolution and regulation of mammalian globin gene clusters. Mol. Biol. Evol. 1993, 10, 73–102. [Google Scholar] [PubMed]
- Zhang, Z.D.; Frankish, A.; Hunt, T.; Harrow, J.; Gerstein, M. Identification and analysis of unitary pseudogenes: Historic and contemporary gene losses in humans and other primates. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Vanin, E.F. Processed pseudogenes: Characteristics and evolution. Annu. Rev. Genet. 1985, 19, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Pavlicek, A.; Paces, J.; Zika, R.; Hejnar, J. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: Implications for retrotransposition and pseudogene detection. Gene 2002, 300, 189–194. [Google Scholar] [CrossRef]
- Sanmiguel, P.; Bennetzen, J.L. Evidence that a recent increase in maize genome size was caused by the massive amplification of intergene retrotransposons. Ann. Bot. 1998, 82, 37–44. [Google Scholar] [CrossRef]
- Li, W.; Zhang, P.; Fellers, J.P.; Friebe, B.; Gill, B.S. Sequence composition, organization, and evolution of the core Triticeae genome. Plant J. 2004, 40, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Maestre, J.; Heidmann, T. Human LINE retrotransposons generate processed pseudogenes. Nat. Genet. 2000, 24, 363–367. [Google Scholar] [PubMed]
- Schacherer, J.; Tourrette, Y.; Souciet, J.L.; Potier, S.; De Montigny, J. Recovery of a function involving gene duplication by retroposition in Saccharomyces cerevisiae. Genome Res. 2004, 14, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Hattori, M.; Yada, T.; Gojobori, T.; Sakaki, Y.; Okada, N. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003, 4. [Google Scholar] [CrossRef] [PubMed]
- Thibaud-Nissen, F.; Ouyang, S.; Buell, C.R. Identification and characterization of pseudogenes in the rice gene complement. BMC Genom. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Baertsch, R.; Diekans, M.; Kent, W.J.; Haussler, D.; Brosius, J. Retrocopy contributions to the evolution of the human genome. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Gerstein, M.B. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biol. 2006, 7. [Google Scholar] [CrossRef] [PubMed]
- Kuryshev, V.Y.; Vorobyov, E.; Zink, D.; Schmitz, J.; Rozhdestvensky, T.S.; Munstermann, E.; Ernst, U.; Wellenreuther, R.; Moosmayer, P.; Bechtel, S.; et al. An anthropoid-specific segmental duplication on human chromosome 1q22. Genomics 2006, 88, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Ho-Huu, J.; Ronfort, J.; De Mita, S.; Bataillon, T.; Hochu, I.; Weber, A.; Chantret, N. Contrasted patterns of selective pressure in three recent paralogous gene pairs in the Medicago genus (L.). BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Slightom, J.L. Isolation and nucleotide sequence analysis of the β-type globin pseudogene from human, gorilla and chimpanzee. J. Mol. Biol. 1984, 180, 767–784. [Google Scholar] [CrossRef]
- Michelmore, R.W.; Meyers, B.C. Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res. 1998, 8, 1113–1130. [Google Scholar] [PubMed]
- Lozano, R.; Ponce, O.; Ramirez, M.; Mostajo, N.; Orjeda, G. Genome-wide identification and mapping of NBS-encoding resistance genes in Solanum tuberosum group phureja. PLoS ONE 2012, 7, e34775. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, Y.; Hu, Q.; Chen, J.; Li, K.; Lu, C.; Liu, H.; Wang, W.; Kuang, H. Dynamic nucleotide-binding site and leucine-rich repeat-encoding genes in the grass family. Plant Physiol. 2012, 159, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Langham, R.J.; Walsh, J.; Dunn, M.; Ko, C.; Goff, S.A.; Freeling, M. Genomic duplication, fractionation and the origin of regulatory novelty. Genetics 2004, 166, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Hufton, A.L.; Panopoulou, G. Polyploidy and Genome Restructuring: A variety of outcomes. Curr. Opin. Genet. Dev. 2009, 19, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Force, A. The probability of duplicate gene preservation by subfunctionalization. Genetics 2000, 154, 459–473. [Google Scholar] [PubMed]
- Zhang, J.; Webb, D.M. Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc. Natl. Acad. Sci. USA 2003, 100, 8337–8341. [Google Scholar] [CrossRef] [PubMed]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [PubMed]
- Otto, S.P.; Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 2000, 34, 401–437. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Vision, T.J.; Brown, D.G.; Tanksley, S.D. The origins of genomic duplications in Arabidopsis. Science 2000, 290, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Tan, J.; Lee, S.; Kong, L.; Heger, A.; Ponting, C.P. Evidence for conserved post-transcriptional roles of unitary pseudogenes and for frequent bifunctionality of mRNAs. Genome Biol. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Rouchka, E.C.; Cha, I.E. Current trends in pseudogene detection and characterization. Curr. Bioinform. 2009, 4, 112–119. [Google Scholar] [CrossRef]
- Lafontaine, I.; Dujon, B. Origin and fate of pseudogenes in Hemiascomycetes: A comparative analysis. BMC Genom. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.O.; Andersson, S.G. Pseudogenes, junk DNA, and the dynamics of Rickettsia genomes. Mol. Biol. Evol. 2001, 18, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.Z.; Zheng, L.L.; Qu, L.H.; Ayala, F.J.; Lun, Z.R. Pseudogenes are not pseudo any more. RNA Biol. 2012, 9, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulou, E.; Bartsakoulia, M.; Tafrali, C.; Kourakli, A.; Poulas, K.; Stavrou, E.F.; Papachatzopoulou, A.; Georgitsi, M.; Patrinos, G.P. A single nucleotide polymorphism in the HBBP1 gene in the human β-globin locus is associated with a mild β-thalassemia disease phenotype. Hemoglobin 2012, 36, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Svensson, O.; Arvestad, L.; Lagergren, J. Genome-wide survey for biologically functional pseudogenes. PLoS Comput. Biol. 2006, 2, e46. [Google Scholar] [CrossRef] [PubMed]
- Pei, B.; Sisu, C.; Frankish, A.; Howald, C.; Habegger, L.; Mu, X.J.; Harte, R.; Balasubramanian, S.; Tanzer, A.; Diekhans, M.; et al. The GENCODE pseudogene resource. Genome Biol. 2012, 13, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Frankish, A.; Baertsch, R.; Kapranov, P.; Reymond, A.; Choo, S.W.; Lu, Y.; Denoeud, F.; Antonarakis, S.E.; Snyder, M.; et al. Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription, and evolution. Genome Res. 2007, 17, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Korneev, S.A.; Park, J.H.; O’Shea, M. Neuronal expression of neural nitric oxide synthase (nNOS) protein is suppressed by an antisense RNA transcribed from an NOS pseudogene. J. Neurosci. 1999, 19, 7711–7720. [Google Scholar] [PubMed]
- Piehler, A.P.; Hellum, M.; Wenzel, J.J.; Kaminski, E.; Haug, K.B.; Kierulf, P.; Kaminski, W.E. The human ABC transporter pseudogene family: Evidence for transcription and gene-pseudogene interference. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Kandouz, M.; Bier, A.; Carystinos, G.D.; Alaoui-Jamali, M.A.; Batist, G. Connexin43 pseudogene is expressed in tumor cells and inhibits growth. Oncogene 2004, 23, 4763–4770. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Gerstein, M.B. The ambiguous boundary between genes and pseudogenes: The dead rise up or do they? Trends Genet. 2007, 23, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Harrison, P.; Gerstein, M. Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome. Genome Res. 2002, 12, 1466–1482. [Google Scholar] [CrossRef] [PubMed]
- Gerstein, M.; Zheng, D. The real life of pseudogenes. Sci. Am. 2006, 295, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gerstein, M. The human genome has 49 cytochrome C pseudogenes, including a relic of a primordial gene that still functions in mouse. Gene 2003, 312, 61–72. [Google Scholar] [CrossRef]
- Podlaha, O.; Zhang, J. Processed pseudogenes: The “fossilized footprints” of past gene expression. Trends Genet. 2009, 25, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gerstein, M. Large-scale analysis of pseudogenes in the human genome. Curr. Opin. Genet. Dev. 2004, 14, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Kozik, A.; Griego, A.; Kuang, H.; Michelmore, R.W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 2003, 15, 809–834. [Google Scholar] [CrossRef] [PubMed]
- Ameline-Torregrosa, C.; Wang, B.B.; O’Bleness, M.S.; Deshpande, S.; Zhu, H.; Roe, B.; Young, N.D.; Cannon, S.B. Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 2008, 146, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Tao, Y.; Chen, X.; Zou, Y.; Lei, C.; Wang, J.; Li, X.; Zhao, X.; Zhang, M.; Lu, Z.; et al. Identification of a new rice blast resistance gene, Pid3, by genomewide comparison of paired nucleotide-binding site-leucine-rich repeat genes and their pseudogene alleles between the two sequenced rice genomes. Genetics 2009, 182, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cheng, Y.; Ma, W.; Zhao, Y.; Jiang, H.; Zhang, M. Identification and characterization of NBS-encoding disease resistance genes in Lotus japonicus. Plant Syst. Evol. 2010, 289, 101–110. [Google Scholar] [CrossRef]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.S.; Brent, M.R. Using multiple alignments to improve gene prediction. J. Comput. Biol. 2006, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Korf, I.; Flicek, P.; Duan, D.; Brent, M.R. Integrating genomic homology into gene structure prediction. Bioinformatics 2001, 17, S140–S148. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Guo, L.; Fu, Y.; Borsuk, L.A.; Wen, T.J.; Skibbe, D.S.; Cui, X.; Scheffler, B.E.; Cao, J.; Emrich, S.J.; et al. Evaluation of five ab initio gene prediction programs for the discovery of maize genes. Plant Mol. Biol. 2005, 57, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.R.; Liu, B.; Delcher, A.L.; Pop, M.; Salzberg, S.L. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res. 2012, 40, e9. [Google Scholar] [CrossRef] [PubMed]
- Mewes, H.W.; Amid, C.; Arnold, R.; Frishman, D.; Güldener, U.; Mannhaupt, G.; Münsterkötter, M.; Pagel, P.; Strack, N.; Stümpflen, V.; et al. MIPS: Analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 2004, 32, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Leroy, P.; Guilhot, N.; Sakai, H.; Bernard, A.; Choulet, F.; Theil, S.; Reboux, S.; Amano, N.; Flutre, T.; Pelegrin, C.; et al. TriAnnot: A versatile and high performance pipeline for the automated annotation of plant genomes. Front. Plant Sci. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Zhang, Z.; Harrison, P.M.; Karro, J.; Carriero, N.; Gerstein, M. Integrated pseudogene annotation for human chromosome 22: Evidence for transcription. J. Mol. Biol. 2005, 349, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Yao, A.; Charlab, R.; Li, P. Systematic identification of pseudogenes through whole genome expression evidence profiling. Nucleic Acids Res. 2006, 34, 4477–4485. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Ouyang, S.; Liu, J.; Suh, B.; Cheung, F.; Sultana, R.; Lee, D.; Quackenbush, J.; Buell, C.R. The TIGR rice genome annotation resource: Annotating the rice genome and creating resources for plant biologists. Nucleic Acids Res. 2003, 31, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Wood, T.; Zhang, Z.; Miller, W. Comparison of DNA sequences with protein sequences. Genomics 1997, 46, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.M.; Echols, N.; Gerstein, M.B. Digging for dead genes: An analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res. 2001, 29, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Koyanagi, K.O.; Itoh, T.; Imanishi, T.; Gojobori, T. Detection of processed pseudogenes based on cDNA mapping to the human genome. Genome Inform. 2003, 14, 452–453. [Google Scholar]
- Ng, P.; Wei, C.L.; Sung, W.K.; Chiu, K.P.; Lipovich, L.; Ang, C.C.; Gupta, S.; Shahab, A.; Ridwan, A.; Wong, C.H.; et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat. Methods 2005, 2, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Harrington, E.; Bork, P.; Torrents, D. Identification and analysis of genes and pseudogenes within duplicated regions in the human and mouse genomes. PLoS Comput. Biol. 2006, 2, e76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Carriero, N.; Gerstein, M. Comparative analysis of processed pseudogenes in the mouse and human genomes. Trends Genet. 2004, 20, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.Y.; Khurana, E.; Fang, G.; Cayting, P.; Carriero, N.; Cheung, K.H.; Gerstein, M.B. Pseudofam: The pseudogene families database. Nucleic. Acids Res. 2009, 37, D738–D743. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Mayer, K.F.; Gundlach, H.; Martis, M.; Steuernagel, B.; Scholz, U.; Simkova, H.; Kubalakova, M.; Choulet, F.; Taudien, S.; et al. Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell 2011, 23, 1706–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choulet, F.; Alberti, A.; Theil, S.; Glover, N.; Barbe, V.; Daron, J.; Pingault, L.; Sourdille, P.; Couloux, A.; Paux, E.; et al. Structural and functional partitioning of bread wheat chromosome 3B. Science 2014, 345. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.F.X.; Rogers, J.; Dolezel, J.; Pozniak, C.; Eversole, K.; Feuillet, C.; Gill, B.; Friebe, B.; Lukaszewski, A.J.; Sourdille, P.; et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Processed Pseudogenes | Duplicated Pseudogenes | |
|---|---|---|
| 1 | Arise from mRNA that was reverse-transcribed and re-integrated into the genome | Arise from gene duplication |
| 2 | Lack of non-coding intervening sequences: introns and promoters | Possess promoters, exon-intron structure and other upstream regulatory sequences |
| 3 | Possess a poly-A tail at 3′ end | No 3′ poly-A tail |
| 4 | Possess flanking direct repeats associated with TE insertion sites | No flanking direct repeats |
| 5 | Mostly present at different loci from its parent genes | Some are present as a cluster with their parent gene as a consequence of tandem segmental duplication |
| 6 | Have 3′ or 5′ truncations | Have 3′ truncations |
| 7 | Generally shorter | Comparatively longer |
| Pipeline/Method | Input Data | Brief Description | Availability | * Ref. |
|---|---|---|---|---|
| Harrison’s Approach | Protein and genome sequence, annotation information | Using protein sequences to find pseudogenes in intergenic regions by FASTA alignment; refinement of alignments for validation and classification | Method, not a pipeline tool | [78] |
| Sakai’s Approach | cDNA and genome sequence | Using cDNA to search and extract corresponding regions from genome sequence by BLASTn; realignment of cDNAs to extract sequence; using est2genome for classification | Method, not a pipeline tool | [79] |
| PPFINDER (Processed Pseudogene Finder) | Gene model and cDNA database | Using cDNA as evidence to determine parent genes in gene models; using parent genes to detect locus missing introns by BLASTN search; removing false candidates | http://www.mybiosoftware.com/ | [10] |
| PseudoFinder | Functional genes and genome sequence | Finding homologues of functional genes in a genome; classification into either pseudo or functional categories using Support Vector Machines (SVMs) based on a combination of features by BLASTz analysis | Not available online | [48] |
| RetroFinder | GenBank mRNA and genome sequence | Alignment of mRNAs from GenBank to genome sequence by BLASTz; detection of biological features; heuristic weighting for known PPGs | Not available online | [48] |
| GIS-PET (Gene identification signature-paired end tag) method | mRNA and genome sequence | Using 5′ and 3′ paired-end-tag (PET) of mRNAs to select candidates based on homology; using the shortest candidate to search the genome by BLAT | Method, not a pipeline tool | [80] |
| PseudoPipe | Genome sequence (repeat marked), parent proteins and their exon coordinates | Using protein sequence to find pseudogenes in repeat-masked intergenic regions by tBLASTn; realignment of candidates to corresponding parent(s) by FASTA to validate and classify pseudogenes | http://www.pseudogene.org/pseudopipe/ | [8] |
| Shiu’s pipeline | Parent proteins and genome sequence (repeat-masked and intergenic) | Using protein sequence to find pseudogenes in repeat-masked intergenic regions by tBLASTn; realignment of candidates to corresponding parent(s) by FASTA to validate pseudogenes. Similar to PseudoPipe | http://shiulab.plantbiology.msu.edu | [7] |
| PSF (Pseudogene Finder) | Same as Shiu’s pipeline | Using protein sequence to find pseudogenes in repeat-masked intergenic regions directly by Pro-map to detect disruption events and classify pseudogenes | http://molquest.com/ | [9] |
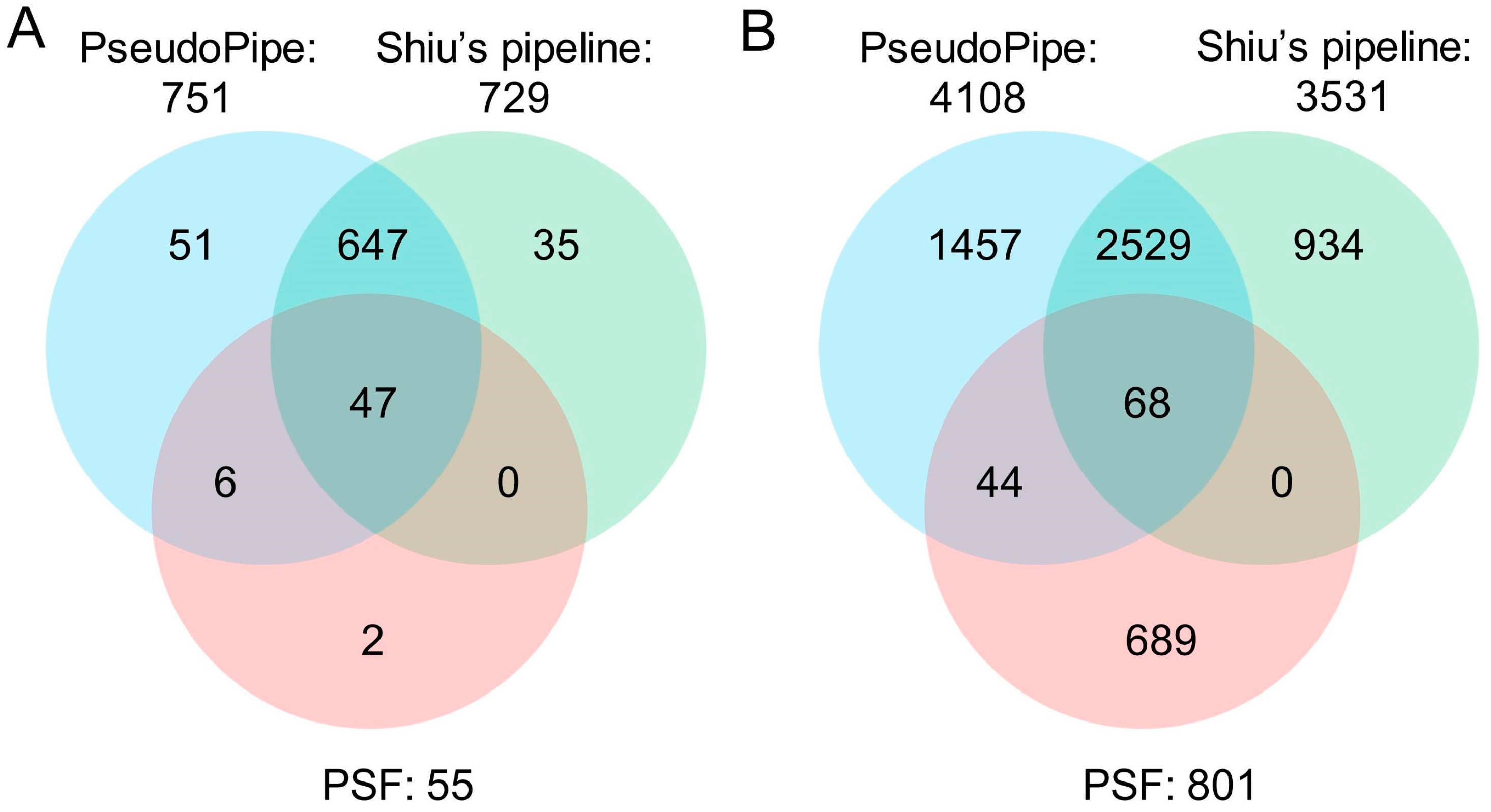
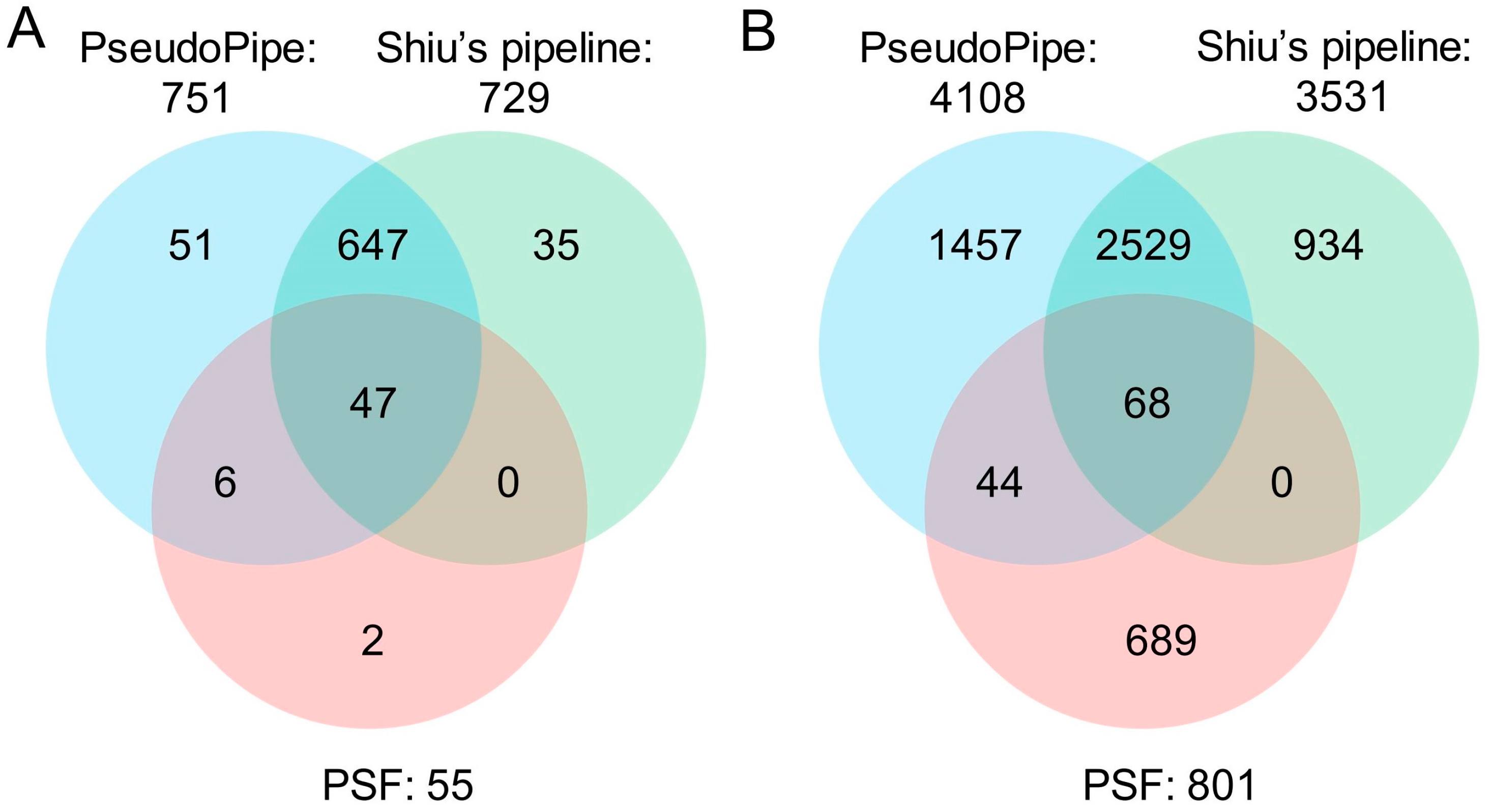
| Tool | No. of Total Pseudogenes Identified | No. of Parents Associated | No. of Known Pseudogenes Identified | Known Pseudogenes Identified (%) |
|---|---|---|---|---|
| PseudoPipe | 4108 | 2550 | 751 | 81.3 |
| Shiu’s pipeline | 3531 | 2317 | 729 | 78.9 |
| PSF | 801 | 604 | 55 | 6.0 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Sekhwal, M.K.; Li, P.; Ragupathy, R.; Cloutier, S.; Wang, X.; You, F.M. Pseudogenes and Their Genome-Wide Prediction in Plants. Int. J. Mol. Sci. 2016, 17, 1991. https://doi.org/10.3390/ijms17121991
Xiao J, Sekhwal MK, Li P, Ragupathy R, Cloutier S, Wang X, You FM. Pseudogenes and Their Genome-Wide Prediction in Plants. International Journal of Molecular Sciences. 2016; 17(12):1991. https://doi.org/10.3390/ijms17121991
Chicago/Turabian StyleXiao, Jin, Manoj Kumar Sekhwal, Pingchuan Li, Raja Ragupathy, Sylvie Cloutier, Xiue Wang, and Frank M. You. 2016. "Pseudogenes and Their Genome-Wide Prediction in Plants" International Journal of Molecular Sciences 17, no. 12: 1991. https://doi.org/10.3390/ijms17121991