
New Treatment Strategies for Alcohol-Induced Heart Damage

Abstract
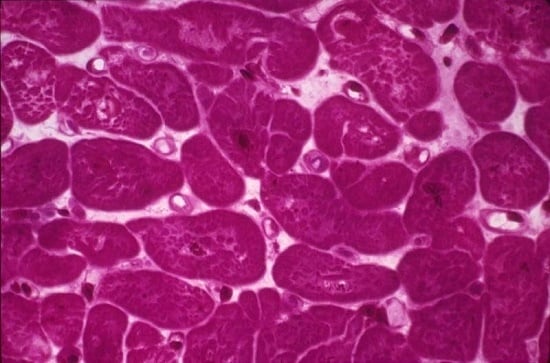
:
1. Introduction
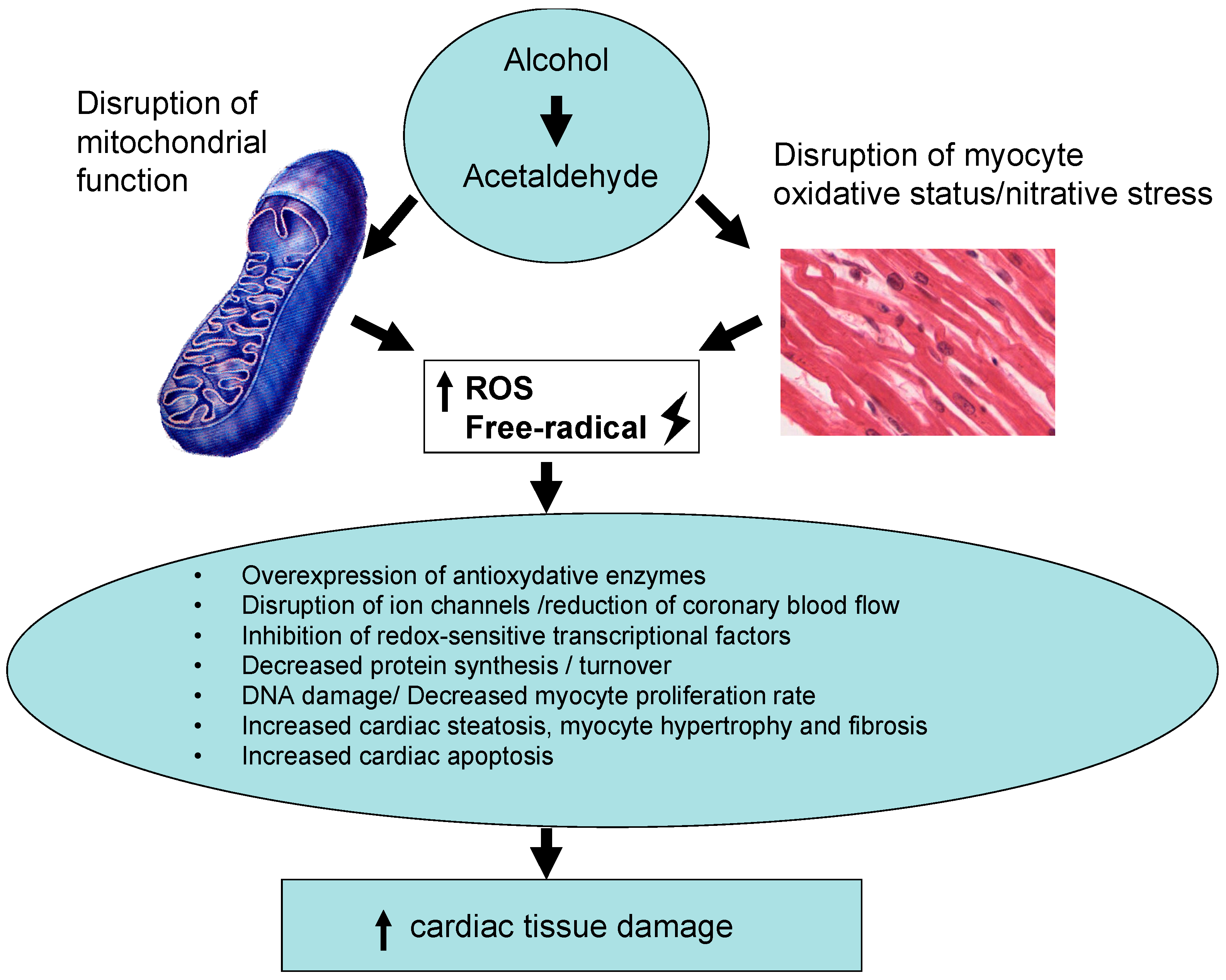
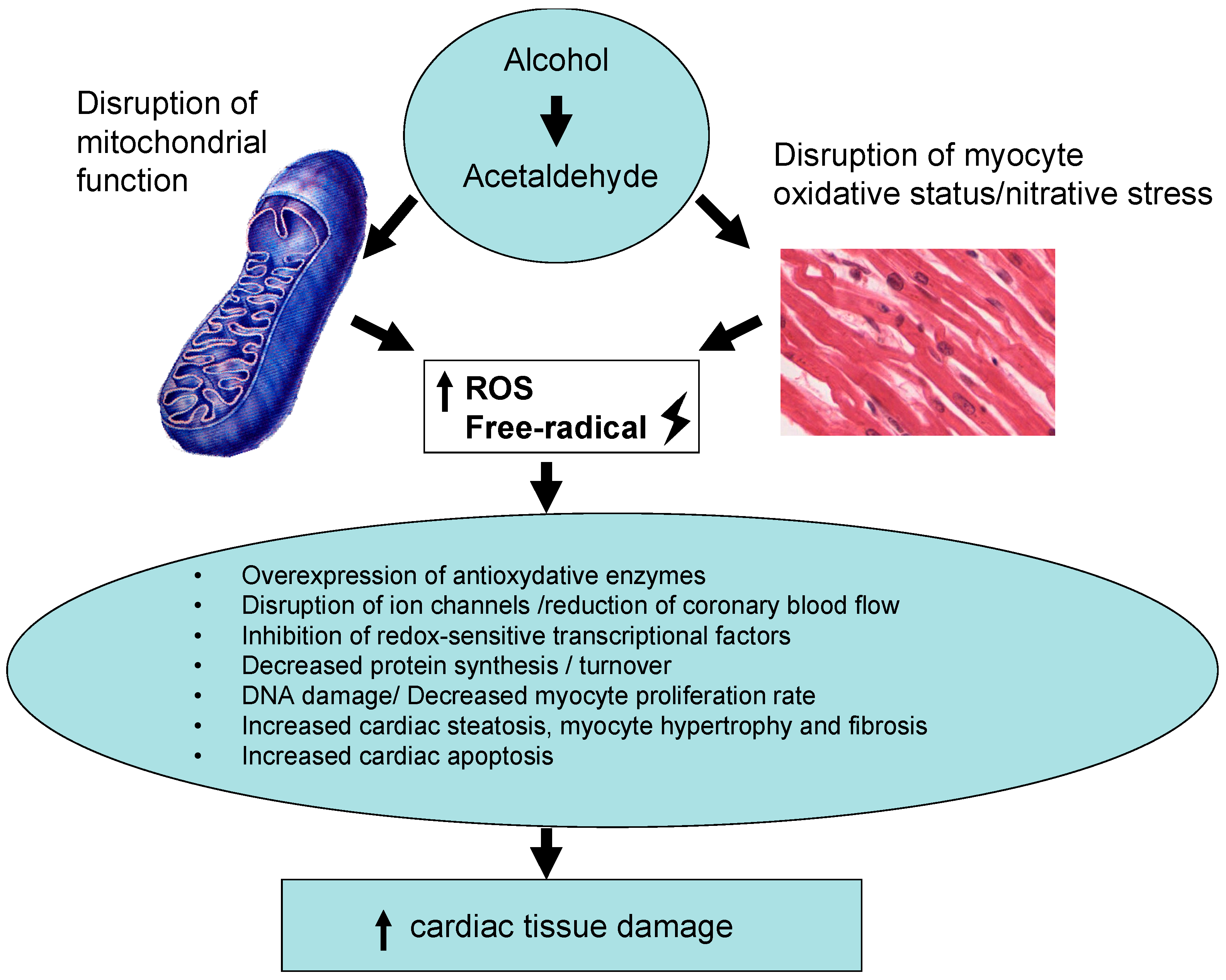
2. Mechanism of Alcohol-Induced Cardiac Damage
3. Strategies to Decrease Factors Inducing Heart Damage
3.1. Control of Alcohol Consumption
3.2. Comorbid Factors
3.3. Therapy against Alcohol-Induced Non-Cardiac Systemic Damage
3.4. Therapeutic Approaches against Myocyte Hypertrophy and Cell Loss
3.4.1. Myostatin (Mstn)
3.4.2. Adrenergic Receptors (AR)
3.4.3. Oxidative/Nitrative Stress
3.4.4. Myocardium RhoA/ROCK Pathway
3.4.5. Sirtuins
3.4.6. Caspase Inhibition
3.4.7. Suppressor of IKKε (SIKE)
3.4.8. Micro RNA (miRNAs)
3.4.9. G-Protein Signaling Pathway
3.4.10. Fibroblast Growth-Factor 21 (FGF21)
3.4.11. Peroxisome Proliferator Activated Receptor Agonists (PPAR)
3.4.12. Resveratrol
3.4.13. Alpha-Lipoic Acid (ALA)
3.4.14. BNIP-3
3.5. Control of Cardiac Fibrosis
3.5.1. ROCK Inhibitors
3.5.2. miRNA
3.5.3. TGF-β Antagonists
3.5.4. Cytokines and Chemokines
3.5.5. Relaxin
3.5.6. Myostatin (Mstn).
3.5.7. ω-3 Polyunsaturated Fatty Acids (ω-3 PUFAs)
3.5.8. Pioglitazone
3.5.9. Anthrocyanin
3.6. Control of Oxidative-Energy Damage
3.6.1. Novel Cardiomyokines
3.6.2. Leptin
3.6.3. Pioglitazone
3.6.4. Ghrelin
3.6.5. Phenolic Compounds
4. Strategies to Improve Cell Regeneration and Repair
4.1. Ki-67 and Myostatin
4.2. Telocytes
4.3. Stem Cell Therapy
4.4. Heart Transplantion
5. Conclusions and Future Trends
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Urbano-Márquez, A.; Fernández-Solà, J. Effects of alcohol on skeletal and cardiac muscle. Muscle Nerve 2004, 30, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Guzzo-Merello, G.; Segovia, J.; Dominguez, F.; Cobo-Marcos, M.; Gomez-Bueno, M.; Avellana, P.; Millan, I.; Alonso-Pulpon, L.; Garcia-Pavia, P. Natural history and prognostic factors in alcoholic cardiomyopathy. JACC Heart Fail. 2015, 3, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Iacovoni, A.; de Maria, R.; Gavazzi, A. Alcoholic cardiomyopathy. J. Cardiovasc. Med. 2010, 11, 884–892. [Google Scholar] [CrossRef] [PubMed]
- WHO Global Status Report on Alcohol and Health, 2014 ed.; World Health Organization: Geneva, Switzerland, 2014; Available online: http://www.who.int/substance_abuse/publications/global_alcohol_report/en/ (accessed on 22 September 2016).
- United Nations World Mortality Report 2013. Department of Economic and Social Affairs Population Division ST/ESA/SER.A/347 New York, 2013. Available online: http://www.un.org/en/development/desa/population/publications/pdf/mortality/WMR2013/World_Mortality_2013_Report.pdf (accessed on 22 September 2016).
- Klatsky, A.L. Alcohol and cardiovascular diseases: A historical overview. Ann. N. Y. Acad. Sci. 2002, 957, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Urbano-Márquez, A.; Estruch, R.; Navarro-López, F.; Grau, J.M.; Mont, L.; Rubin, E. The effects of alcoholism on skeletal and cardiac muscle. N. Engl. J. Med. 1989, 320, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wold, L.E. Mechanisms of alcoholic heart disease. Ther. Adv. Cardiovasc. Dis. 2008, 2, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Solà, J. Cardiovascular risk and benefits of moderate and heavy alcohol consumption. Nat. Rev. Cardiol. 2015, 12, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Jakubczyk, A.; Wojnar, M. Total abstinence or harm reduction—Different strategies of alcohol treatment in research studies and international guidelines. Psychiatr. Polska 2012, 46, 373–386. [Google Scholar]
- Muckle, W.; Muckle, J.; Welch, V.; Tugwell, P. Managed alcohol as a harm reduction intervention for alcohol addiction in populations at high risk for substance abuse. Cochrane Database Syst. Rev. 2012, 12, CD006747. [Google Scholar] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Keoleian, V.; Polcin, D.; Galloway, G.P. Text messaging for addiction: A review. J. Psychoact. Drugs 2015, 47, 158–176. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.K.; Cousins, V.M.; Umoh, N.A.; Jeffress, M.A.; Taghipour, D.; Al-Rubaiee, M.; Haddad, G.E. The good, the bad, and the ugly with alcohol use and abuse on the heart. Alcohol Clin. Exp. Res. 2013, 37, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; D’Souza El-Guindy, N.B.; Apte, M.; Mari, M.; Dooley, S.; Neuman, M.; Haber, P.S.; Kundu, G.C.; Darwanto, A.; de Villiers, W.J.; et al. Alcohol, signaling, and ECM turnover. Alcohol Clin. Exp. Res. 2010, 34, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Leibing, E.; Meyer, T. Enzymes and signal pathways in the pathogenesis of alcoholic cardiomyopathy. Herz 2016, 41, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.P.; Sass, E.J.; Tun-Kirchmann, T.T.; Rubin, E. Ethanol inhibits electrically-induced calcium transients in isolated rat cardiac myocytes. Mol. Cell. Cardiol. 1989, 21, 555–565. [Google Scholar] [CrossRef]
- Nicolás, J.M.; Rubin, E.; Thomas, A.P. Ethanol and cocaine cause additive inhibitory effects on the calcium transients and contraction in single cardiomyocytes. Alcohol Clin. Exp. Res. 1996, 20, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Matyas, C.; Varga, Z.V.; Mukhopadhyay, P.; Paloczi, J.; Lajtos, T.; Erdelyi, K.; Nemeth, B.T.; Nan, M.; Hasko, G.; Gao, B.; et al. Chronic plus binge ethanol feeding induces myocardial oxidative stress, mitochondrial and cardiovascular dysfunction, and steatosis. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1658–H1670. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, Y. Biomarkers in heart failure. Int. Heart J. 2014, 55, 474–481. [Google Scholar] [CrossRef] [PubMed]
- González-Reimers, E.; Santolaria-Fernández, F.; Martín-González, M.C.; Fernández-Rodríguez, C.M.; Quintero-Platt, G. Alcoholism: A systemic proinflammatory condition. World J. Gastroenterol. 2014, 20, 14660–14671. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Chawla, K.; Umoh, N.A.; Cousins, V.M.; Ketegou, A.; Reddy, M.G.; AlRubaiee, M.; Haddad, G.E.; Burke, M.W. Alcohol and apoptosis: Friends or foes? Biomolecules 2015, 5, 3193–3203. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Solà, J.; Fatjó, F.; Sacanella, E.; Estruch, R.; Bosch, X.; Urbano-Márquez, A.; Nicolás, J.M. Evidence of apoptosis in alcoholic cardiomyopathy. Hum. Pathol. 2006, 37, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcohol and fibrogenesis. Alcohol Alcohol. Suppl. 1991, 1, 339–344. [Google Scholar] [PubMed]
- Niemelä, O.; Parkkila, S.; Worrall, S.; Emery, P.W.; Preedy, V.R. Generation of aldehyde-derived protein modifications in ethanol-exposed heart. Alcohol Clin. Exp. Res. 2003, 27, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Ji, C. Advances and new concepts in alcohol-induced organelle stress, unfolded protein responses and organ damage. Biomolecules 2015, 5, 1099–1121. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Solà, J.; Estruch, R.; Grau, J.M.; Paré, J.C.; Rubin, E.; Urbano-Márquez, A. The relation of alcoholic myopathy to cardiomyopathy. Ann. Intern. Med. 1994, 120, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, O.V. The morphological changes in the myocardial tissue after sudden cardiac death from alcoholic cardiomyopathy. Sud. Med. Ekspert. 2016, 59, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Ceron, C.S.; Marchi, K.C.; Muniz, J.J.; Tirapelli, C.R. Vascular oxidative stress: A key factor in the development of hypertension associated with ethanol consumption. Curr. Hypertens. Rev. 2014, 10, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Borriser-Pairó, F.; Fernández-Solà, J.; Antúnez, E.; Tobías, E. Insulin-like growth-factor 1 myocardial expression decreases in chronic alcohol consumption. Reg. Med. Res. 2013, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Mancone, M.; Chilian, W.M.; Severino, P.; Canali, E.; Logan, S.; de Marchis, M.L.; Volterrani, M.; Palmirotta, R.; Guadagni, F. Role of genetic polymorphisms of ion channels in the pathophysiology of coronary microvascular dysfunction and ischemic heart disease. Basic Res. Cardiol. 2013, 108, 387. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo, I.; Hondares, E.; Vinciguerra, M.; Munts, C.; Iglesias, R.; Gabrielli, L.A.; Sitges, M.; Giralt, M.; van Bilsen, M.; et al. Fibroblast growth factor 21 protects against cardiac hypertrophy in mice. Nat. Commun. 2013, 4, 2019. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Redondo-Angulo, I.; Ribas, F.; Garrabou, G.; Casademont, J.; Giralt, M.; Villarroya, F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc. Res. 2015, 106, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Lerman, D.A.; Alotti, N.; Ume, K.L.; Péault, B. Cardiac repair and regeneration: The value of cell therapies. Eur. Cardiol. 2016, 11, 43–48. [Google Scholar] [CrossRef]
- Fernández-Solà, J.; Preedy, V.R.; Lang, C.H.; González-Reimers, E.; Arno, M.; Lin, J.C.; Wiseman, H.; Zhou, S.; Emery, P.W.; Nakahara, T.; et al. Molecular and cellular events in alcohol-induced muscle disease. Alcohol Clin. Exp. Res. 2007, 31, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S. Mechanisms of cardiotoxicity and the development of heart failure. Crit. Care Nurs. Clin. N. Am. 2015, 27, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Casado Cerrada, J.; Zabaleta Camino, J.P.; Fontecha Ortega, M. Target organ damage in acute heart failure. Rev. Clin. Esp. 2016, 216, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Scolletta, S.; Biagioli, B. Energetic myocardial metabolism and oxidative stress: Let’s make them our friends in the fight against heart failure. Biomed. Pharmacother. 2010, 64, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Chivite, D.; Formiga, F.; Corbella, X. Organ-protection therapy. A new therapeutic approach for acute heart failure? Med. Clin. 2014, 142, 66–71. [Google Scholar] [CrossRef]
- Von Haehling, S. Recent developments in the treatment of heart failure: Highlights from the American Heart Association’s Scientific Sessions, Los Angeles, California, 3–7 December 2012. Expert Opin. Investig. Drugs 2013, 22, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, J.M.; Fernández-Solà, J.; Estruch, R.; Paré, J.C.; Urbano-Márquez, A.; Rubin, E. The effect of controlled drinking in alcoholic cardiomyopathy. Ann. Intern. Med. 2002, 136, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, M.T.; Jude, R. Defining “binge” drinking as five drinks per occasion or drinking to a 0.08% BAC: Which is more sensitive to risk? Am. J. Addict. 2011, 20, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Lesch, O.M.; Walter, H.; Wetschka, C.; Hesselbrock, M.; Hesselbrock, V. Alcohol and Tobacco Medical and Sociological Aspects of Use, Abuse and Addiction; Spinger: Viena, Austria, 2011; pp. 1–353. [Google Scholar]
- Ait-Daoud, N.; Wiesbeck, G.A.; Bienkowski, P.; Li, M.D.; Pfützer, R.H.; Singer, M.V.; Lesch, O.M.; Johnson, B.A. Comorbid alcohol and nicotine dependence: From the biomolecular basis to clinical consequences. Alcohol Clin. Exp. Res. 2005, 29, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Volpe, M.; Battistoni, A.; Tocci, G.; Rosei, E.A.; Catapano, A.L.; Coppo, R.; del Prato, S.; Gentile, S.; Mannarino, E.; Novo, S.; et al. Cardiovascular risk assessment beyond systemic coronary risk estimation: A role for organ damage markers. J. Hypertens. 2012, 30, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Severino, P.; Calcagno, S.; Mancone, M. Heart failure: TNM-like classification. J. Am. Coll. Cardiol. 2014, 63, 1959–1960. [Google Scholar] [CrossRef] [PubMed]
- Sehestedt, T.; Jeppesen, J.; Hansen, T.W.; Wachtell, K.; Ibsen, H.; Torp-Pedersen, C.; Hildebrandt, P.; Olsen, M.H. Risk prediction is improved by adding markers of subclinical organ damage to SCORE. Eur. Heart J. 2010, 31, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Marteles, M.; Rubio Gracia, J.; Giménez López, I. Pathophysiology of acute heart failure: A world to know. Rev. Clin. Esp. 2016, 216, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Fernández-Solà, J.; Sacanella, E.; Paré, C.; Rubin, E.; Urbano-Márquez, A. Relationship between cardiomyopathy and liver disease in chronic alcoholism. Hepatology 1995, 22, 532–538. [Google Scholar] [PubMed]
- Fernández-Solà, J. Treatment of extrahepatic manifestations of alcohol abuse. In Alcohol and Liver Disease, 1st ed.; Neuberger, J., DiMartini, A., Eds.; Wiley-Blackwell: Regis West Sussex, UK, 2015; pp. 187–196. [Google Scholar]
- Keil, V.C.; Greschus, S.; Schneider, C.; Hadizadeh, D.R.; Schild, H.H. The whole spectrum of alcohol-related changes in the CNS: Practical MR and CT imaging guidelines for daily clinical use. RöFo 2015, 187, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Chisholm, D.; Parikh, R.; Charlson, F.J.; Degenhardt, L.; Dua, T.; Ferrari, A.J.; Hyman, S.; Laxminarayan, R.; Levin, C.; et al. Global Priorities for Addressing the Burden of Mental, Neurological, and Substance Use Disorders. In Mental, Neurological, and Substance Use Disorders: Disease Control Priorities, 3rd ed.; Patel, V., Chisholm, D., Dua, T., Laxminarayan, R., Medina-Mora, M.E., Eds.; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2016. [Google Scholar]
- Weidler, D.J. Myocardial damage and cardiac arrhythmias after intracranial hemorrhage. A critical review. Stroke 1974, 5, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Samuels, M.A. The brain-heart connection. Circulation 2007, 116, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Guidot, D.M.; Hart, C.M. Alcohol abuse and acute lung injury: Epidemiology and pathophysiology of a recently recognized association. J. Investig. Med. 2005, 53, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Aytacoglu, B.N.; Calikoglu, M.; Tamer, L.; Coşkun, B.; Sucu, N.; Köse, N.; Aktas, S.; Dikmengil, M. Alcohol-induced lung damage and increased oxidative stress. Respiration 2006, 73, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Bekfani, T.; Pellicori, P.; Morris, D.A.; Ebner, N.; Valentova, M.; Steinbeck, L.; Wachter, R.; Elsner, S.; Sliziuk, V.; Schefold, J.C.; et al. Sarcopenia in patients with heart failure with preserved ejection fraction: Impact on muscle strength, exercise capacity and quality of life. Int. J. Cardiol. 2016, 222, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Moriya, N.; Nihira, M.; Sato, S.; Watanabe, T. Ultrastructural changes of liver, heart, lung and kidney of mice in a large dose of ethanol injection. Arukoru Kenkyuto Yakubutsu Ison 1992, 27, 189–200. [Google Scholar] [PubMed]
- Epstein, M. Alcohol’s impact on kidney function. Alcohol Health Res. World 1997, 21, 84–92. [Google Scholar] [PubMed]
- López-Gómez, J.M.; Jofré, R.; Cases, A. Cardiovascular risk factors in chronic renal failure. Nefrologia 2002, 22, 59–67. [Google Scholar] [PubMed]
- Morris, N.L.; Ippolito, J.A.; Curtis, B.J.; Chen, M.M.; Friedman, S.L.; Hines, I.N.; Haddad, G.E.; Chang, S.L.; Brown, L.A.; Waldschmidt, T.J.; et al. Alcohol and inflammatory responses: Summary of the 2013 alcohol and immunology research interest group (AIRIG) meeting. Alcohol 2015, 49, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nicolás, J.M.; García, G.; Fatjó, F.; Sacanella, E.; Tobías, E.; Badía, E.; Estruch, R.; Fernández-Solà, J. Influence of nutritional status on alcoholic myopathy. Am. J. Clin. Nutr. 2003, 78, 326–333. [Google Scholar] [PubMed]
- Estruch, R.; Sacanella, E.; Fernández-Solà, J.; Nicolas, J.M. Nutritional Status in Alcoholics. In The Handbook of Alcohol Related Pathology; Preedy, V.R., Ed.; Elseiver Science Pub.: London, UK, 2005; pp. 363–377. [Google Scholar]
- Cotecchia, S.; del Vescovo, C.D.; Colella, M.; Caso, S.; Diviani, D. The α1-adrenergic receptors in cardiac hypertrophy: Signaling mechanisms and functional implications. Cell Signal. 2015, 27, 1984–1993. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in cardiac hypertrophy and failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, S. Cardiac hypertrophy as early adjustments to a chronically sustained mechanical overload. Jpn. Circ. J. 1985, 49, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Engel, F.B. The cardiomyocyte cell cycle in hypertrophy, tissue homeostasis, and regeneration. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 67–96. [Google Scholar] [PubMed]
- Corbalan, J.; Vatner, D.E.; Vatner, S.F. Myocardial apoptosis in heart disease: Does the emperor have clothes? Basic Res. Cardiol. 2016, 111, 31. [Google Scholar] [CrossRef] [PubMed]
- Gaussin, V.; Depre, C. Myostatin, the cardiac chalone of insulin-like growth factor-1. Cardiovasc. Res. 2005, 68, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Joulia-Ekaza, D.; Cabello, G. Myostatin regulation of muscle development: Molecular basis, natural mutations, physiopathological aspects. Exp. Cell Res. 2006, 312, 2401–2414. [Google Scholar] [PubMed]
- Biesemann, N.; Mendler, L.; Wietelmann, A.; Hermann, S.; Schäfers, M.; Krüger, M.; Boettger, T.; Borchardt, T.; Braun, T. Myostatin regulates energy homeostasis in the heart and prevents heart failure. Circ. Res. 2014, 115, 296–310. [Google Scholar] [CrossRef] [PubMed]
- McPerson, A.C. Through thick and thin: A circulating growth factor inhibits age-related cardiac hypertrophy. Circ. Res. 2013, 113, 487–491. [Google Scholar]
- Loffredo, F.S.; Steinhause, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H.; Frost, R.A.; Svanberg, E.; Vary, T.C. IGF-I/IGFBP-3 ameliorates alterations in protein synthesis, eIF4E availability, and myostatin in alcohol-fed rats. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E916–E926. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Solà, J.; Borrisser-Pairó, F.; Antúnez, E.; Tobías, E. Myostatin and insulin-like growth factor-1 in hypertensive heart disease: A prospective study in human heart donors. J. Hypertens. 2015, 33, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; de Gorter, D.J.; Aartsma-Rus, A.; van Ommen, G.J.; ten Dijke, P.; Hoen, P.A.; Hoogaars, W.M. Cell-type specific regulation of myostatin signaling. FASEB J. 2012, 26, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.R.; Harrison, D.G.; Bhatnagar, A. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: A scientific statement from the American heart association. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yao, F.; Chen, S.; Huang, H.; Wu, L.; He, J.; Dong, Y. Endogenous BNP attenuates cardiomyocyte hypertrophy induced by Ang II via p38 MAPK/Smad signaling. Pharmazie 2014, 69, 833–837. [Google Scholar] [PubMed]
- Fordjour, P.A.; Wang, L.; Gao, H.; Li, L.; Wang, Y.; Nyagblordzro, M.; Agyemang, K.; Fan, G. Targeting BNIP3 in inflammation-mediated heart failure: A novel concept in heart failure therapy. Heart Fail. Rev. 2016, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M. Cardiac remodelling and RAS inhibition. Adv. Cardiovasc. Dis. 2016, 10, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, D.; Savion, N.; Kolodgie, F.; Simon, A.; Fisch, S.; Schäfer, K.; Bachner-Hinenzon, N.; Cao, X.; Gertler, A.; Solomon, G.; et al. Local application of leptin antagonist attenuates angiotensin II-induced ascending aortic aneurysm and cardiac remodeling. J. Am. Heart Assoc. 2016, 5, e003474. [Google Scholar] [CrossRef] [PubMed]
- Balligand, J.L. Cardiac salvage by tweaking with β-3-adrenergic receptors. Cardiovasc. Res. 2016, 111, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chi, R.F.; Qin, F.Z.; Guo, X.F. Distinct changes of myocyte autophagy during myocardial hypertrophy and heart failure: Association with oxidative stress. Exp. Physiol. 2016, 101, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Huang, H.; Jiang, J.; Wu, L.; Lin, C.; Tang, A.; Dai, G.; He, J.; Chen, Y. AVE 0991 attenuates cardiac hypertrophy through reducing oxidative stress. Biochem. Biophys. Res. Commun. 2016, 474, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Xuan, F.; Jian, J. Epigallocatechin gallate exerts protective effects against myocardial ischemia/reperfusion injury through the PI3K/Akt pathway-mediated inhibition of apoptosis and the restoration of the autophagic flux. Int. J. Mol. Med. 2016, 38, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Liao, J.K. Rho kinases and cardiac remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Surma, M.; Wei, L.; Shi, J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011, 7, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.K.; Lal, H.; Golden, H.B.; Gerilechaogetu, F.; Smith, M.; Guleria, R.S.; Foster, D.M.; Lu, G.; Dostal, D.E. Rac1 and RhoA differentially regulate angiotensinogen gene expression in stretched cardiac fibroblasts. Cardiovasc. Res. 2011, 90, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.; Schirok, H.; Figueroa-Perez, S.; Mittendorf, J.; Gnoth, M.J.; Apeler, H.; Lenz, J.; Franz, J.K.; Knorr, A.; Hütter, J.; et al. Cardiovascular effects of a novel potent and highly selective azaindole-based inhibitor of Rho-kinase. Br. J. Pharmacol. 2007, 152, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Doe, C.; Bentley, R.; Behm, D.J.; Lafferty, R.; Stavenger, R.; Jung, D.; Bamford, M.; Panchal, T.; Grygielko, E.; Wright, L.L.; et al. Novel Rho kinase inhibitors with anti-inflammatory and vasodilatory activities. J. Pharmacol. Exp. Ther. 2007, 320, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Pillai, V.B.; Gupta, M.P. Role of sirtuins in regulating pathophysiology of the heart. Trends Endocrinol. Metab. 2016, 27, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Ting, W.J.; Huang, C.Y.; Yang, J.Y.; Lin, W.T. Resveratrol attenuated hydrogen peroxide-induced myocardial apoptosis by autophagic flux. Food Nutr. Res. 2016, 60, 30511. [Google Scholar] [CrossRef] [PubMed]
- Bagul, P.K.; Dinda, A.K.; Banerjee, S.K. Effect of resveratrol on sirtuins expression and cardiac complications in diabetes. Biochem. Biophys. Res. Commun. 2015, 468, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Y.; Deng, W.; Dai, J.; Li, F.; Yuan, Y.; Wu, Q.; Zhou, H.; Bian, Z.; Tang, Q. Hesperetin attenuates mitochondria-dependent apoptosis in lipopolysaccharide-induced H9C2 cardiomyocytes. Mol. Med. Rep. 2014, 9, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Deng, K.Q.; Wang, A.; Ji, Y.X.; Zhang, X.J.; Fang, J.; Zhang, Y.; Zhang, P.; Jiang, X.; Gao, L.; Zhu, X.Y.; et al. Suppressor of IKKε is an essential negative regulator of pathological cardiac hypertrophy. Nat. Commun. 2016, 7, 11432. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y.T. Overview of microRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int. J. Mol. Sci. 2016, 17, 749. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Gao, X.-M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Simonetta, I.; Pinto, A. microRNA and receptor mediated signaling pathways as potential therapeutic targets in heart failure. Expert Opin. Ther. Targets 2016, 25, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ooi, J.Y.; Bernardo, B.C.; Singla, S.; Patterson, N.L.; Lin, R.C.; McMullen, J.R. Identification of miR-34 regulatory networks in settings of disease and anti-miR-therapy: Implications for treating cardiac pathology and other diseases. RNA Biol. 2016, 28, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Liu, W.; Zhang, J.; Xiang, D. miR-153 regulates apoptosis and autophagy of cardiomyocytes by targeting Mcl-1. Mol. Med. Rep. 2016, 14, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Vegter, E.L.; van der Meer, P.; de Windt, L.J.; Pinto, Y.M.; Voors, A.A. microRNAs in heart failure: From biomarker to target for therapy. Eur. J. Heart Fail. 2016, 18, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, L.; Yao, Y.; Tang, C.; Ding, J.; Fu, C.; Li, H.; Ma, G. Pivotal role of regulator of G-protein signaling 12 in pathological cardiac hypertrophy. Hypertension 2016, 67, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Lu, Y.; Xing, X.; Li, Y.; Huang, Z.; Zhong, H.; Huang, Y.; Chen, A.F.; Tang, X.; Li, H.; et al. Regulator of G-Protein signaling 10 negatively regulates cardiac remodeling by blocking mitogen-activated protein kinase-extracellular signal-regulated protein kinase 1/2 signaling. Hypertension 2016, 67, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Maity, B.; Anderegg, S.P.; Allamargot, C.; Yang, J.; Fisher, R.A. Regulator of G protein signaling 6 is a critical mediator of both reward-related behavioral and pathological responses to alcohol. Proc. Natl. Acad. Sci. USA 2015, 112, E786–E795. [Google Scholar] [CrossRef] [PubMed]
- Smeets, P.J.; Teunissen, B.E.; Planavila, A.; de Vogel-van den Bosch, F.A.; Willemsen, P.H.; van der Vusse, G.J.; van Bilsen, M. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARα and PPARδ. Cardiovasc. Res. 2008, 90, 276–284. [Google Scholar]
- Wei, W.Y.; Ma, Z.G.; Xu, S.C.; Zhang, N.; Tang, Q.Z. Pioglitazone protected against cardiac hypertrophy via inhibiting AKT/GSK3β and MAPK signaling pathways. PPAR Res. 2016, 2016, 9174190. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Soltys, C.L.; Rogan, K.J.; Chan, A.Y.; Nagendran, J.; Wang, S.; Dyck, J.R. Resveratrol prevents pathological but not physiological cardiac hypertrophy. J. Mol. Med. 2015, 93, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Gan, X.; Zhou, H.; Chen, X.; Guo, Y.; Chen, J.; Yang, X.; Lei, J. Alpha-lipoic acid attenuates cardiac hypertrophy via inhibition of C/EBPβ activation. Mol. Cell. Endocrinol. 2015, 399, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zou, J.; Chai, E.; Qi, Y.; Zhang, Y. Alpha-lipoic acid attenuates cardiac hypertrophy via downregulation of PARP2 and subsequent activation of SIRT-1. Eur. J. Pharmacol. 2014, 744, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblast awakens. Circ. Res. 2016, 118, 1021–1240. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, A.C.; Richardson, W.J.; Holmes, J.W.; Saucerman, J.J. Computational modeling of cardiac fibroblasts and fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Stempien-Otero, A.; Kim, D.H.; Davis, J. Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol. 2016, 97, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Piek, A.; de Noer, R.A.; Silljé, H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Honda, E.; Park, A.M.; Yoshida, K.; Tabuchi, M.; Munakata, H. Myofibroblasts: Biochemical and proteomic approaches to fibrosis. Tohoku J. Exp. Med. 2013, 230, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGF-β, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-β. Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Cavalera, M.; Frangogiannis, N.G. Targeting the chemokines in cardiac repair. Curr. Pharm. Des. 2014, 20, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.S.; Du, X.J.; Bathgate, R.A.; Summers, R.J. “Relaxin” the stiffened heart and arteries: The therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol. Ther. 2006, 112, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.S.; Lekgabe, E.D.; Mookerjee, I. The effects of relaxin on extracellular matrix remodeling in health and fibrotic disease. Adv. Exp. Med. Biol. 2007, 612, 88–103. [Google Scholar] [PubMed]
- Biesemann, N.; Mendler, L.; Kostin, S.; Wietelmann, A.; Borchardt, T.; Braun, T. Myostatin induces interstitial fibrosis in the heart via TAK1 and p38. Cell Tissue Res. 2015, 361, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; O’Horo, J. Antifibrotic effects of ω-3 fatty acids in the heart: One possible treatment for diastolic heart failure. Trends Cardiovasc. Med. 2011, 21, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Shibu, M.A.; Fan, M.J.; Chen, M.C.; Viswanadha, V.P.; Lin, Y.L.; Lai, C.H.; Lin, K.H.; Ho, T.J.; Kuo, W.W.; et al. Purple rice anthocyanin extract protects cardiac function in STZ-induced diabetes rat hearts by inhibiting cardiac hypertrophy and fibrosis. J. Nutr. Biochem. 2016, 31, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ströhle, A.; Wolters, M.; Hahn, A. Alcohol intake—A two-edged sword. Part 1: Metabolism and pathogenic effects of alcohol. Med. Monatsschr. Pharm. 2012, 35, 281–292. [Google Scholar] [PubMed]
- Liu, W.; Wang, B.; Ding, H.; Wang, D.W.; Zeng, H. A potential therapeutic effect of CYP2C8 overexpression on anti-TNF-α activity. Int. J. Mol. Med. 2014, 34, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Sundaresan, N.R.; Jeevanandam, V.; Gupta, M.P. Mitochondrial SIRT3 and heart disease. Cardiovasc. Res. 2010, 88, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Morales-González, J.A.; Madrigal-Santillán, E.; Morales-González, Á.; Bautista, M.; Gayosso-Islas, E.; Sánchez-Moreno, C. What is known regarding the participation of factor Nrf-2 in liver regeneration? Cells 2015, 4, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Fatjó, F.; Fernández-Solà, J.; Lluís, M.; Elena, M.; Badía, E.; Sacanella, E.; Estruch, R.; Nicolás, J.M. Myocardial antioxidant status in chronic alcoholism. Alcohol Clin. Exp. Res. 2005, 29, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.C. A new pathway for sympathetic cardioprotection in heart failure. Circ. Res. 2015, 117, 592–595. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Severino, P.; Bruno, N.; Stio, R.; Caira, C.; D’Ambrosio, A.; Brasolin, B.; Ohanyan, V.; Mancone, M. Role of ion channels in coronary microcirculation: A review of the literature. Future Cardiol. 2013, 9, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ren, Y.; Sorokin, V.; Poh, K.K.; Ho, H.H.; Lee, C.N.; de Kleijn, D.; Lim, S.K.; Tam, J.P.; Sze, S.K. Quantitative profiling of the rat heart myoblast secretome reveals differential responses to hypoxia and re-oxygenation stress. J. Proteom. 2014, 98, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Pedersenm, B.K. Muscle as a secretory organ. Compr. Physiol. 2013, 3, 1337–1362. [Google Scholar]
- Planavila, A.; Redondo-Angulo, I.; Villarroya, F. FGF21 and cardiac physiopathology. Front. Endocrinol. 2015, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Passino, C.; Barison, A.; Vergaro, G.; Gabutti, A.; Borrelli, C.; Emdin, M.; Clerico, A. Markers of fibrosis, inflammation, and remodeling pathways in heart failure. Clin. Chim. Acta 2015, 443, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolás, J.M.; Fernández-Solà, J.; Fatjó, F.; Casamitjana, R.; Bataller, R.; Sacanella, E.; Tobías, E.; Badía, E.; Estruch, R. Increased circulating leptin levels in chronic alcoholism. Alcohol Clin. Exp. Res. 2001, 25, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Jin, X.; Crawford, B.H.; Cheng, H.; Saafir, T.B.; Wagner, M.B.; Yuan, Z.; Ding, G. Cardioprotection from oxidative stress in the newborn heart by activation of PPARγ is mediated by catalase. Free Radic. Biol. Med. 2012, 53, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.G.; Cai, H.Q.; Li, Y.H.; Sui, Y.B.; Zhang, J.S.; Chang, J.R.; Ning, M.; Wu, Y.; Tang, C.S.; Qi, Y.F.; et al. Ghrelin protects heart against ERS-induced injury and apoptosis by activating AMP-activated protein kinase. Peptides 2013, 48, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Virdis, A.; Lerman, L.O.; Regoli, F.; Ghiadoni, L.; Lerman, A.; Taddei, S. Human ghrelin: A gastric hormone with cardiovascular properties. Curr. Pharm. Des. 2016, 22, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef] [PubMed]
- Bernatoniene, J.; Kopustinskiene, D.M.; Jakstas, V.; Majiene, D.; Baniene, R.; Kuršvietiene, L.; Masteikova, R.; Savickas, A.; Toleikis, A.; Trumbeckaite, S. The effect of Leonurus cardiaca herb extract and some of its flavonoids on mitochondrial oxidative phosphorylation in the heart. Planta Med. 2014, 80, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.E.; Bonacasa, B.; Ishii, T.; Siow, R.C. Targeting the redox sensitive Nrf2-Keap1 defense pathway in cardiovascular disease: Protection afforded by dietary isoflavones. Curr. Opin. Pharmacol. 2009, 9, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Vidavalur, R.; Otani, H.; Singal, P.K.; Maulik, N. Significance of wine and resveratrol in cardiovascular disease: French paradox revisited. Exp. Clin. Cardiol. 2006, 11, 217–225. [Google Scholar] [PubMed]
- Vasanthi, H.R.; Parameswari, R.P.; DeLeiris, J.; Das, D.K. Health benefits of wine and alcohol from neuroprotection to heart health. Front. Biosci. 2012, 4, 1505–1512. [Google Scholar] [CrossRef]
- Krga, I.; Milenkovic, D.; Morand, C.; Monfoulet, L.E. An update on the role of nutrigenomic modulations in mediating the cardiovascular protective effect of fruit polyphenols. Food Funct. 2016, 7, 3656–3676. [Google Scholar] [CrossRef] [PubMed]
- Tresserra-Rimbau, A.; Rimm, E.B.; Medina-Remón, A.; Martínez-González, M.A.; de la Torre, R.; Corella, D.; Salas-Salvadó, J.; Gómez-Gracia, E.; Lapetra, J.; Arós, F.; et al. Inverse association between habitual polyphenol intake and incidence of cardiovascular events in the PREDIMED study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Cordova, A.C.; Sumpio, B.E. Polyphenols are medicine: Is it time to prescribe red wine for our patients? Int. J. Angiol. 2009, 18, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Bloomekatz, J.; Galvez-Santisteban, M.; Chi, N.C. Myocardial plasticity: Cardiac development, regeneration and disease. Curr. Opin. Genet. Dev. 2016, 4, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Leor, J.; Palevski, D.; Amit, U.; Konfino, T. Macrophages and regeneration: Lessons from the heart. Semin. Cell Dev. Biol. 2016, 58, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Rubin, N.; Harrison, M.R.; Krainock, M.; Kim, R.; Lien, C.L. Recent advancements in understanding endogenous heart regeneration-insights from adult zebrafish and neonatal mice. Semin. Cell Dev. Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Lluís, M.; Fernández-Solà, J.; Castellví-Bel, S.; Sacanella, E.; Estruch, R.; Urbano-Márquez, A. Evaluation of myocyte proliferation in alcoholic cardiomyopathy: Telomerase enzyme activity (TERT) compared with Ki-67 expression. Alcohol Alcohol. 2011, 46, 534–541. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Bicknell, K.A.; Patel, K.; Brooks, G. Developmental expression of myostatin in cardiomyocytes and its effect on faetal and neonatal rat cardiomyocyte proliferation. Cardiovasc. Res. 2007, 74, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Lu, M.J.; Wang, B.W.; Sun, H.Y.; Chang, H. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur. J. Clin. Investig. 2006, 36, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S. Cardiac telocytes in normal and diseased hearts. Semin. Cell Dev. Biol. 2016, 55, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Gerbin, K.A.; Murry, C.E. The winding road to regenerating the human heart. Cardiovasc. Pathol. 2015, 24, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Behfar, A.; Crespo-Diaz, R.; Terzic, A.; Gerhrs, B.J. Cell therapy for cardiac repair—Lessons from clinical trials. Nat. Rev. Cardiol. 2014, 11, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, A. Cardiac cell therapy—Mixed results from mixed cells. N. Engl. J. Med. 2006, 355, 1274–1277. [Google Scholar] [CrossRef] [PubMed]
- Suerder, D.; Manka, R.; Moccetti, T.; Lo Cicero, V.; Emmert, M.Y.; Klersy, C.; Soncin, S.; Turchetto, L.; Radrizzani, M.; Zuber, M.; et al. The effect of bone marrow derived mononuclear cell treatment, early or late after acute myocardial infarction: Twelve months CMR and long-term clinical results. Circ. Res. 2016, 119, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Singh, A.; Sen, D. Mesenchymal stem cells in cardiac regeneration: A detailed progress report of the last 6 years (2010–2015). Stem. Cell Res. Ther. 2016, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Psaltis, P.J.; Schwarz, N.; Toledo-Flores, D.; Nicholls, S.J. Cellular therapy for heart failure. Curr. Cardiol. Rev. 2016. [Google Scholar] [CrossRef]
- Madonna, R.; van Laake, L.W.; Davidson, S.M.; Engel, F.B.; Hausenloy, D.J.; Lecour, S.; Leor, J.; Perrino, C.; Schulz, R.; Ytrehus, K.; et al. Position paper of the European Society of Cardiology Working Group Cellular Biology of the Heart: Cell-based therapies for myocardial repair and regeneration in ischemic heart disease and heart failure. Eur. Heart J. 2016, 37, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Brinkley, D.M.; Novak, E.; Topkara, V.K.; Geltman, E.M. Graft survival after cardiac transplantation for alcohol cardiomyopathy. Transplantation 2014, 98, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Lippai, D. Converging actions of alcohol on liver and brain immune signaling. Int. Rev. Neurobiol. 2014, 118, 359–380. [Google Scholar] [PubMed]
- Wang, H.J.; Zakhari, S.; Jung, M.K. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J. Gastroenterol. 2010, 16, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Pachunka, J.M.; Mott, J.L. Role of microRNAs in alcohol-induced multi-organ injury. Biomolecules 2015, 5, 3309–3338. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J. Application of mitochondria-targeted pharmaceuticals for the treatment of heart disease. Curr. Pharm. Des. 2016. [Google Scholar] [CrossRef]
- Panchenko, L.F.; Moiseev, V.S.; Pirozhkov, S.V.; Terebilina, N.N.; Naumova, T.A.; Baronets, V.; Goncharov, A.S. Blood content of markers of inflammation and cytokines in patients with alcoholic cardiomyopathy and ischemic heart disease at various stages of heart failure. Kardiologiia 2015, 55, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ye, D.; Wang, Y. Caspase-3 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2013, 1, 255–263. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Mechanisms | Effectors |
|---|---|
| Interference with cell signaling and calcium transients [16,17] | MAPK, TGF-β, PKC, PPARγ, MMPs, NF-κβ, PAI-1 |
| Decrease in excitation-contraction coupling mechanisms [17,18,19] | intracellular [Ca]2+ transients, L-type Ca2+ channel |
| Induction of oxidative damage [20,21] | ROS, SOD, acetaldehyde |
| Pro-inflammatory effect [22] | IL-2, TNF-α, NF-κβ |
| Induction of apoptosis [23,24] | FAS, TNF-α, TGF-β, Bax-Bcl-2, caspases 3,6 |
| Induction of fibrosis [25] | TLR-4, TGF-β |
| Protein-adduct formation [26] | protein-ethanol-adducts |
| malondialdehyde-DNA adducts | |
| Disruption in protein synthesis [27] | decrease in ribosomal protein synthesis, actin, myosin, troponin, titin |
| Increased glycogen deposition [28,29] | glycogen synthase kinase-3β, PARP |
| Renin-angiotensin-aldosterone activation [30] | renin, angiotensin, aldosterone, p38 MAPK/Smad |
| Interference in hormone-growth factors [31,32] | myostatin, ghrelin, leptin, IGF-1 |
| Interference in regulatory cardiomyokines [33,34] | FGF21 |
| Decrease in myocyte regeneration [35] | myostatin, IGF-1 |
| Impairment of extracellular matrix turnover [16] | cytoskeletal structure, connexin channel, desmosome contacts |
| Imbalance between cardiac lesions/repair mechanisms [9] | cell apoptosis and necrosis increased myocardial fibrosis decreased myocyte regeneration |
| Short-Term Effects on the Heart | Long-Term Effects on the Heart | Long-Term Effects on the Vascular System | ||
|---|---|---|---|---|
| Dysfunction of cardiac contractility | Ventricular dysfunction | Diastolic dysfunction | Increased systemic atherosclerosis | |
| Systolic dysfunction | ||||
| Acute arrhythmias supraventricular ventricular (holiday heart syndrome) | Atrial dysfunction | Arterial hypertension | ||
| Arterial hypertension | Chronic arrhythmias | Peripheral artery disease | ||
| Transitory ischemic cerebral attack | Alcoholic cardiomyopathy | Subclinical cardiomyopathy | Changes in lipid profile | Increase in LDL cholesterol |
| Clinical congestive heart failure | Increase in triglycerides | |||
| Low-output dilated cardiomyopathy | ||||
| Sudden death | Coronary heart disease | Angina | Increased risk of diabetes | |
| Myocardial infarction | ||||
| Increased cardiovascular mortality | Interference with other cardiotoxic drugs (tobacco, cocaine) | |||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Solà, J.; Planavila Porta, A. New Treatment Strategies for Alcohol-Induced Heart Damage. Int. J. Mol. Sci. 2016, 17, 1651. https://doi.org/10.3390/ijms17101651
Fernández-Solà J, Planavila Porta A. New Treatment Strategies for Alcohol-Induced Heart Damage. International Journal of Molecular Sciences. 2016; 17(10):1651. https://doi.org/10.3390/ijms17101651
Chicago/Turabian StyleFernández-Solà, Joaquim, and Ana Planavila Porta. 2016. "New Treatment Strategies for Alcohol-Induced Heart Damage" International Journal of Molecular Sciences 17, no. 10: 1651. https://doi.org/10.3390/ijms17101651