
Effect of Post-Translational Amidation on Islet Amyloid Polypeptide Conformational Ensemble: Implications for Its Aggregation Early Steps


Abstract
:
1. Introduction
2. Results
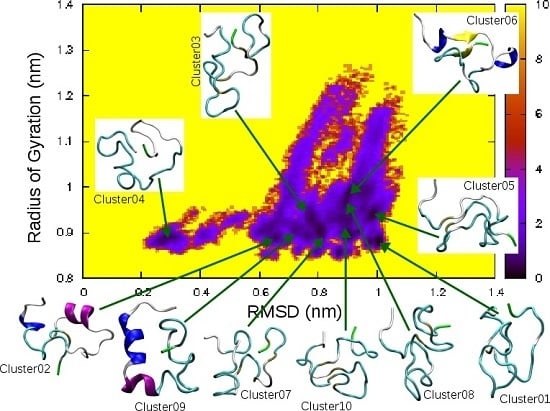
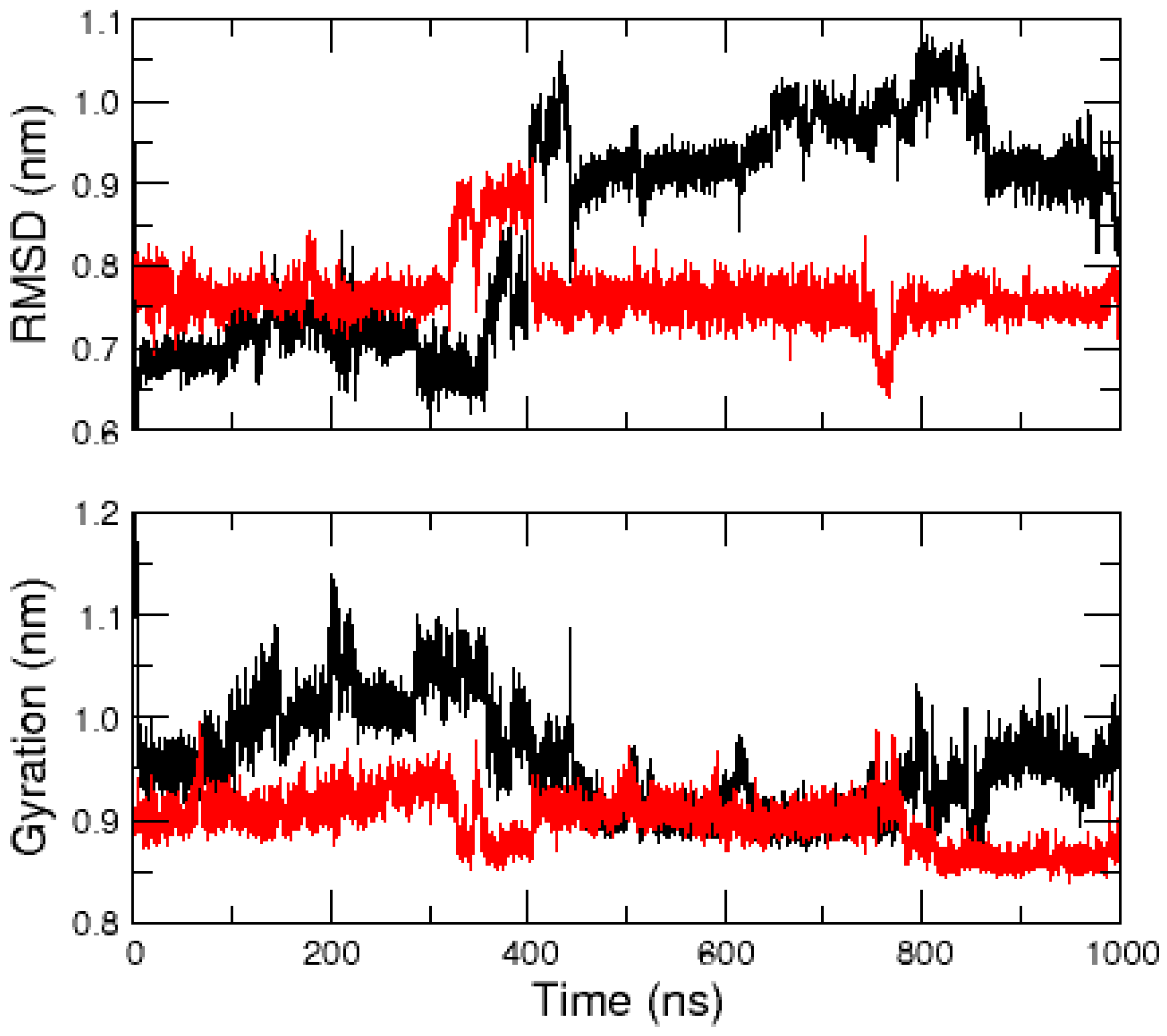
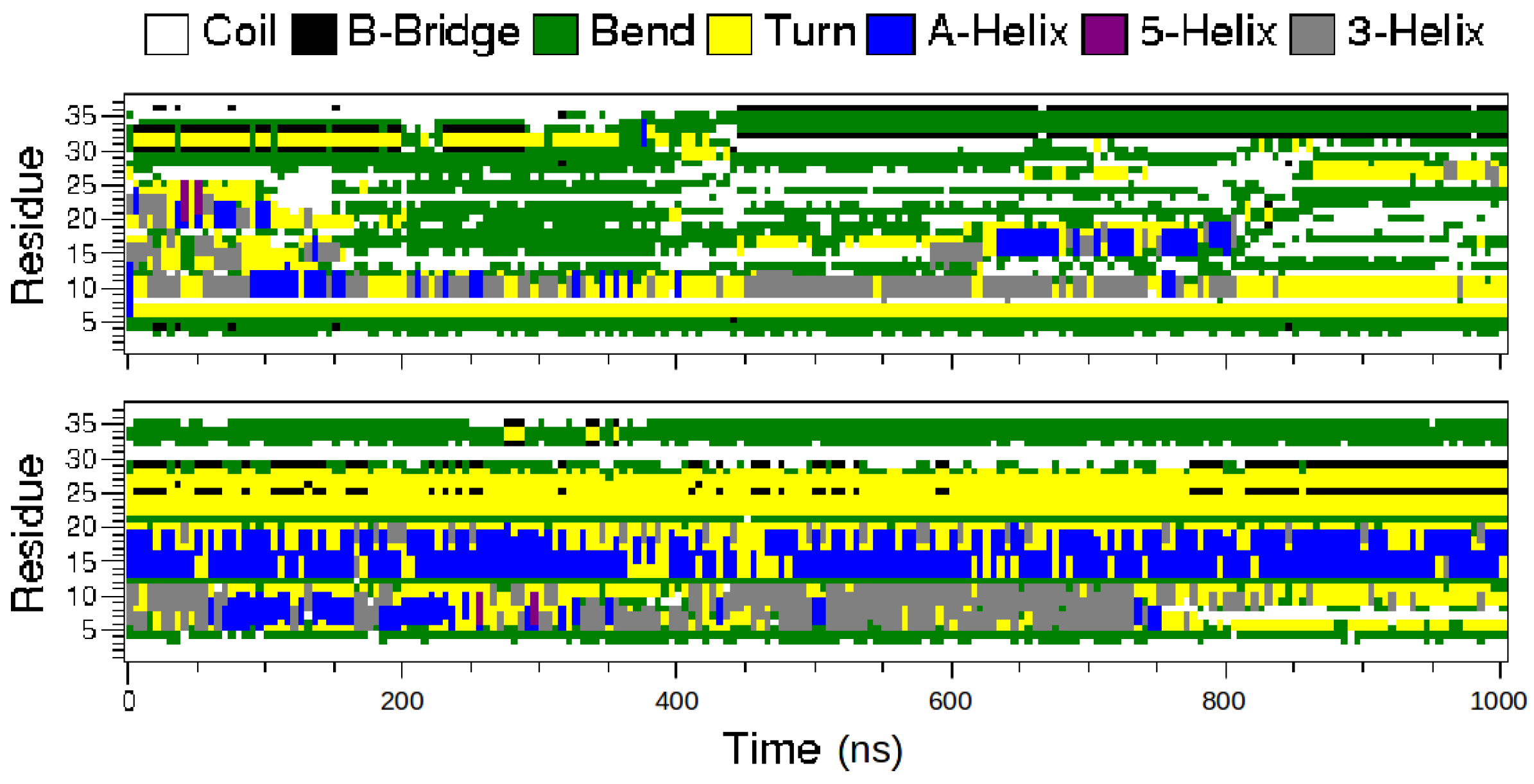
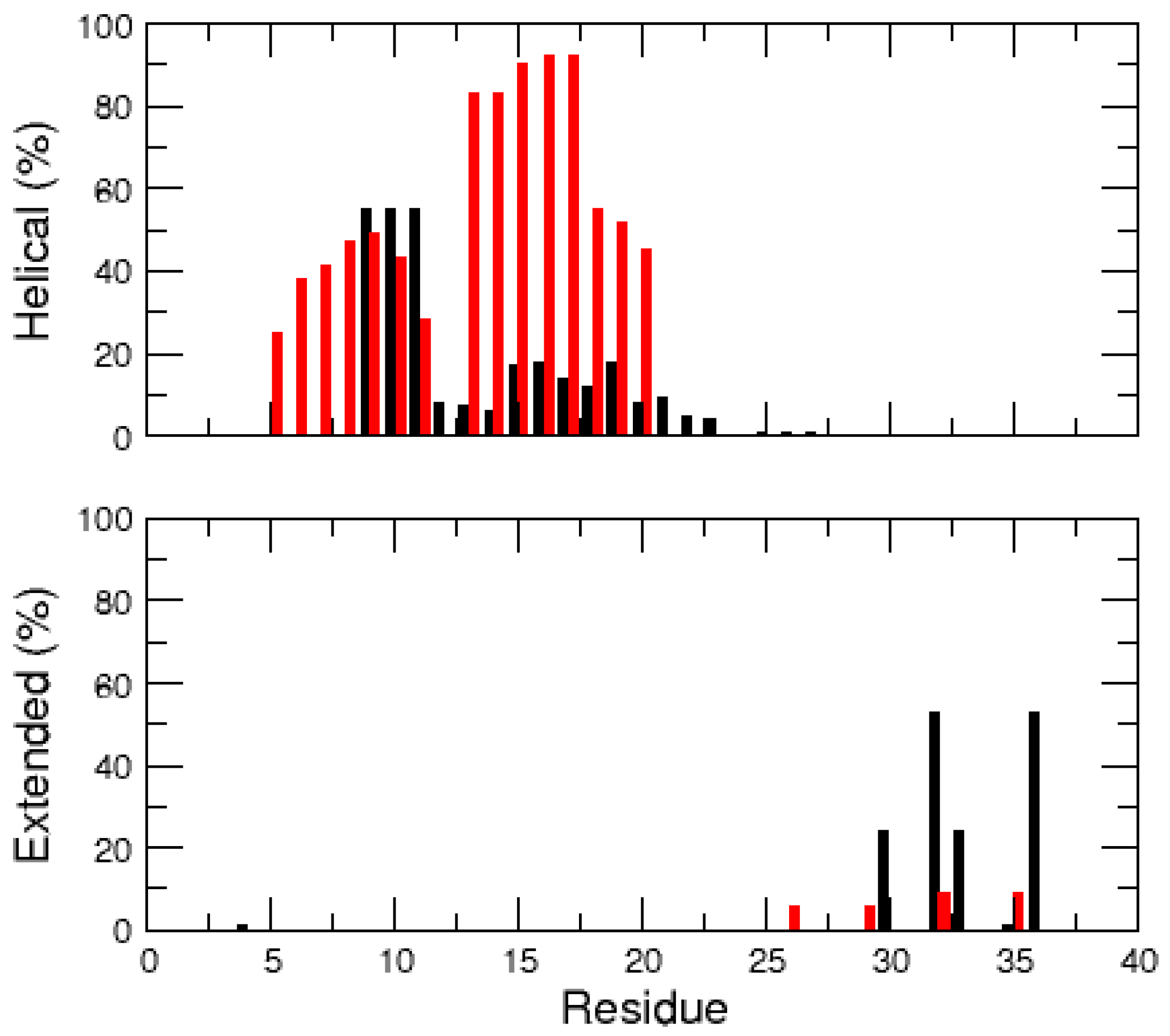
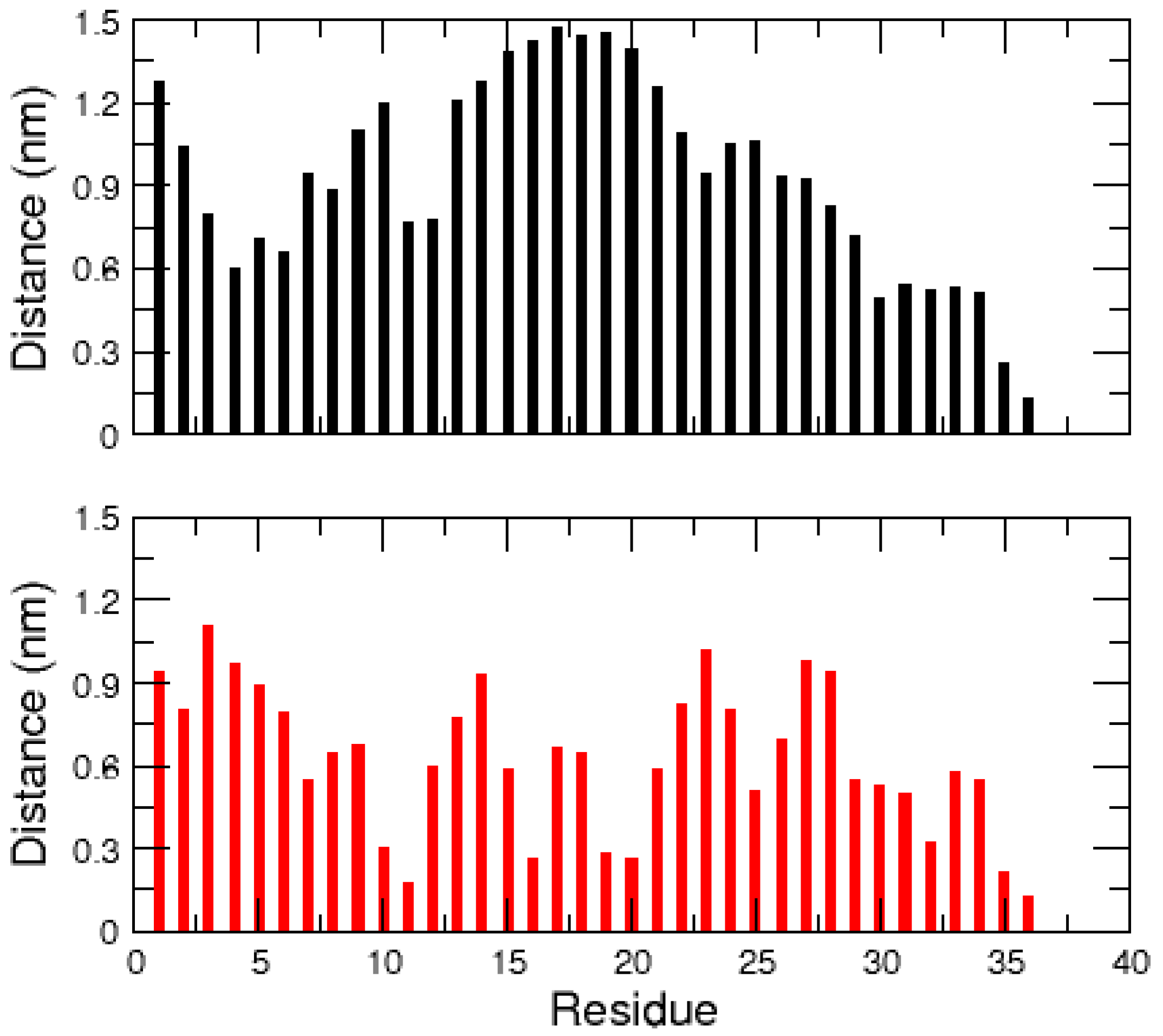
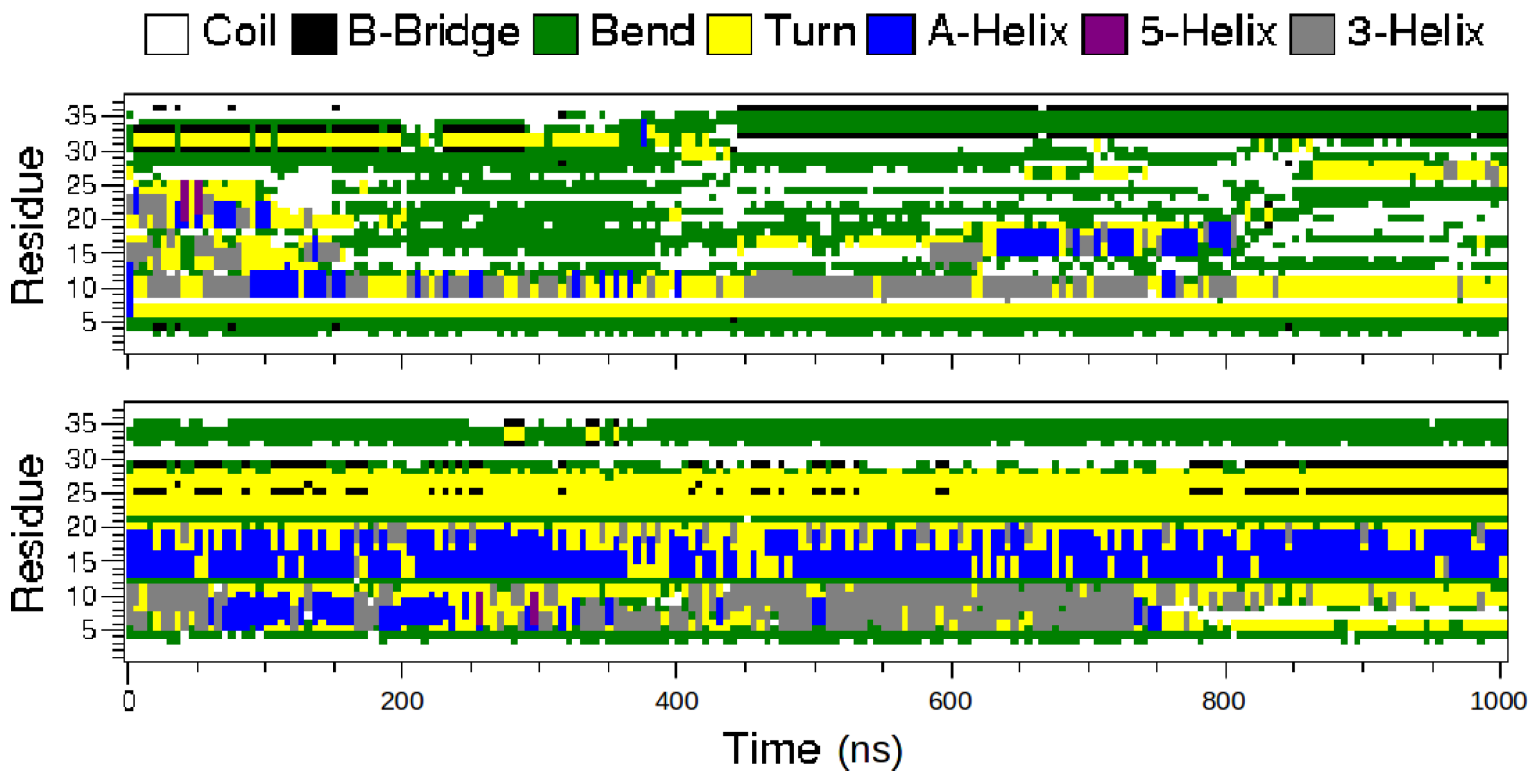
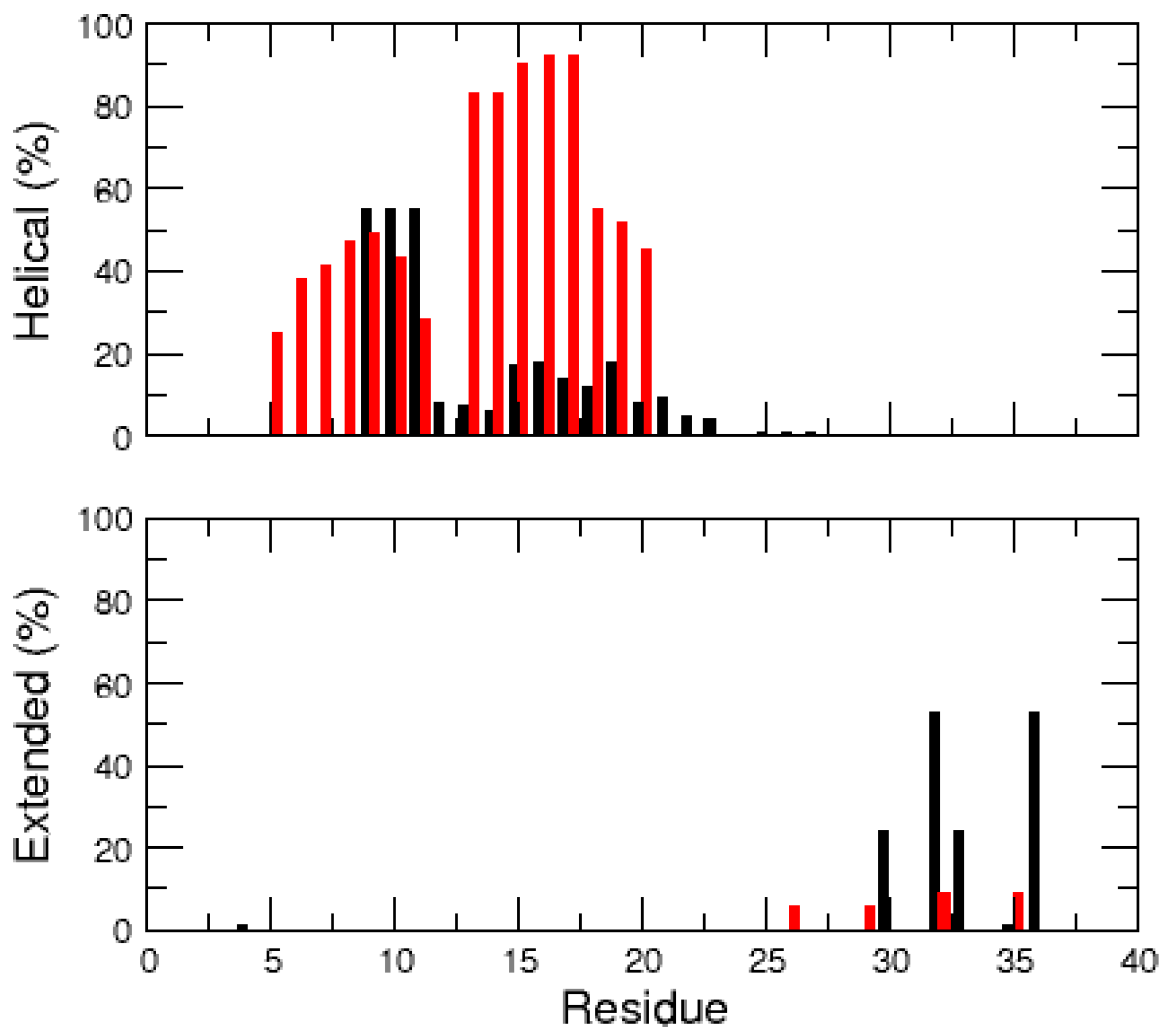
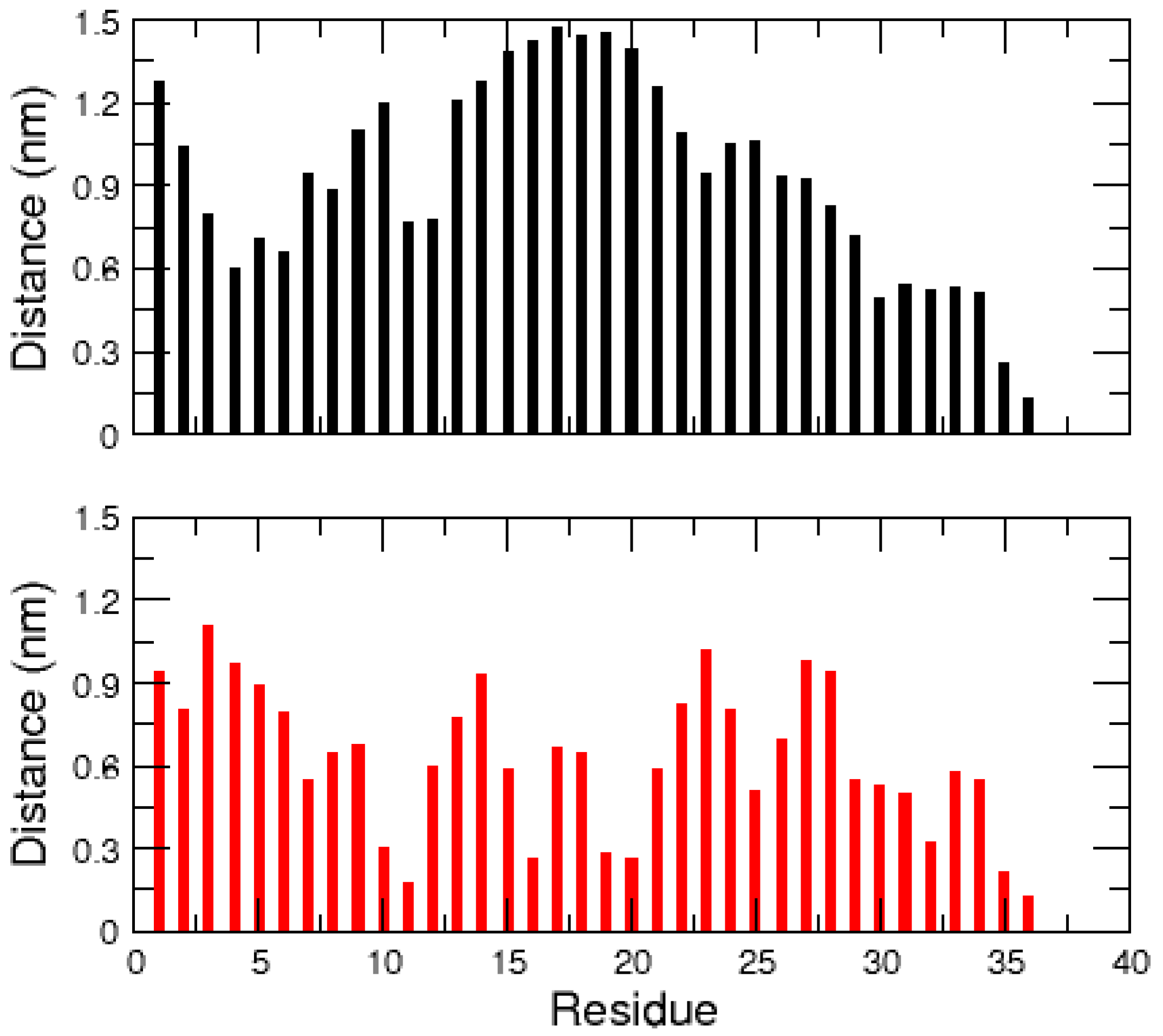
2.1. MD Simulations of the Amidated and Non-Amidated hIAPP
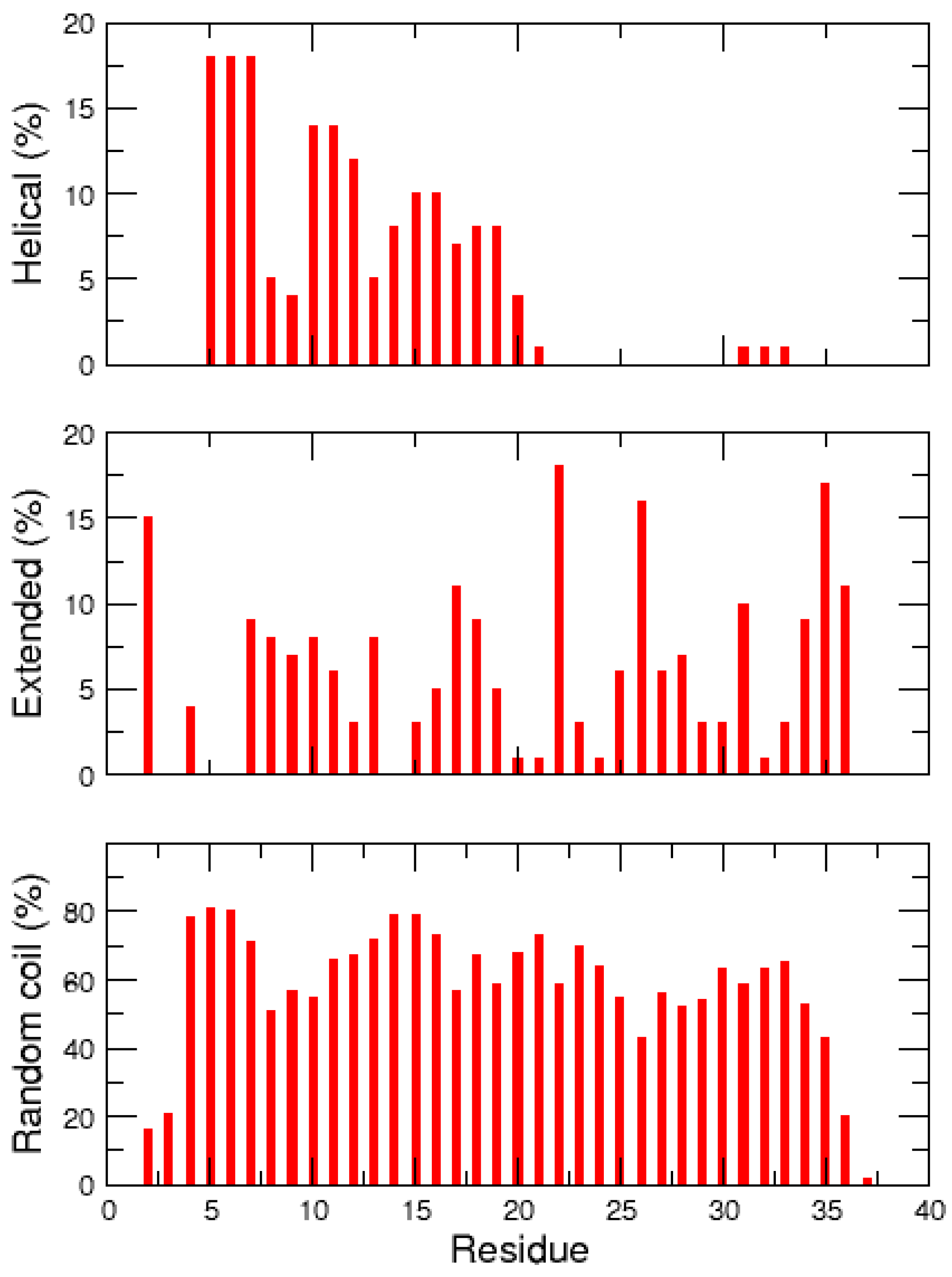
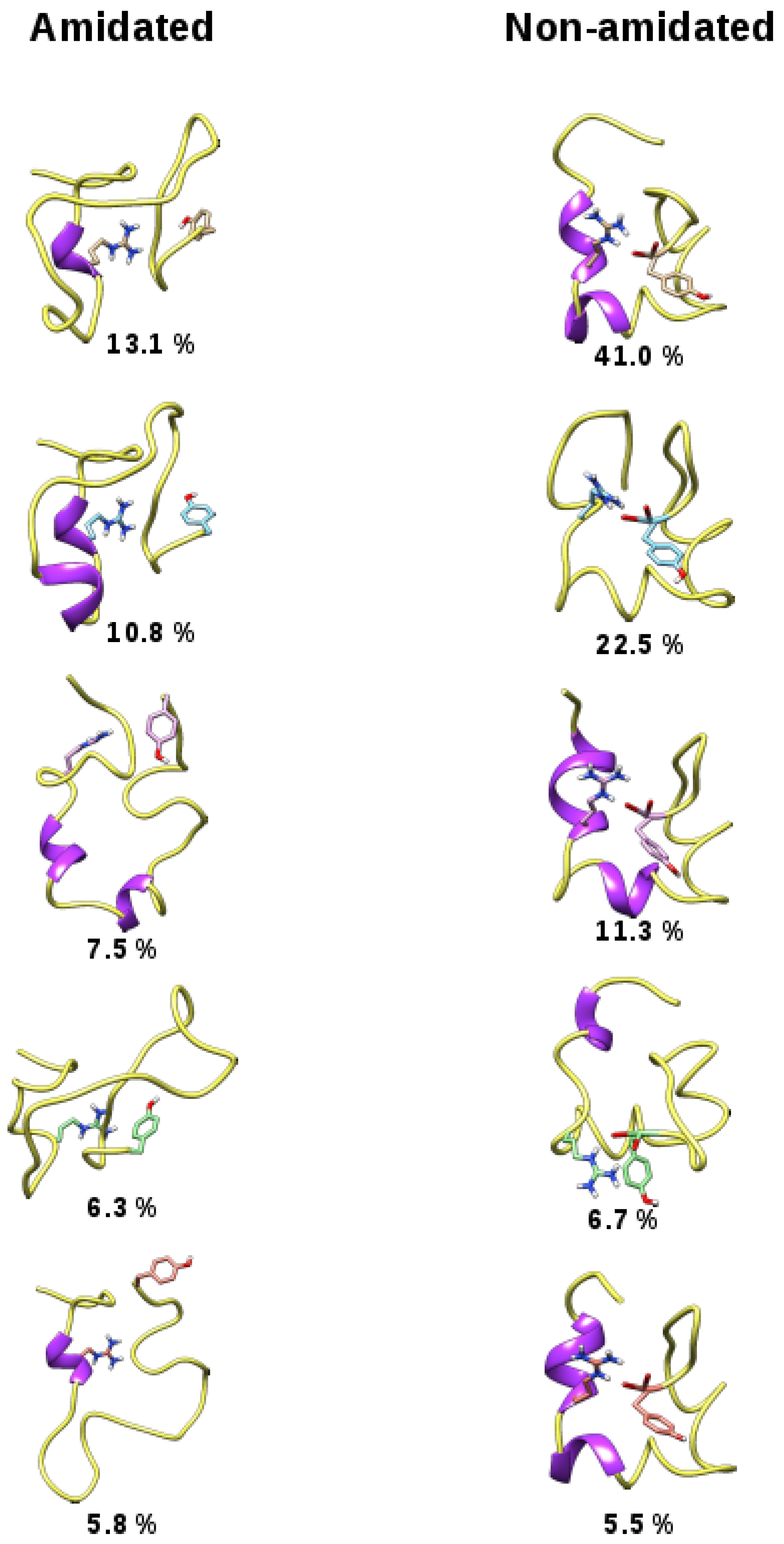
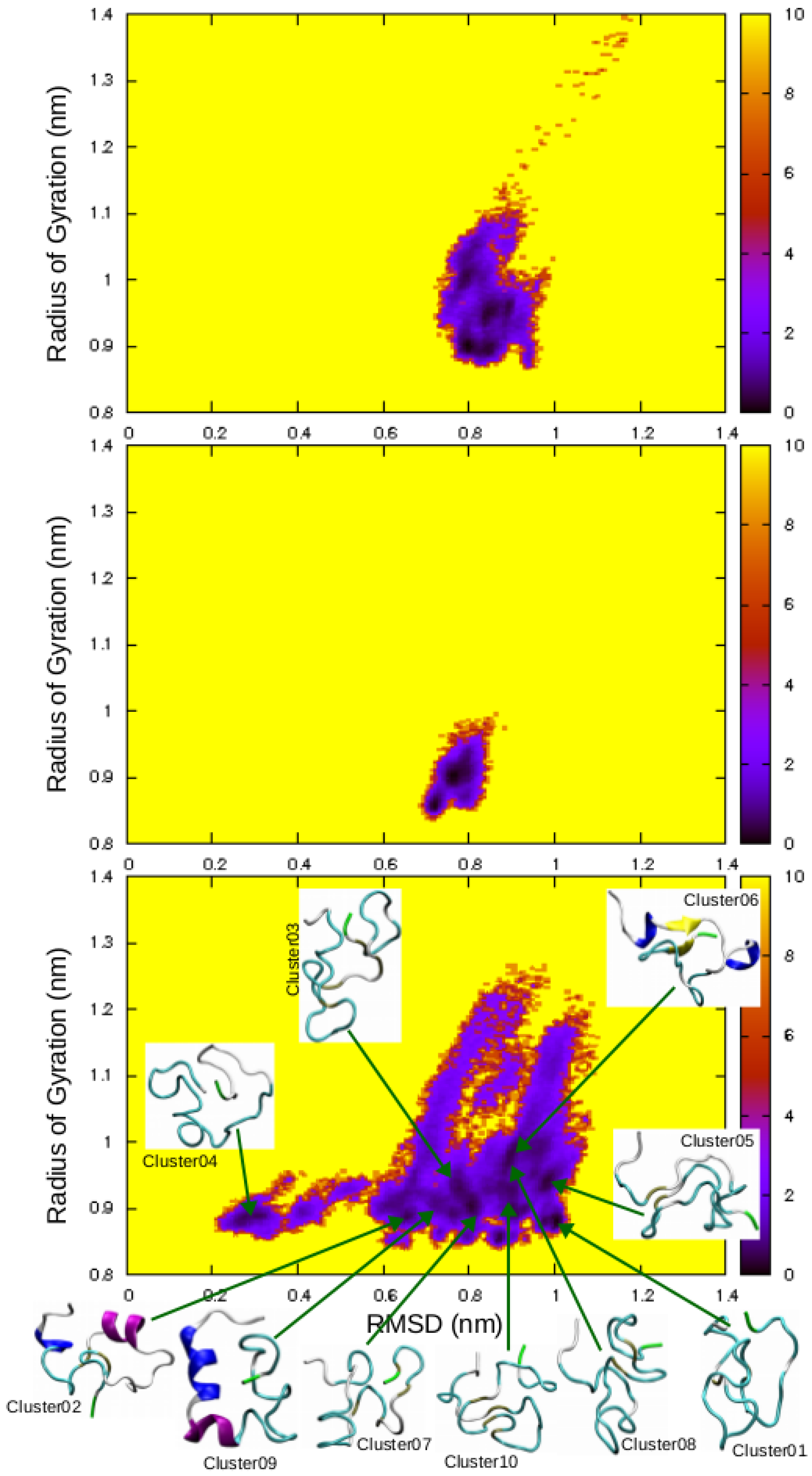
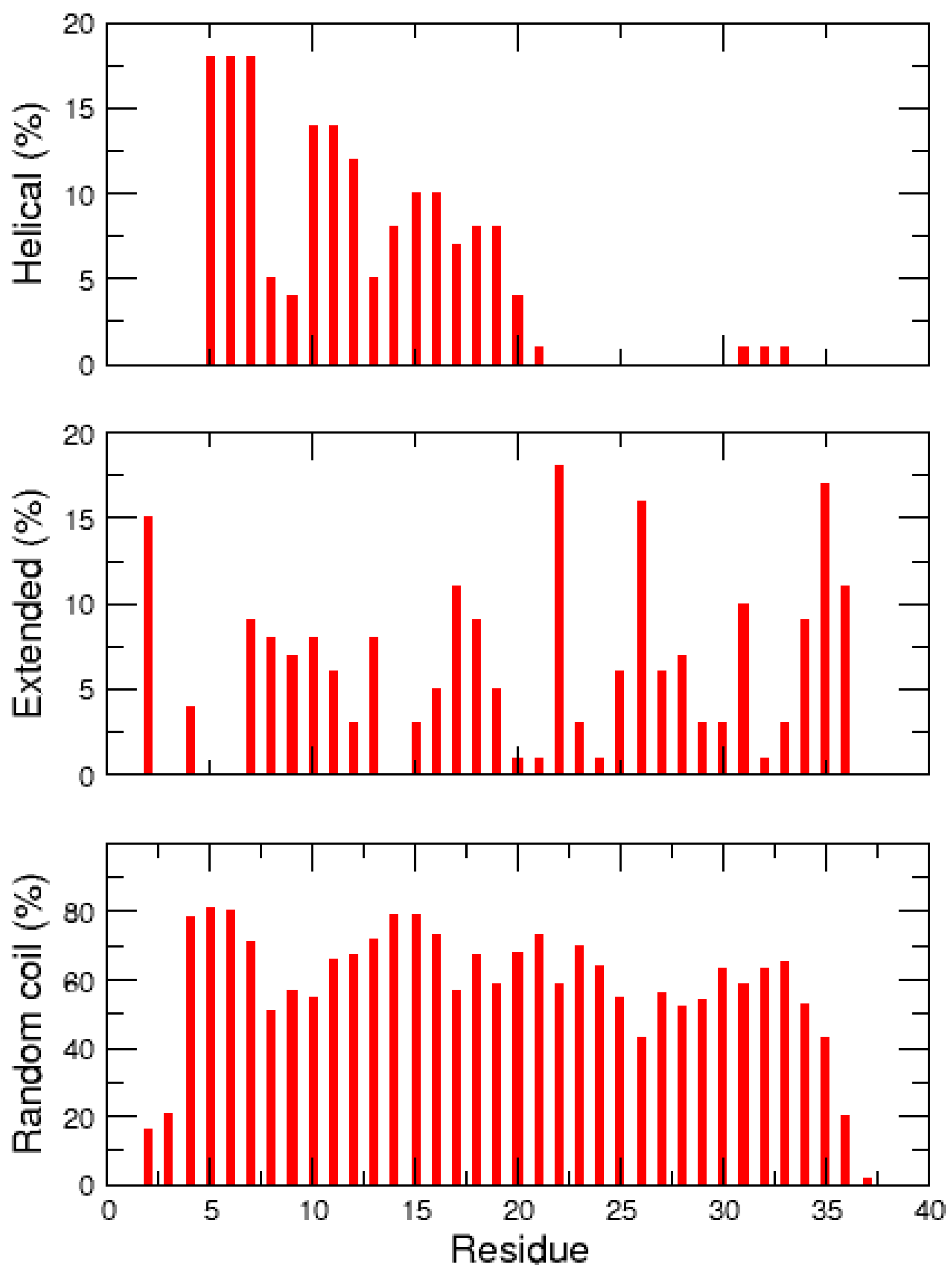
2.2. REMD Simulations of the Non-Amidated hIAPP
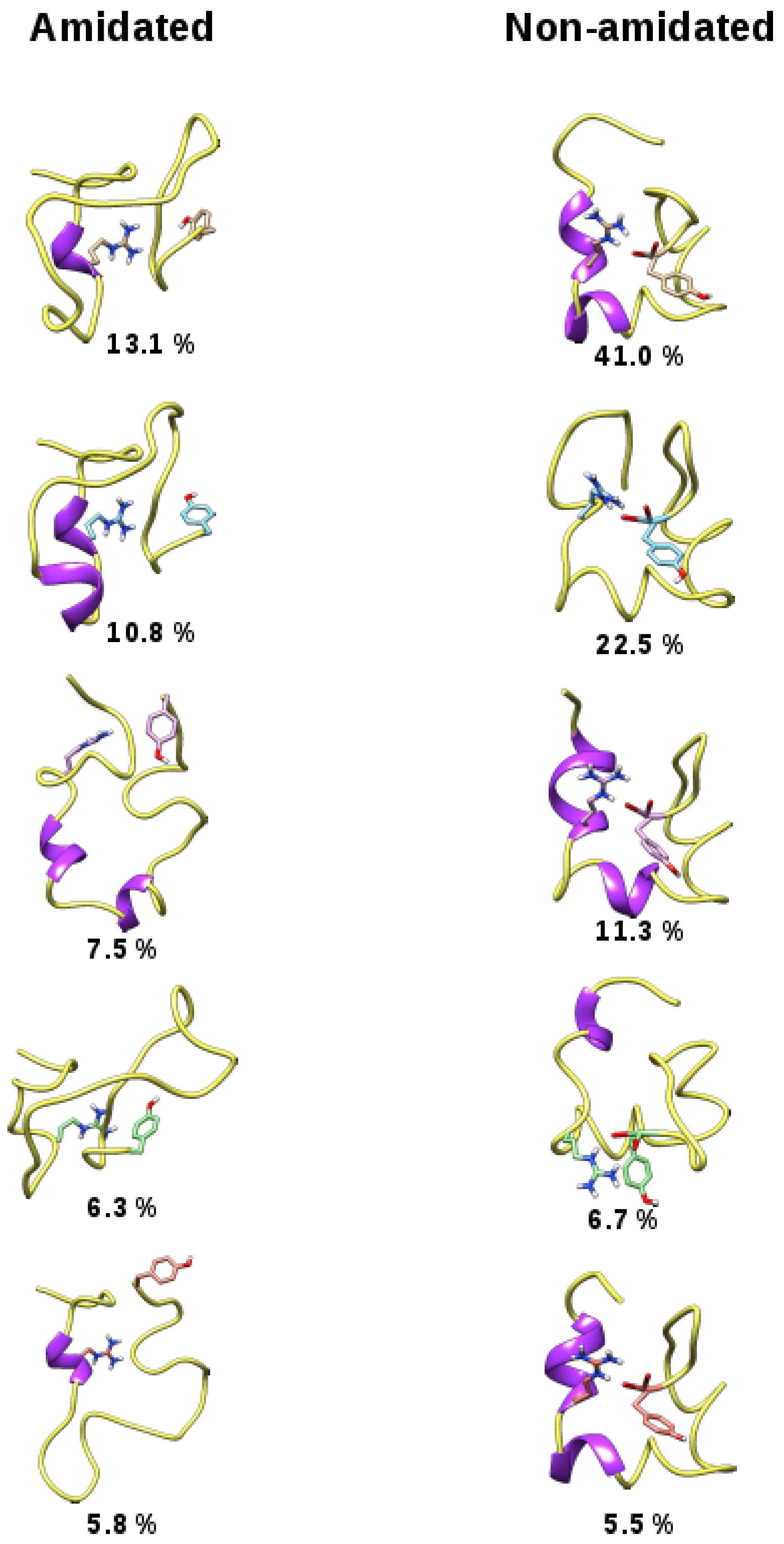
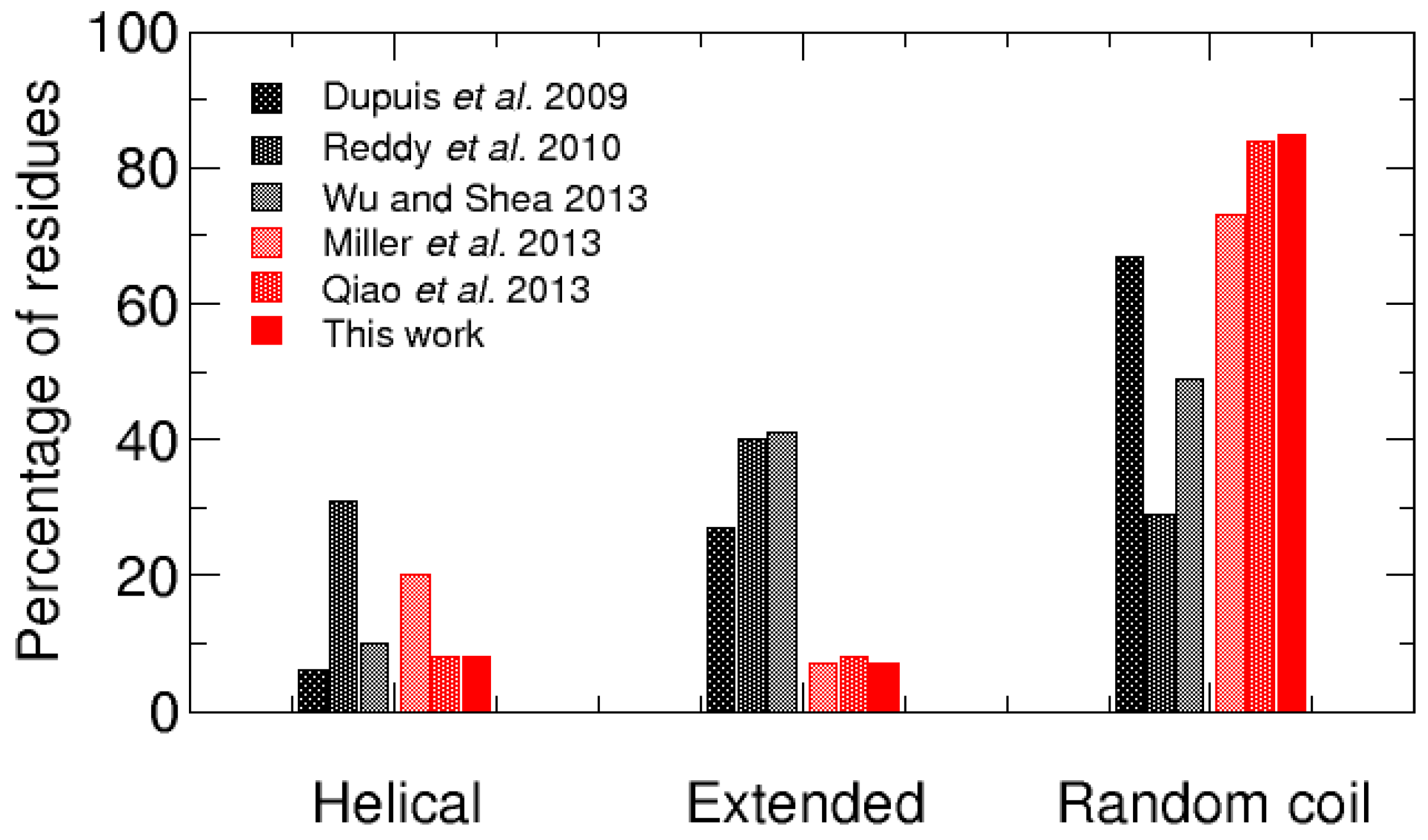
3. Discussion
4. Materials and Methods
4.1. Molecular Simulations
4.2. Structural Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CINES | Centre Informatique National de l’Enseignement Supérieur (Montpellier, France) |
| GENCI | Grand Équipement National de Calcul Intensif (Paris, France) |
| GROMACS | GROningen MAchine for Chemical Simulations |
| IAPP | Islet Amyloid Polypeptide |
| HPC | High Performance Computer |
| LINCS | LINear Constraint Solver |
| MD | Molecular Dynamics |
| NPT | constant Number of particles Pressure and Temperature |
| OPLS-AA | Optimized Potentials for Liquid Simulations - All Atom |
| PME | Particle Mesh Ewald |
| REMD | Replica Exchange Molecular Dynamics |
| RMSD | Root Mean Square Deviation |
| SDS | Sodium Dodecyl Sulfate |
| SETTLE | ShakE and raTTLE |
| SPC/E | Simple Point Charge / Extended |
| STRIDE | STRucture IDEntification |
References
- Cooper, G.J.; Leighton, B.; Dimitriadis, G.D.; Parry-Billings, M.; Kowalchuk, J.M.; Howland, K.; Rothbard, J.B.; Willis, A.C.; Reid, K.B. Amylin found in amyloid deposits in human type 2 diabetes mellitus may be a hormone that regulates glycogen metabolism in skeletal muscle. Proc. Natl. Acad. Sci. USA 1988, 85, 7763–7766. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.N.; Leighton, B.; Todd, J.A.; Cockburn, D.; Schofield, P.N.; Sutton, R.; Holt, S.; Boyd, Y.; Day, A.J.; Foot, E.A. Molecular and functional characterization of amylin, a peptide associated with type 2 diabetes mellitus. Proc. Natl. Acad. Sci. USA 1989, 86, 9662–9666. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.D.; Scognamiglio, P.L.; Cascella, R.; Cecchi, C.; Russo, A.; Leone, M.; Penco, A.; Relini, A.; Federici, L.; Matteo, A.D.; et al. Nucleophosmin contains amyloidogenic regions that are able to form toxic aggregates under physiological conditions. FASEB J. 2015, 29, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Natale, C.D.; Leone, M.; Cascella, R.; Cecchi, C.; Lirussi, L.; Antoniali, G.; Riccardi, D.; Morelli, G.; Tell, G.; et al. Destabilisation, aggregation, toxicity and cytosolic mislocalisation of nucleophosmin regions associated with acute myeloid leukemia. Oncotarget 2016, 7, 59129–59143. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.; Cooper, G.J.; Lewis, C.E.; Morris, J.F.; Willis, A.C.; Reid, K.B.; Turner, R.C. Islet amyloid formed from diabetes-associated peptide may be pathogenic in type-2 diabetes. Lancet 1987, 2, 231–234. [Google Scholar] [CrossRef]
- Kahn, S.E.; Andrikopoulos, S.; Verchere, C.B. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999, 48, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Hoppener, J.W.; Ahrén, B.; Lips, C.J. Islet Amyloid and Type 2 Diabetes Mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Zraika, S.; Hull, R.L.; Verchere, C.B.; Clark, A.; Potter, K.J.; Fraser, P.E.; Raleigh, D.P.; Kahn, S.E. Toxic oligomers and islet beta cell death: Guilty by association or convicted by circumstantial evidence? Diabetologia 2010, 53, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Jaikaran, E.T.A.S.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Nagel-Steger, L.; Owen, M.C.; Strodel, B. An Account of Amyloid Oligomers: Facts and Figures Obtained from Experiments and Simulations. ChemBioChem 2016, 17, 657–676. [Google Scholar] [CrossRef] [PubMed]
- Wiltzius, J.J.W.; Sievers, S.A.; Sawaya, M.R.; Eisenberg, D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009, 18, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Abedini, A.; Raleigh, D.P. A critical assessment of the role of helical intermediates in amyloid formation by natively unfolded proteins and polypeptides. Protein Eng. Des. Sel. 2009, 22, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Prabhakar, A.; Aucoin, D.; Simon, M.; Sparks, S.; Robbins, K.J.; Sheen, A.; Petty, S.A.; Lazo, N.D. Mechanistic Studies of Peptide Self-Assembly: Transient α-Helices to Stable β-Sheets. J. Am. Chem. Soc. 2010, 132, 18223–18232. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.H.; Gupta, R.; Ling, Y.L.; Strasfeld, D.B.; Raleigh, D.P.; Zanni, M.T. Two-dimensional IR spectroscopy and isotope labeling defines the pathway of amyloid formation with residue-specific resolution. Proc. Natl. Acad. Sci. USA 2009, 106, 6614–6619. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.F.; Wu, C.; Shea, J.E.; Bowers, M.T. The Amyloid Formation Mechanism in Human IAPP: Dimers Have β-Strand Monomer-Monomer Interfaces. J. Am. Chem. Soc. 2011, 133, 7240–7243. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, L.E.; Dunkelberger, E.B.; Tran, H.Q.; Cheng, P.N.; Chiu, C.C.; Cao, P.; Raleigh, D.P.; de Pablo, J.J.; Nowick, J.S.; Zanni, M.T. Mechanism of IAPP amyloid fibril formation involves an intermediate with a transient β-sheet. Proc. Natl. Acad. Sci. USA 2013, 110, 19285–19290. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.H.J.; Colin, C.; Degaki, T.L.; Sousa, A.C.V.D.; Vieira, M.N.N.; Sebollela, A.; Martinez, A.M.B.; Bloch, C.; Ferreira, S.T.; Sogayar, M.C. Amyloidogenicity and Cytotoxicity of Recombinant Mature Human Islet Amyloid Polypeptide (rhIAPP). J. Biol. Chem. 2004, 279, 42803–42810. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, I.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jiang, P.; Xu, W.; Li, H.; Zhang, H.; Yan, L.; Chan-Park, M.B.; Liu, X.W.; Tang, K.; Mu, Y.; Pervushin, K. The Molecular Basis of Distinct Aggregation Pathways of Islet Amyloid Polypeptide. J. Biol. Chem. 2011, 286, 6291–6300. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S.; Zhao, D.S.; Yu, Y.P.; Li, W.W.; Chen, Y.X.; Zhao, Y.F.; Li, Y.M. Characterizing the assembly behaviors of human amylin: A perspective derived from C-terminal variants. Chem. Commun. 2013, 49, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.H.; Serrano, A.L.; Zanni, M.T.; Raleigh, D.P. Mutational Analysis of Preamyloid Intermediates: The Role of His-Tyr Interactions in Islet Amyloid Formation. Biophys. J. 2014, 106, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology, Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2015, 2016, e2798269. [Google Scholar]
- Patil, S.M.; Xu, S.; Sheftic, S.R.; Alexandrescu, A.T. Dynamic α-Helix Structure of Micelle-bound Human Amylin. J. Biol. Chem. 2009, 284, 11982–11991. [Google Scholar] [CrossRef] [PubMed]
- Nanga, R.P.R.; Brender, J.R.; Vivekanandan, S.; Ramamoorthy, A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 2011, 1808, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Bernhagen, J.; Greenfield, N.; Sweimeh, K.; Brunner, H.; Voelter, W.; Kapurniotu, A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in Vitro1. J. Mol. Biol. 1999, 287, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Higham, C.E.; Jaikaran, E.T.A.S.; Fraser, P.E.; Gross, M.; Clark, A. Preparation of synthetic human islet amyloid polypeptide (IAPP) in a stable conformation to enable study of conversion to amyloid-like fibrils. FEBS Lett. 2000, 470, 55–60. [Google Scholar] [CrossRef]
- Williamson, J.A.; Loria, J.P.; Miranker, A.D. Helix Stabilization Precedes Aqueous and Bilayer-Catalyzed Fiber Formation in Islet Amyloid Polypeptide. J. Mol. Biol. 2009, 393, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Cort, J.R.; Liu, Z.; Lee, G.M.; Huggins, K.; Janes, S.; Prickett, K.; Andersen, N.H. Solution state structures of human pancreatic amylin and pramlintide. Protein Eng. Des. Sel. 2009, 22, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.F.; Wu, C.; Shea, J.E.; Bowers, M.T. Human Islet Amyloid Polypeptide Monomers Form Ordered β-hairpins: A Possible Direct Amyloidogenic Precursor. J. Am. Chem. Soc. 2009, 131, 18283–18292. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Ha-Duong, T. Insights into the conformational ensemble of human islet amyloid polypeptide from molecular simulations. Curr. Pharm. Des. 2016, 22, 3601–3607. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.N.; Winter, R. Comparing the structural properties of human and rat islet amyloid polypeptide by MD computer simulations. Biophys. Chem. 2011, 156, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.A.; Miranker, A.D. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007, 16, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Shea, J.E. Structural Similarities and Differences between Amyloidogenic and Non-Amyloidogenic Islet Amyloid Polypeptide (IAPP) Sequences and Implications for the Dual Physiological and Pathological Activities of These Peptides. PLoS Comput. Biol. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.; Zerze, G.H.; Mittal, J. Molecular Simulations Indicate Marked Differences in the Structure of Amylin Mutants, Correlated with Known Aggregation Propensity. J. Phys. Chem. B 2013, 117, 16066–16075. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhao, J.; Yu, X.; Zheng, J. Comparative Molecular Dynamics Study of Human Islet Amyloid Polypeptide (IAPP) and Rat IAPP Oligomers. Biochemistry 2013, 52, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.C.; Singh, S.; de Pablo, J.J. Effect of Proline Mutations on the Monomer Conformations of Amylin. Biophys. J. 2013, 105, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Wang, L.; Singh, S.; Ling, Y.L.; Buchanan, L.; Zanni, M.T.; Skinner, J.L.; de Pablo, J.J. Stable and Metastable States of Human Amylin in Solution. Biophys. J. 2010, 99, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Bowman, G.R.; Huang, X. Dynamics of an Intrinsically Disordered Protein Reveal Metastable Conformations That Potentially Seed Aggregation. J. Am. Chem. Soc. 2013, 135, 16092–16101. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Yoda, T.; Sugita, Y.; Okamoto, Y. Comparisons of force fields for proteins by generalized-ensemble simulations. Chem. Phys. Lett. 2004, 386, 460–467. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Patriksson, A.; Spoel, D.V.D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Heinig, M.; Frishman, D. STRIDE: A web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 2004, 32, W500–W502. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
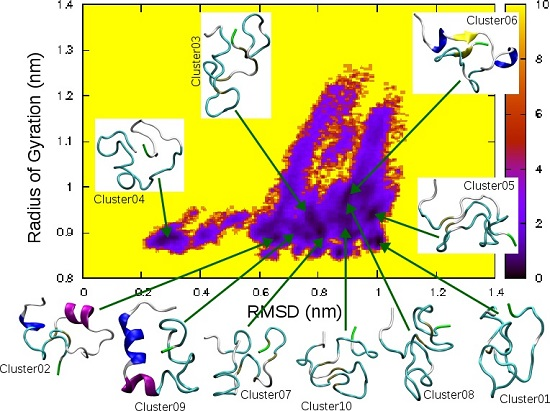
| Cluster | Population (%) | RMSD (nm) | Gyration (nm) | α-Helix | β-Strand |
|---|---|---|---|---|---|
| 01 | 4.6 | 1.00 | 0.88 | ||
| 02 | 4.5 | 0.65 | 0.90 | 5–7, 16–21 | |
| 03 | 3.7 | 0.78 | 0.93 | ||
| 04 | 3.6 | 0.30 | 0.89 | ||
| 05 | 2.7 | 0.99 | 0.94 | ||
| 06 | 2.3 | 0.88 | 0.97 | 5–7, 14–16 | 9–10, 34–35 |
| 07 | 2.1 | 0.87 | 0.86 | ||
| 08 | 2.0 | 0.91 | 0.88 | ||
| 09 | 1.9 | 0.75 | 0.89 | 5–11, 13–17 | |
| 10 | 1.9 | 0.90 | 0.90 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, L.; Ha-Duong, T. Effect of Post-Translational Amidation on Islet Amyloid Polypeptide Conformational Ensemble: Implications for Its Aggregation Early Steps. Int. J. Mol. Sci. 2016, 17, 1896. https://doi.org/10.3390/ijms17111896
Tran L, Ha-Duong T. Effect of Post-Translational Amidation on Islet Amyloid Polypeptide Conformational Ensemble: Implications for Its Aggregation Early Steps. International Journal of Molecular Sciences. 2016; 17(11):1896. https://doi.org/10.3390/ijms17111896
Chicago/Turabian StyleTran, Linh, and Tâp Ha-Duong. 2016. "Effect of Post-Translational Amidation on Islet Amyloid Polypeptide Conformational Ensemble: Implications for Its Aggregation Early Steps" International Journal of Molecular Sciences 17, no. 11: 1896. https://doi.org/10.3390/ijms17111896