
Intracellular Protein Shuttling: A Mechanism Relevant for Myelin Repair in Multiple Sclerosis?


Abstract
:1. Introduction
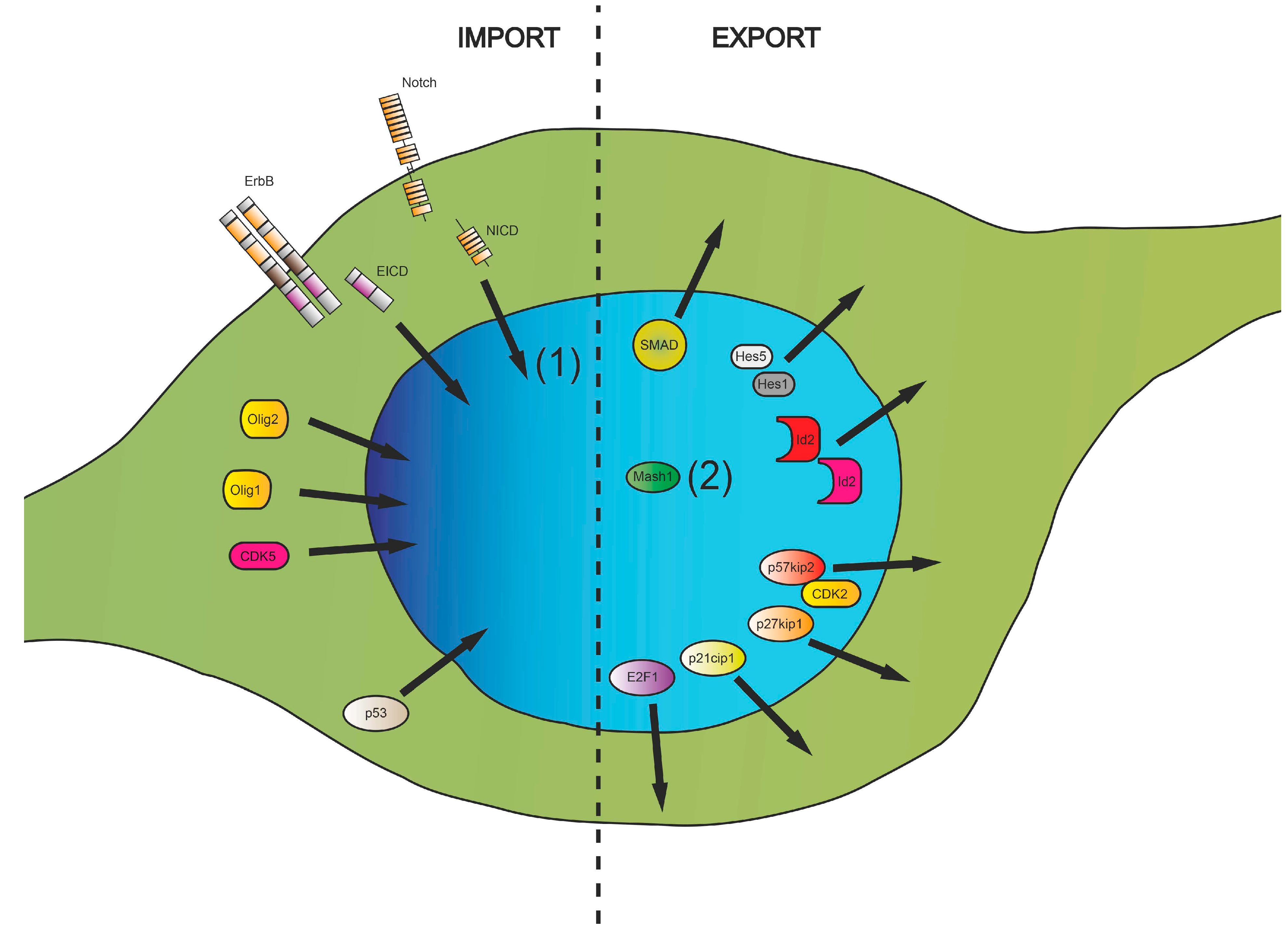
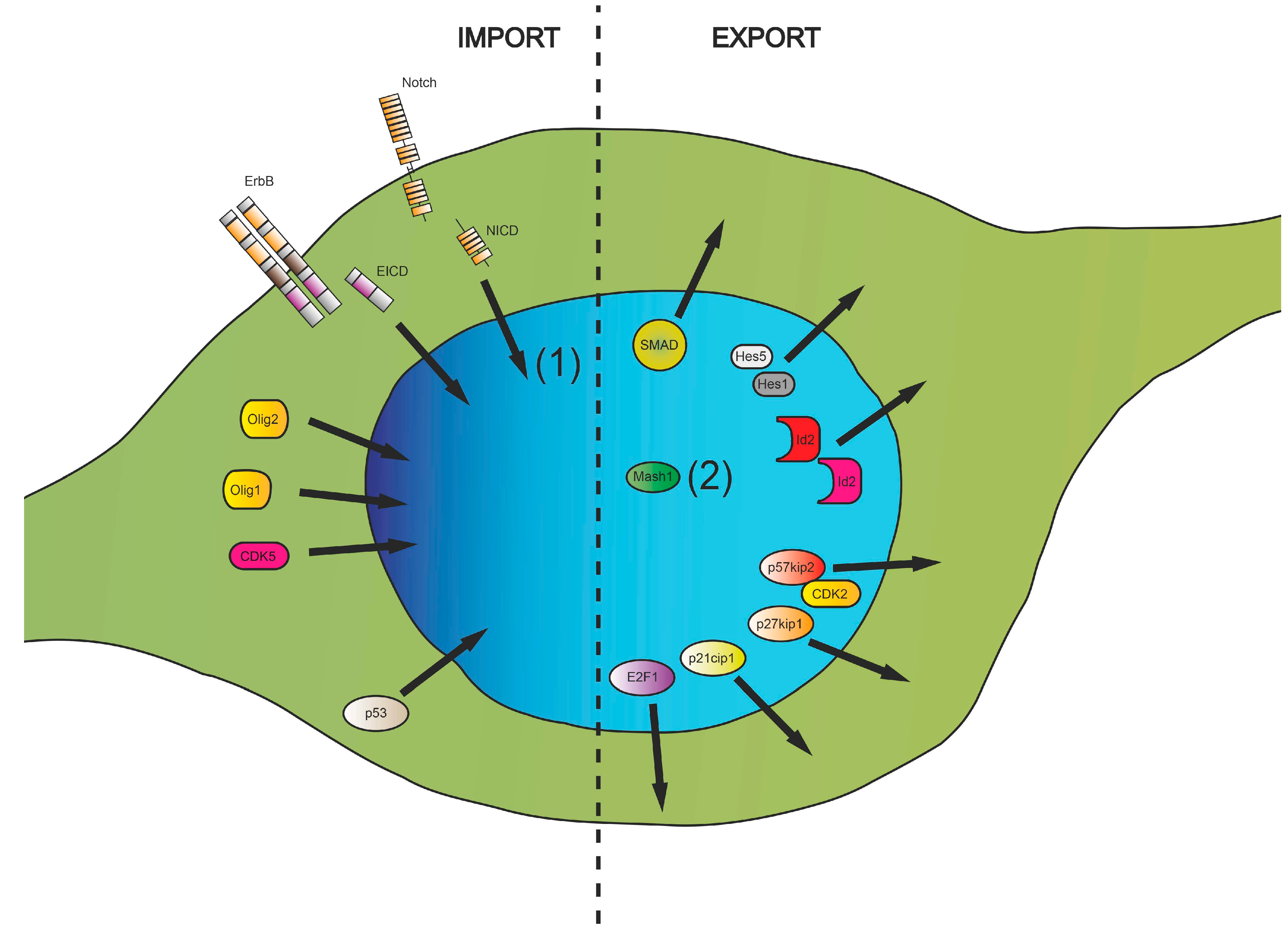
2. Intracellular Protein Shuttling—A Mechanism Involved in Neurodegenerative Diseases?
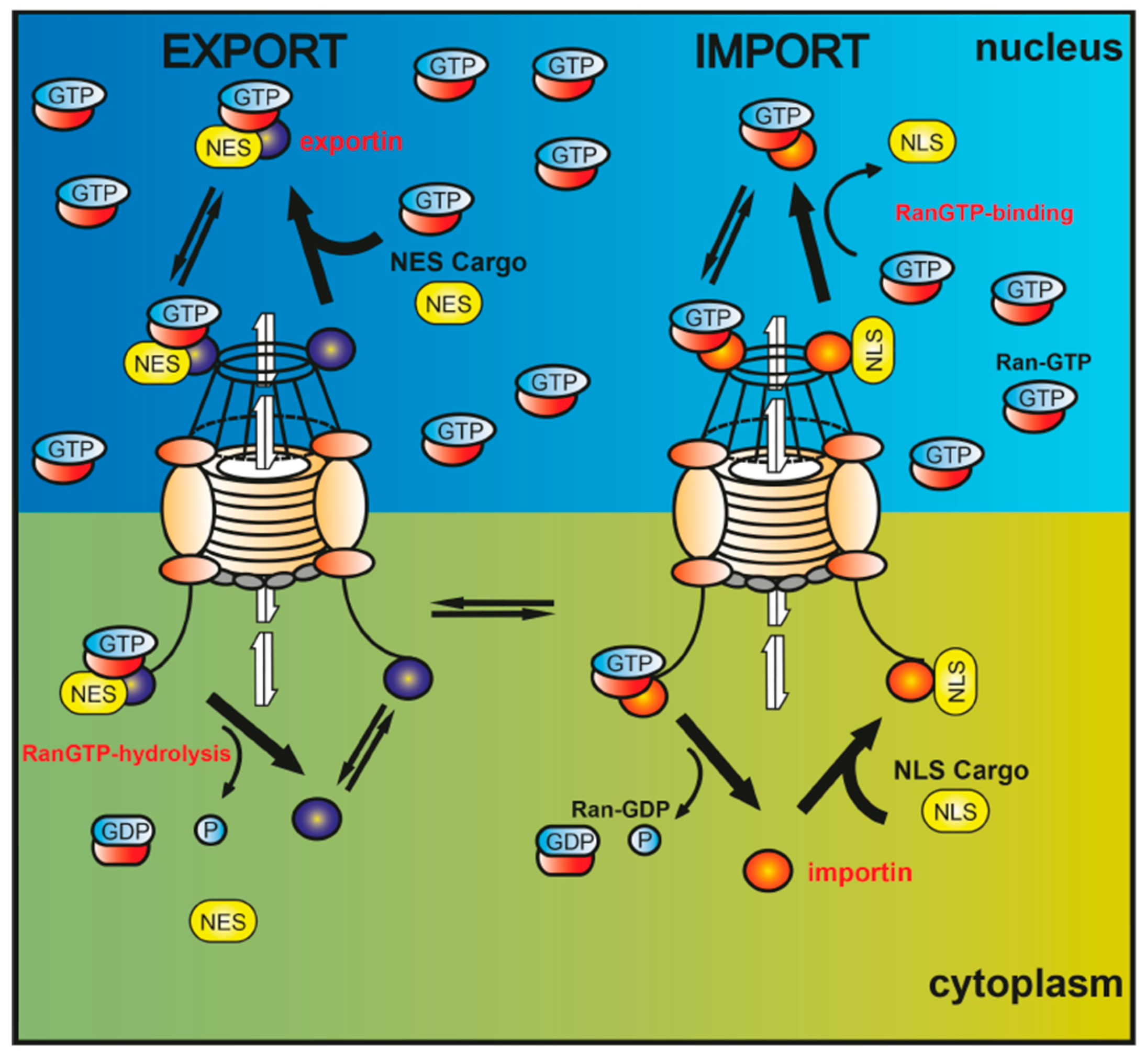
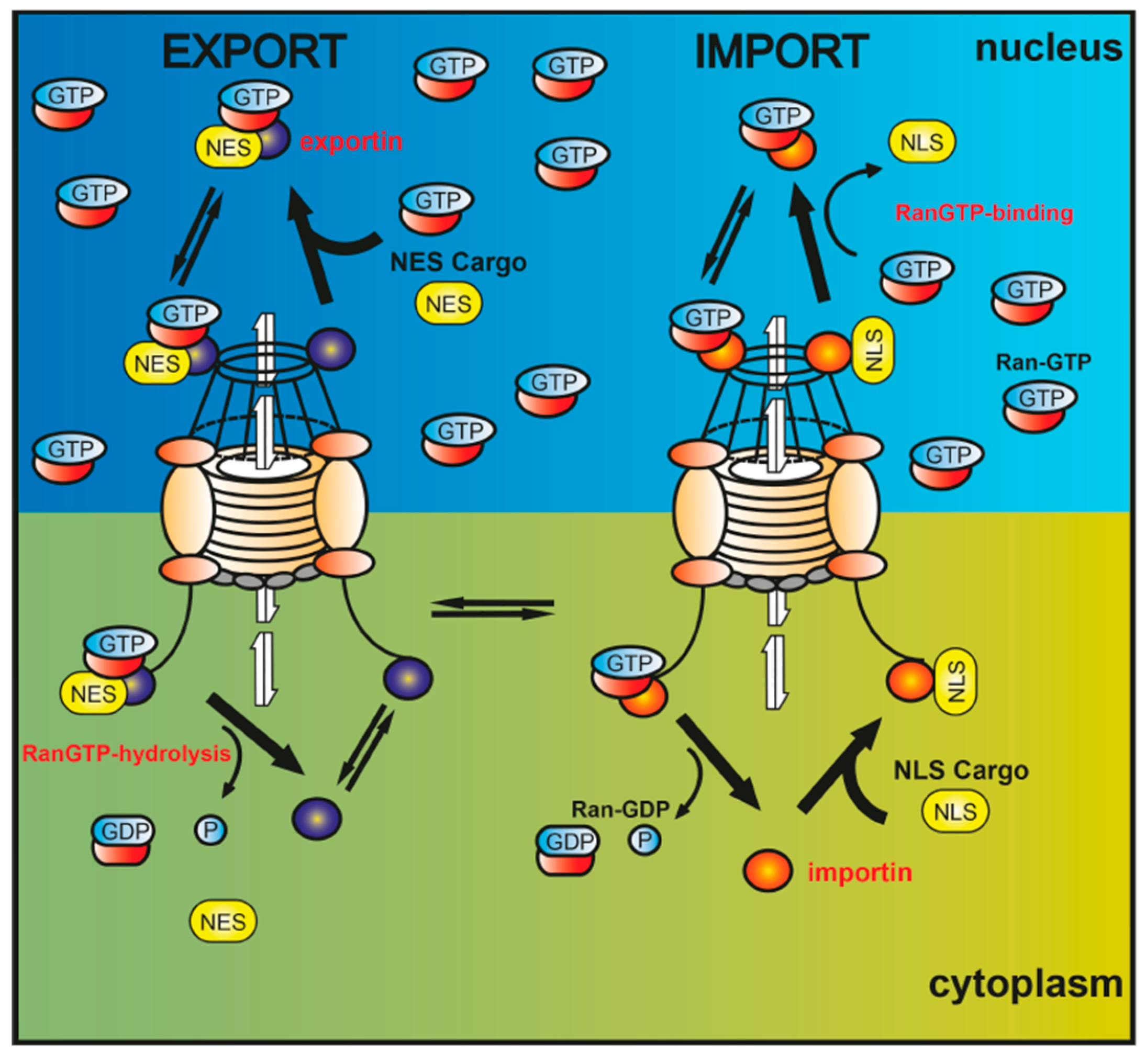
3. Nucleocytoplasmic Translocation Activities of Myelinating Glial Cells
3.1. Cell Cycle-Associated Proteins
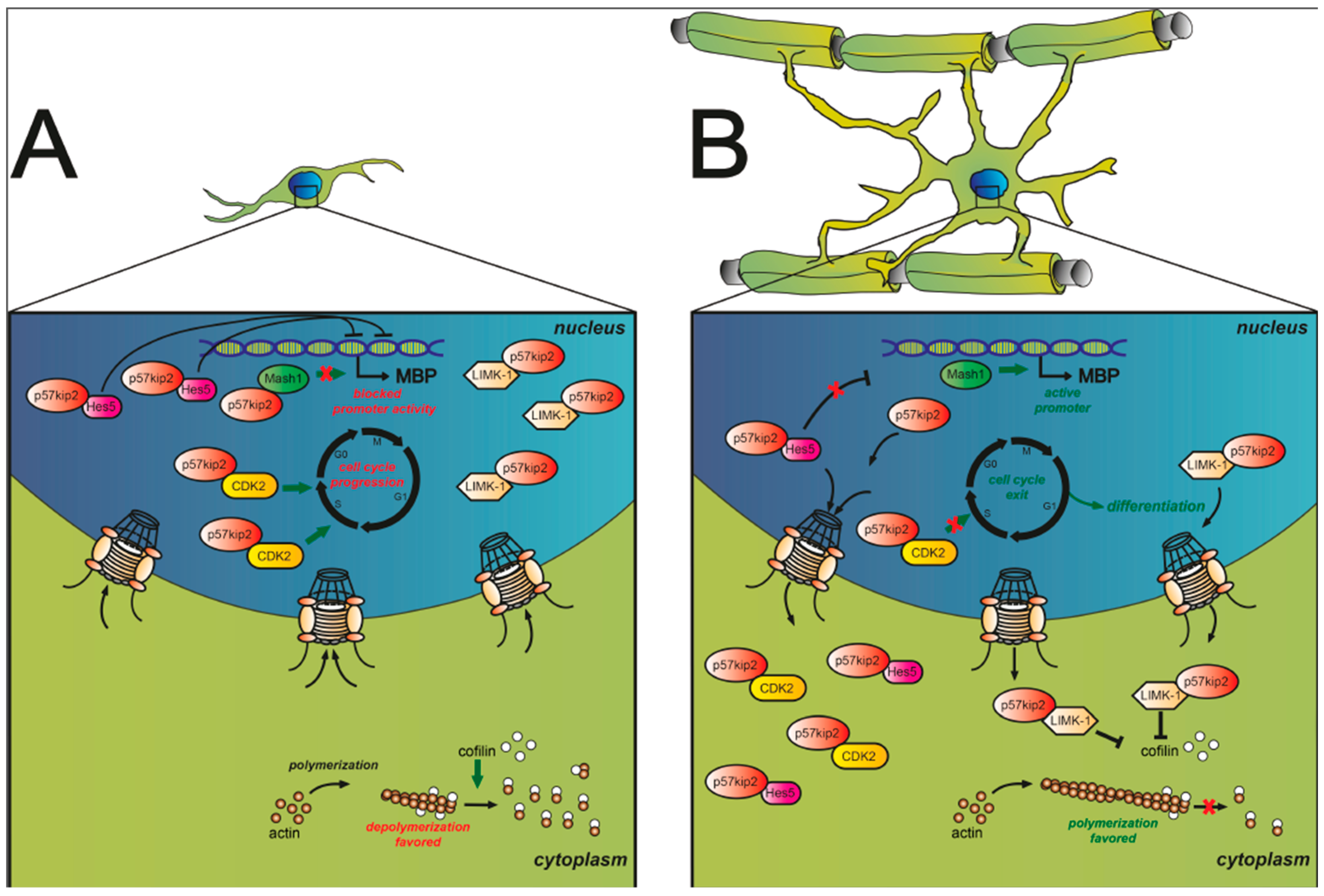

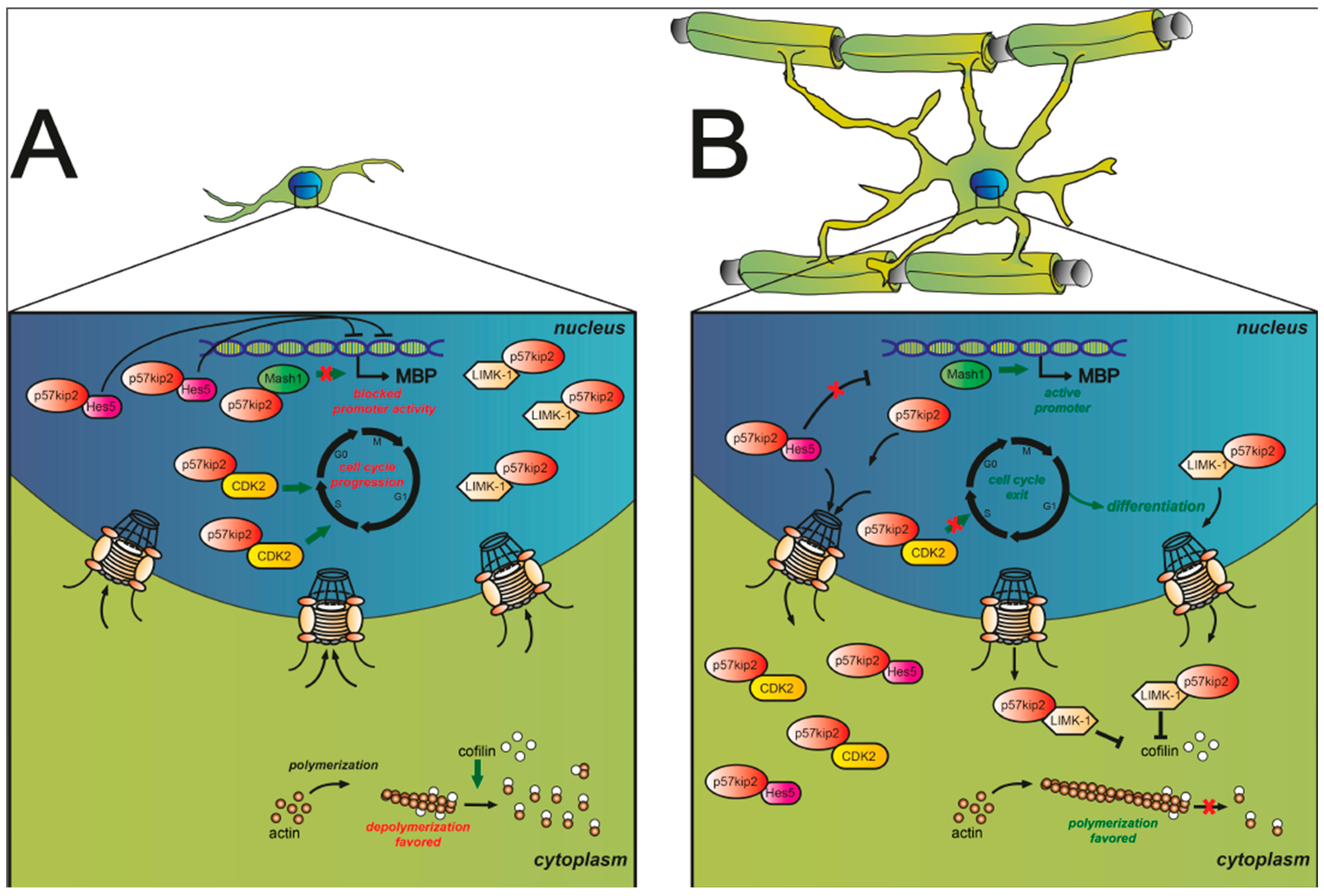
{kind=link}
{kind=link}
{kind=link}
| Molecules | Major Function in OPCs | Role in MS/MS-Models | References |
|---|---|---|---|
| CDK5 | migration, differentiation and myelination, mRNA transport | – | Miyamoto et al., 2007/2008 [59,60]; Yang et al., 2013 [57]; Zhou et al., 2015 [56] |
| CDK2 | cell cycle progression | LPC: alters adult OPCs renewal, differentiation, remyelination | Malumbres et al., 2005 [53]; Caillava et al., 2011 [49]; Göttle et al., 2015 [55] |
| E2F1 | modulates chromatin components during transition from proliferation to differentiation | – | Magri et al., 2014 [65] |
| p21cip1 | proliferation, differentiation | – | Ghiani et al., 1999 [50] |
| p53 | proliferation, differentiation | MS lesions: apoptosis | Eizenberg et al., 1996 [77]; Wosik et al., 2003 [80] |
| p27kip1 | proliferation | LPC: proliferative response | Crockett et al., 2005 [76]; Raff et al., 2007 [70]; Durand et al., 1997/1998 [71,74]; Miskimis et al., 2002 [72]; Tamaki et al., 2004 [73] |
| p57kip2 | glial fate decision, differentiation | MS lesions: myelin repair | Kremer et al., 2009 [85]; Jadasz et al., 2012 [84]; Pfeifenbring et al., 2013 [90]; Göttle et al., 2015 [55] |
3.2. Transcriptional Regulators
| Molecules | Major Function in OPCs | Role in MS/MS-Models | References |
|---|---|---|---|
| Olig1/2 | lineage determination, differentiation | cuprizone, MS lesion: activation of OPCs, remyelination | Arnett et al., 2004 [17]; Balabanov et al., 2005 [101]; Cheng et al., 2015 [102] |
| Ascl1/Mash1 | OPC specification, differentiation, myelination | LPC, MS lesions: oligodendrogenesis, promoted remyelination | Gokhan et al., 2005 [109]; Parras et al., 2007 [106]; Göttle et al., 2015 [55]; Sugimori et al., 2008 [107]; Nakatani et al., 2013 [108] |
| GPR17 Id2/4 | inhibited differentiation | LPC, MS lesions: diminished remyelination | Lecca et al., 2008 [115]; Chen et al., 2009 [102] |
| SMAD | OPC specification, inhibited differentiation | cuprizone, MS lesion: diminished remyelination, astrogenesis/gliosis | Grinspan et al., 2000 [130]; Kondo et al., 2004 [129]; Setoguchi et al., 2004 [104]; Fuller et al., 2007 [134]; Ara et al., 2008 [135]; See et al., 2009 [127]; Sabo et al., 2011 [131]; Wang et al., 2011 [132] |
| Notch Hes1/5 | inhibited differentiation | EAE, TMEV-IDD, MS lesion: activation and differentiation of T-helper cells, diminished remyelination | Jarriault et al., 1998 [141]; Wang et al., 1998 [132]; John et al., 2002 [144]; Hu et al., 2003 [143]; Liu et al., 2006 [148]; Elayman et al., 2009 [145]; Nakahara et al., 2009 [47]; Tsugane et al., 2012 [146] |
3.3. Posttranscriptional and Posttranslational Factors
3.4. Nuclear Translocation of Membrane Proteins
| Molecules | Major Function in OPCs | Role in MS/MS-Models | References |
|---|---|---|---|
| NRG | survival, differentiation, myelination | LPC, MS lesions: oligodendrogenesis, immune cells | Barres et al., 1999 [165]; Fernandez et al., 2000 [163]; Lai et al., 2004 [164]; Brinkmann et al., 2008 [168]; Tynyakov-Samra et al., 2011 [169]; Gauthier et al., 2013 [167] |
| RXRs | differentiation | LPC, MS lesions: remyelination | Schrage et al., 2006 [175]; Huang et al., 2011 [176] |
| PPARs | differentiation | SCI, EAE, MS lesions: oligodendrogenesis, immune cells | Saluja 2001 et al., [177]; Woods et al., 2003 [178]; Almad et al., 2010 [180]; Bernardo et al., 2013 [179]; Szalardy et al., 2013 [182]; Unoda et al., 2013 [181] |
3.5. RNA Transport
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Waxman, S.G. Demyelination in spinal cord injury and multiple sclerosis: What can we do to enhance functional recovery? J. Neurotrauma 1992, 9 (Suppl. 1), S105–S117. [Google Scholar] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Bruck, W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000, 123 Pt 6, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Antel, J.; Antel, S.; Caramanos, Z.; Arnold, D.L.; Kuhlmann, T. Primary progressive multiple sclerosis: Part of the MS disease spectrum or separate disease entity? Acta Neuropathol. 2012, 123, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Pomeroy, I.M.; Jordan, E.K.; Frank, J.A.; Matthews, P.M.; Esiri, M.M. Focal and diffuse cortical degenerative changes in a marmoset model of multiple sclerosis. Mult. Scler. 2010, 16, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.; Esiri, M.M.; Chance, S.A.; Palace, J.; Matthews, P.M. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006, 67, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Nishiyama, A.; Peterson, J.; Prineas, J.; Trapp, B.D. Ng2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J. Neurosci. 2000, 20, 6404–6412. [Google Scholar] [PubMed]
- Ffrench-Constant, C.; Raff, M.C. The oligodendrocyte-type-2 astrocyte cell lineage is specialized for myelination. Nature 1986, 323, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Ffrench-Constant, C.; Raff, M.C. Proliferating bipotential glial progenitor cells in adult rat optic nerve. Nature 1986, 319, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (Ng2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Reynolds, R.; Fawcett, J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Levine, J.M.; Reynolds, R. Activation and proliferation of endogenous oligodendrocyte precursor cells during ethidium bromide-induced demyelination. Exp. Neurol. 1999, 160, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Redwine, J.M.; Armstrong, R.C. In vivo proliferation of oligodendrocyte progenitors expressing pdgfalphar during early remyelination. J. Neurobiol. 1998, 37, 413–428. [Google Scholar] [CrossRef]
- Fancy, S.P.; Zhao, C.; Franklin, R.J. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult cns. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Zhao, C.; Penderis, J.; Franklin, R.J. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci. 2002, 22, 2451–2459. [Google Scholar] [PubMed]
- Arnett, H.A.; Fancy, S.P.; Alberta, J.A.; Zhao, C.; Plant, S.R.; Kaing, S.; Raine, C.S.; Rowitch, D.H.; Franklin, R.J.; Stiles, C.D. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the cns. Science 2004, 306, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hadzic, T.; Nishiyama, A. Transient up-regulation of Nkx2.2 expression in oligodendrocyte lineage cells during remyelination. Glia 2004, 46, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Vana, A.C.; Lucchinetti, C.F.; Le, T.Q.; Armstrong, R.C. Myelin transcription factor 1 (Myt1) expression in demyelinated lesions of rodent and human CNS. Glia 2007, 55, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.; Dupree, J.L.; Mangin, J.M.; Gallo, V. A functional role for EGFR signaling in myelination and remyelination. Nat. Neurosci. 2007, 10, 990–1002. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Gallo, V. The translational biology of remyelination: Past, present, and future. Glia 2014, 62, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Bruce, C.C.; Zhao, C.; Franklin, R.J. Remyelination—An effective means of neuroprotection. Horm. Behav. 2010, 57, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.J.; Steffenhagen, C.; Kremer, D.; Kandasamy, M.; Sandner, B.; Couillard-Despres, S.; Weidner, N.; Küry, P.; Aigner, L. Deciphering the oligodendrogenic program of neural progenitors: Cell intrinsic and extrinsic regulators. Stem Cells Dev. 2010, 19, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Aktas, O.; Hartung, H.P.; Küry, P. The complex world of oligodendroglial differentiation inhibitors. Ann. Neurol. 2011, 69, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.; Dawson, M.; Papadopoulos, D.; Polito, A.; di Bello, I.C.; Pham-Dinh, D.; Levine, J. The response of Ng2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J. Neurocytol. 2002, 31, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Kotter, M.R. The biology of CNS remyelination: The key to therapeutic advances. J. Neurol. 2008, 255 (Suppl. 1), 19–25. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.F.; Franklin, R.J. Remyelination in experimental models of toxin-induced demyelination. Curr. Top. Microbiol. Immunol. 2008, 318, 193–212. [Google Scholar] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wolswijk, G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J. Neurosci. 1998, 18, 601–609. [Google Scholar] [PubMed]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Farmer, A.; Chook, Y.M. Recognition of nuclear targeting signals by karyopherin-β proteins. Curr. Opin. Struct. Biol. 2010, 20, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; White, M.A.; Fontoura, B.M. Nuclear trafficking in health and disease. Curr. Opin. Cell Biol. 2014, 28, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ossareh-Nazari, B.; Bachelerie, F.; Dargemont, C. Evidence for a role of CRM1 in signal-mediated nuclear protein export. Science 1997, 278, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Kosugi, S.; Hasebe, M.; Tomita, M.; Yanagawa, H. Nuclear export signal consensus sequences defined using a localization-based yeast selection system. Traffic 2008, 9, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Corbett, A.H.; Silver, P.A. Nucleocytoplasmic transport of macromolecules. Microbiol. Mol. Biol. Rev. 1997, 61, 193–211. [Google Scholar] [PubMed]
- Gorlich, D.; Mattaj, I.W. Nucleocytoplasmic transport. Science 1996, 271, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2, a000562. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.C.; Link, W. Protein localization in disease and therapy. J. Cell Sci. 2011, 124, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ito, H.; Wate, R.; Ohnishi, S.; Nakano, S.; Kusaka, H. Altered distributions of nucleocytoplasmic transport-related proteins in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2006, 112, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Nagara, Y.; Tateishi, T.; Yamasaki, R.; Hayashi, S.; Kawamura, M.; Kikuchi, H.; Iinuma, K.M.; Tanaka, M.; Iwaki, T.; Matsushita, T.; et al. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1α in amyotrophic lateral sclerosis. Brain Pathol. 2013, 23, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, L.G.; Miskiewicz, H.B.; Tannenbaum, L.B.; Mirra, S.S. Nuclear pore complex proteins in alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.C.; Chen, H.M.; Lin, J.T.; Chang, C.P.; Chang, W.C.; Kang, J.J.; Sun, C.P.; Tao, M.H.; Tu, P.H.; Chang, C.; et al. Nuclear translocation of AMPK-α1 potentiates striatal neurodegeneration in huntington’s disease. J. Cell Biol. 2011, 194, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zou, F.; Fu, H.; Cui, G.; Yan, Y.; Wu, Q.; Gu, X. Up-regulation of CRM1 relates to neuronal apoptosis after traumatic brain injury in adult rats. J. Mol. Neurosci. MN 2013, 51, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, J.; Kanekura, K.; Nawa, M.; Aiso, S.; Suzuki, N. Abnormal expression of Tip30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J. Clin. Investig. 2009, 119, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Shen, S.; Dietz, K.; He, Y.; Howell, O.; Reynolds, R.; Casaccia, P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 2010, 13, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Caillava, C.; Baron-Van Evercooren, A. Differential requirement of cyclin-dependent kinase 2 for oligodendrocyte progenitor cell proliferation and differentiation. Cell Div. 2012, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Ghiani, C.; Gallo, V. Inhibition of cyclin e-cyclin-dependent kinase 2 complex formation and activity is associated with cell cycle arrest and withdrawal in oligodendrocyte progenitor cells. J. Neurosci. 2001, 21, 1274–1282. [Google Scholar] [PubMed]
- Frederick, T.J.; Min, J.; Altieri, S.C.; Mitchell, N.E.; Wood, T.L. Synergistic induction of cyclin d1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia 2007, 55, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Frederick, T.J.; Wood, T.L. IGF-I synergizes with FGF-2 to stimulate oligodendrocyte progenitor entry into the cell cycle. Dev. Biol. 2001, 232, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Mammalian cyclin-dependent kinases. Trends Biochem. Sci. 2005, 30, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Caillava, C.; Vandenbosch, R.; Jablonska, B.; Deboux, C.; Spigoni, G.; Gallo, V.; Malgrange, B.; Baron-Van Evercooren, A. CDK2 loss accelerates precursor differentiation and remyelination in the adult central nervous system. J. Cell Biol. 2011, 193, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Göttle, P.; Sabo, J.K.; Heinen, A.; Venables, G.; Torres, K.; Tzekova, N.; Parras, C.M.; Kremer, D.; Hartung, H.P.; Cate, H.S.; et al. Oligodendroglial maturation is dependent on intracellular protein shuttling. J. Neurosci. 2015, 35, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, H.; Li, X.; Zhang, G.; Niu, Y.; Yuan, Z.; Herrup, K.; Zhang, Y.W.; Bu, G.; Xu, H.; et al. The roles of CDK5-mediated subcellular localization of FOXO1 in neuronal death. J. Neurosci. 2015, 35, 2624–2635. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Zhang, J.; Luo, F.; Herrup, K.; Bibb, J.A.; Lu, R.; Miller, R.H. Cyclin dependent kinase 5 is required for the normal development of oligodendrocytes and myelin formation. Dev. Biol. 2013, 378, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Pandey, J.P.; Smith, D.S. A CDK5-dependent switch regulates LIS1/NDEL1/dynein-driven organelle transport in adult axons. J. Neurosci. 2011, 31, 17207–17219. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamauchi, J.; Chan, J.R.; Okada, A.; Tomooka, Y.; Hisanaga, S.; Tanoue, A. CDK5 regulates differentiation of oligodendrocyte precursor cells through the direct phosphorylation of paxillin. J. Cell Sci. 2007, 120, 4355–4366. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamauchi, J.; Tanoue, A. CDK5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase fyn. J. Neurosci. 2008, 28, 8326–8337. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.A.; Vartanian, T.K. WAVE1 and regulation of actin nucleation in myelination. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Balbas, M.A.; Bauer, U.M.; Nielsen, S.J.; Brehm, A.; Kouzarides, T. Regulation of E2F1 activity by acetylation. EMBO J. 2000, 19, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N. The regulation of E2F by prb-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Magri, L.; Swiss, V.A.; Jablonska, B.; Lei, L.; Pedre, X.; Walsh, M.; Zhang, W.; Gallo, V.; Canoll, P.; Casaccia, P. E2F1 coregulates cell cycle genes and chromatin components during the transition of oligodendrocyte progenitors from proliferation to differentiation. J. Neurosci. 2014, 34, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Casaccia-Bonnefil, P.; Tikoo, R.; Kiyokawa, H.; Friedrich, V., Jr.; Chao, M.V.; Koff, A. Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27kip1. Genes Dev. 1997, 11, 2335–2346. [Google Scholar] [CrossRef] [PubMed]
- Ghiani, C.A.; Eisen, A.M.; Yuan, X.; DePinho, R.A.; McBain, C.J.; Gallo, V. Neurotransmitter receptor activation triggers p27(kip1)and p21(cip1) accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development 1999, 126, 1077–1090. [Google Scholar] [PubMed]
- Raff, M. Intracellular developmental timers. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Gao, F.B.; Raff, M. Accumulation of the cyclin-dependent kinase inhibitor p27/kip1 and the timing of oligodendrocyte differentiation. EMBO J. 1997, 16, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Miskimins, R.; Srinivasan, R.; Marin-Husstege, M.; Miskimins, W.K.; Casaccia-Bonnefil, P. P27(kip1) enhances myelin basic protein gene promoter activity. J. Neurosci. Res. 2002, 67, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Tokumoto, Y. Over-expression of cyclin dependent kinase inhibitor p27/kip1 increases oligodendrocyte differentiation from induced pluripotent stem cells. In Vitro Cell. Dev. Biol. Anim. 2014, 50, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Durand, B.; Fero, M.L.; Roberts, J.M.; Raff, M.C. P27kip1 alters the response of cells to mitogen and is part of a cell-intrinsic timer that arrests the cell cycle and initiates differentiation. Curr. Biol. 1998, 8, 431–440. [Google Scholar] [CrossRef]
- Shen, A.; Liu, Y.; Zhao, J.; Qin, J.; Shi, S.; Chen, M.; Gao, S.; Xiao, F.; Lu, Q.; Cheng, C. Temporal-spatial expressions of p27kip1 and its phosphorylation on serine-10 after acute spinal cord injury in adult rat: Implications for post-traumatic glial proliferation. Neurochem. Int. 2008, 52, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Crockett, D.P.; Burshteyn, M.; Garcia, C.; Muggironi, M.; Casaccia-Bonnefil, P. Number of oligodendrocyte progenitors recruited to the lesioned spinal cord is modulated by the levels of the cell cycle regulatory protein p27kip-1. Glia 2005, 49, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Eizenberg, O.; Faber-Elman, A.; Gottlieb, E.; Oren, M.; Rotter, V.; Schwartz, M. P53 plays a regulatory role in differentiation and apoptosis of central nervous system-associated cells. Mol. Cell. Biol. 1996, 16, 5178–5185. [Google Scholar] [PubMed]
- Franklin, D.S.; Godfrey, V.L.; Lee, H.; Kovalev, G.I.; Schoonhoven, R.; Chen-Kiang, S.; Su, L.; Xiong, Y. Cdk inhibitors p18(ink4c) and p27(kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998, 12, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, Y.M.; Apperly, J.A.; Gao, F.B.; Raff, M.C. Posttranscriptional regulation of p18 and p27 CDK inhibitor proteins and the timing of oligodendrocyte differentiation. Dev. Biol. 2002, 245, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Wosik, K.; Antel, J.; Kuhlmann, T.; Bruck, W.; Massie, B.; Nalbantoglu, J. Oligodendrocyte injury in multiple sclerosis: A role for p53. J. Neurochem. 2003, 85, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Coqueret, O. New roles for p21 and p27 cell-cycle inhibitors: A function for each cell compartment? Trends Cell Biol. 2003, 13, 65–70. [Google Scholar] [CrossRef]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK inhibitors: Cell cycle regulators and beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Heinen, A.; Kremer, D.; Göttle, P.; Kruse, F.; Hasse, B.; Lehmann, H.; Hartung, H.P.; Küry, P. The cyclin-dependent kinase inhibitor p57kip2 is a negative regulator of schwann cell differentiation and in vitro myelination. Proc. Natl. Acad. Sci. USA 2008, 105, 8748–8753. [Google Scholar] [CrossRef] [PubMed]
- Jadasz, J.J.; Rivera, F.J.; Taubert, A.; Kandasamy, M.; Sandner, B.; Weidner, N.; Aktas, O.; Hartung, H.P.; Aigner, L.; Küry, P. p57kip2 regulates glial fate decision in adult neural stem cells. Development 2012, 139, 3306–3315. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Heinen, A.; Jadasz, J.; Göttle, P.; Zimmermann, K.; Zickler, P.; Jander, S.; Hartung, H.P.; Küry, P. p57kip2 is dynamically regulated in experimental autoimmune encephalomyelitis and interferes with oligodendroglial maturation. Proc. Natl. Acad. Sci. USA 2009, 106, 9087–9092. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Reynisdottir, I.; Massague, J. Cloning of p57kip2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995, 9, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Apostolopoulou, K.; Niforou, K.; Kotsinas, A.; Gorgoulis, V.G. p57kip2: “Kip”ing the cell under control. Mol. Cancer Res. 2009, 7, 1902–1919. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, T.; Toyoshima, H.; Miura, M.; Wang, Y.; Iida, K.T.; Suzuki, H.; Sone, H.; Shimano, H.; Gotoda, T.; Nishimori, S.; et al. p57kip2 regulates actin dynamics by binding and translocating lim-kinase 1 to the nucleus. J. Biol. Chem. 2003, 278, 52919–52923. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Barbayannis, F.A.; Hanser, H.; Schneider, C.; Stanyon, C.A.; Bernard, O.; Caroni, P. Regulation of actin dynamics through phosphorylation of cofilin by lim-kinase. Nature 1998, 393, 805–809. [Google Scholar] [PubMed]
- Pfeifenbring, S.; Metz, I.; Kremer, D.; Küry, P.; Hartung, H.P.; Brück, W. Oligodendroglial lineage cells express nuclear p57kip2 in multiple sclerosis lesions. Glia 2013, 61, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, Y.; Richardson, W.D.; Casaccia, P. Two-tier transcriptional control of oligodendrocyte differentiation. Curr. Opin. Neurobiol. 2009, 19, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.R.; Yuk, D.; Alberta, J.A.; Zhu, Z.; Pawlitzky, I.; Chan, J.; McMahon, A.P.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog—Regulated oligodendrocyte lineage genes encoding BHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef]
- Wu, M.; Hernandez, M.; Shen, S.; Sabo, J.K.; Kelkar, D.; Wang, J.; O’Leary, R.; Phillips, G.R.; Cate, H.S.; Casaccia, P. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J. Neurosci. 2012, 32, 6651–6664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, S.; Anderson, D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25, 331–343. [Google Scholar] [CrossRef]
- Lu, Q.R.; Sun, T.; Zhu, Z.; Ma, N.; Garcia, M.; Stiles, C.D.; Rowitch, D.H. Common developmental requirement for olig function indicates a motor neuron/oligodendrocyte connection. Cell 2002, 109, 75–86. [Google Scholar] [CrossRef]
- Takebayashi, H.; Nabeshima, Y.; Yoshida, S.; Chisaka, O.; Ikenaka, K. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr. Biol. 2002, 12, 1157–1163. [Google Scholar] [CrossRef]
- Li, H.; Lu, Y.; Smith, H.K.; Richardson, W.D. Olig1 and Sox10 interact synergistically to drive myelin basic protein transcription in oligodendrocytes. J. Neurosci. 2007, 27, 14375–14382. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Yue, T.; Ma, Z.; Wu, F.F.; Gow, A.; Lu, Q.R. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J. Neurosci. 2005, 25, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Frim, D.M.; Polak, P.; Vujicic, S.; Arnason, B.G.; Boullerne, A.I. Olig1 is expressed in human oligodendrocytes during maturation and regeneration. Glia 2011, 59, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Mei, F.; Wang, L.; Liu, S.; Tian, Y.; Mo, W.; Li, H.; Lu, Q.R.; Xiao, L. Phosphorylated Olig1 localizes to the cytosol of oligodendrocytes and promotes membrane expansion and maturation. Glia 2012, 60, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, R.; Popko, B. Myelin repair: Developmental myelination redux? Nat. Neurosci. 2005, 8, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Xue, X.; Fu, J. Effect of olig1 on the development of oligodendrocytes and myelination in a neonatal rat pvl model induced by hypoxia-ischemia. Mol. Med. Rep. 2015, 11, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Kondo, T.; Takebayashi, H.; Taga, T. Negative regulatory effect of an oligodendrocytic bhlh factor Olig2 on the astrocytic differentiation pathway. Cell Death Differ. 2004, 11, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, T.; Kondo, T. Nuclear export of Olig2 in neural stem cells is essential for ciliary neurotrophic factor-induced astrocyte differentiation. J. Cell Biol. 2004, 166, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Magnus, T.; Coksaygan, T.; Korn, T.; Xue, H.; Arumugam, T.V.; Mughal, M.R.; Eckley, D.M.; Tang, S.C.; Detolla, L.; Rao, M.S.; et al. Evidence that nucleocytoplasmic Olig2 translocation mediates brain-injury-induced differentiation of glial precursors to astrocytes. J. Neurosci. Res. 2007, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Parras, C.M.; Hunt, C.; Sugimori, M.; Nakafuku, M.; Rowitch, D.; Guillemot, F. The proneural gene Mash1 specifies an early population of telencephalic oligodendrocytes. J. Neurosci. 2007, 27, 4233–4242. [Google Scholar] [CrossRef] [PubMed]
- Sugimori, M.; Nagao, M.; Parras, C.M.; Nakatani, H.; Lebel, M.; Guillemot, F.; Nakafuku, M. Ascl1 is required for oligodendrocyte development in the spinal cord. Development 2008, 135, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, H.; Martin, E.; Hassani, H.; Clavairoly, A.; Maire, C.L.; Viadieu, A.; Kerninon, C.; Delmasure, A.; Frah, M.; Weber, M.; et al. Ascl1/Mash1 promotes brain oligodendrogenesis during myelination and remyelination. J. Neurosci. 2013, 33, 9752–9768. [Google Scholar] [CrossRef] [PubMed]
- Gokhan, S.; Marin-Husstege, M.; Yung, S.Y.; Fontanez, D.; Casaccia-Bonnefil, P.; Mehler, M.F. Combinatorial profiles of oligodendrocyte-selective classes of transcriptional regulators differentially modulate myelin basic protein gene expression. J. Neurosci. 2005, 25, 8311–8321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Armstrong, R.C.; v Agoston, D.; Robinsky, A.; Wiese, C.; Nagle, J.; Hudson, L.D. Myelin transcription factor 1 (Myt1) of the oligodendrocyte lineage, along with a closely related cchc zinc finger, is expressed in developing neurons in the mammalian central nervous system. J. Neurosci. Res. 1997, 50, 272–290. [Google Scholar] [CrossRef]
- Armstrong, R.C.; Kim, J.G.; Hudson, L.D. Expression of myelin transcription factor 1 (Myt1), a “zinc-finger” DNA-binding protein, in developing oligodendrocytes. Glia 1995, 14, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Samanta, J.; Kessler, J.A. Interactions between id and olig proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development 2004, 131, 4131–4142. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci. 2009, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sdrulla, A.; Johnson, J.E.; Yokota, Y.; Barres, B.A. A role for the helix-loop-helix protein ID2 in the control of oligodendrocyte development. Neuron 2001, 29, 603–614. [Google Scholar] [CrossRef]
- Lecca, D.; Trincavelli, M.L.; Gelosa, P.; Sironi, L.; Ciana, P.; Fumagalli, M.; Villa, G.; Verderio, C.; Grumelli, C.; Guerrini, U.; et al. The recently identified p2y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS ONE 2008, 3, e3579. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Bae, Y.K.; Muraoka, O.; Hibi, M. Interaction of Wnt and caudal-related genes in zebrafish posterior body formation. Dev. Biol. 2005, 279, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Baranzini, S.E.; Zhao, C.; Yuk, D.I.; Irvine, K.A.; Kaing, S.; Sanai, N.; Franklin, R.J.; Rowitch, D.H. Dysregulation of the wnt pathway inhibits timely myelination and remyelination in the mammalian cns. Genes Dev. 2009, 23, 1571–1585. [Google Scholar] [CrossRef] [PubMed]
- Lurbke, A.; Hagemeier, K.; Cui, Q.L.; Metz, I.; Bruck, W.; Antel, J.; Kuhlmann, T. Limited Tcf7l2 expression in MS lesions. PLoS ONE 2013, 8, e72822. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Liu, Y.I. Wnt signaling: Complexity at the surface. J. Cell Sci. 2006, 119, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Malbon, C.C.; Wang, H.Y. Dishevelled: A mobile scaffold catalyzing development. Curr. Top. Dev. Biol. 2006, 72, 153–166. [Google Scholar] [PubMed]
- Azim, K.; Butt, A.M. Gsk3beta negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Feigenson, K.; Reid, M.; See, J.; Crenshaw, E.B., 3rd.; Grinspan, J.B. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell. Neurosci. 2009, 42, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the β-catenin-tcf interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Harrington, E.P.; Yuen, T.J.; Silbereis, J.C.; Zhao, C.; Baranzini, S.E.; Bruce, C.C.; Otero, J.J.; Huang, E.J.; Nusse, R.; et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nat. Neurosci. 2011, 14, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.L.; Weis, W.I. Beta-catenin directly displaces groucho/tle repressors from TCF/LEF in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 2005, 12, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Labbe, E.; Letamendia, A.; Attisano, L. Association of SMADs with lymphoid enhancer binding factor 1/t cell-specific factor mediates cooperative signaling by the transforming growth factor-β and wnt pathways. Proc. Natl. Acad. Sci. USA 2000, 97, 8358–8363. [Google Scholar] [CrossRef] [PubMed]
- See, J.M.; Grinspan, J.B. Sending mixed signals: Bone morphogenetic protein in myelination and demyelination. J. Neuropathol. Exp. Neurol. 2009, 68, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Raff, M.C. A role for noggin in the development of oligodendrocyte precursor cells. Dev. Biol. 2004, 267, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Grinspan, J.B.; Edell, E.; Carpio, D.F.; Beesley, J.S.; Lavy, L.; Pleasure, D.; Golden, J.A. Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J. Neurobiol. 2000, 43, 1–17. [Google Scholar] [CrossRef]
- Sabo, J.K.; Aumann, T.D.; Merlo, D.; Kilpatrick, T.J.; Cate, H.S. Remyelination is altered by bone morphogenic protein signaling in demyelinated lesions. J. Neurosci. 2011, 31, 4504–4510. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, X.; He, Q.; Zheng, Y.; Kim, D.H.; Whittemore, S.R.; Cao, Q.L. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J. Neurosci. 2011, 31, 6053–6058. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, T.; Nakashima, K.; Takizawa, T.; Yanagisawa, M.; Ochiai, W.; Okabe, M.; Yone, K.; Komiya, S.; Taga, T. Treatment of spinal cord injury by transplantation of fetal neural precursor cells engineered to express BMP inhibitor. Exp. Neurol. 2004, 189, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.L.; DeChant, A.K.; Rothstein, B.; Caprariello, A.; Wang, R.; Hall, A.K.; Miller, R.H. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann. Neurol. 2007, 62, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Ara, J.; See, J.; Mamontov, P.; Hahn, A.; Bannerman, P.; Pleasure, D.; Grinspan, J.B. Bone morphogenetic proteins 4, 6, and 7 are up-regulated in mouse spinal cord during experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2008, 86, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Meyermann, R.; Schluesener, H. Detection of two transforming growth factor-β-related morphogens, bone morphogenetic proteins-4 and -5, in RNA of multiple sclerosis and creutzfeldt-jakob disease lesions. Acta Neuropathol. 1995, 90, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, K.; Yanagisawa, M.; Arakawa, H.; Kimura, N.; Hisatsune, T.; Kawabata, M.; Miyazono, K.; Taga, T. Synergistic signaling in fetal brain by Stat3-Smad1 complex bridged by p300. Science 1999, 284, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Wotton, D. Transcriptional control by the TGF-β/smad signaling system. EMBO J. 2000, 19, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Manzini, I.; Le, W.; Krieglstein, K.; Spittau, B. Thapsigargin-induced Ca2+ increase inhibits TGFβ1-mediated Smad2 transcriptional responses via Ca2+/calmodulin-dependent protein kinase II. J. Cell. Biochem. 2010, 111, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Jarriault, S.; le Bail, O.; Hirsinger, E.; Pourquie, O.; Logeat, F.; Strong, C.F.; Brou, C.; Seidah, N.G.; Isra l, A. Delta-1 activation of Notch-1 signaling results in HES-1 transactivation. Mol. Cell. Biol. 1998, 18, 7423–7431. [Google Scholar] [PubMed]
- Wang, S.; Sdrulla, A.D.; diSibio, G.; Bush, G.; Nofziger, D.; Hicks, C.; Weinmaster, G.; Barres, B.A. Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 1998, 21, 63–75. [Google Scholar] [CrossRef]
- Hu, Q.D.; Ang, B.T.; Karsak, M.; Hu, W.P.; Cui, X.Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.K.; et al. F3/contactin acts as a functional ligand for notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef]
- John, G.R.; Shankar, S.L.; Shafit-Zagardo, B.; Massimi, A.; Lee, S.C.; Raine, C.S.; Brosnan, C.F. Multiple sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat. Med. 2002, 8, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Bradshaw, E.M.; Wang, Y.; Oukka, M.; Kivisakk, P.; Chiba, S.; Yagita, H.; Khoury, S.J. Jagged1 and delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 5990–5998. [Google Scholar] [CrossRef] [PubMed]
- Tsugane, S.; Takizawa, S.; Kaneyama, T.; Ichikawa, M.; Yagita, H.; Kim, B.S.; Koh, C.S. Therapeutic effects of anti-δ1 mab on theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neuroimmunol. 2012, 252, 66–74. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Li, J.; Marin-Husstege, M.; Kageyama, R.; Fan, Y.; Gelinas, C.; Casaccia-Bonnefil, P. A molecular insight of HES5-dependent inhibition of myelin gene expression: Old partners and new players. EMBO J. 2006, 25, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Galiova, G.; Bartova, E.; Raska, I.; Krejci, J.; Kozubek, S. Chromatin changes induced by lamin a/c deficiency and the histone deacetylase inhibitor trichostatin A. Eur. J. Cell Biol. 2008, 87, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Sandoval, J.; Swiss, V.A.; Li, J.; Dupree, J.; Franklin, R.J.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Camelo, S.; Iglesias, A.H.; Hwang, D.; Due, B.; Ryu, H.; Smith, K.; Gray, S.G.; Imitola, J.; Duran, G.; Assaf, B.; et al. Transcriptional therapy with the histone deacetylase inhibitor trichostatin a ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 164, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Cavone, L.; Chiarugi, A. The therapeutic potential of hdac inhibitors in the treatment of multiple sclerosis. Mol. Med. 2011, 17, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Pedre, X.; Mastronardi, F.; Bruck, W.; Lopez-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Mastronardi, F.G.; Wood, D.D.; Mei, J.; Raijmakers, R.; Tseveleki, V.; Dosch, H.M.; Probert, L.; Casaccia-Bonnefil, P.; Moscarello, M.A. Increased citrullination of histone h3 in multiple sclerosis brain and animal models of demyelination: A role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J. Neurosci. 2006, 26, 11387–11396. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011, 25, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dai, J.; Ma, Y.; Mi, Y.; Cui, D.; Ju, G.; Macklin, W.B.; Jin, W. Dynamics of ten-eleven translocation hydroxylase family proteins and 5-hydroxymethylcytosine in oligodendrocyte differentiation. Glia 2014, 62, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, R.; Valentini, E.; Ciccarone, F.; Guastafierro, T.; Bacalini, M.G.; Ricigliano, V.A.; Zampieri, M.; Annibali, V.; Mechelli, R.; Franceschi, C.; et al. Tet2 gene expression and 5-hydroxymethylcytosine level in multiple sclerosis peripheral blood cells. Biochim. Biophys. Acta 2014, 1842, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.Y.; Hu, Q.D.; Tekaya, M.; Shimoda, Y.; Ang, B.T.; Nie, D.Y.; Sun, L.; Hu, W.P.; Karsak, M.; Duka, T.; et al. Nb-3/Notch1 pathway via deltex1 promotes neural progenitor cell differentiation into oligodendrocytes. J. Biol. Chem. 2004, 279, 25858–25865. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, C.F.; John, G.R. Revisiting Notch in remyelination of multiple sclerosis lesions. J. Clin. Investig. 2009, 119, 10–13. [Google Scholar] [CrossRef] [PubMed]
- El Omari, K.; Bird, L.E.; Nichols, C.E.; Ren, J.; Stammers, D.K. Crystal structure of cc3 (Tip30): Implications for its role as a tumor suppressor. J. Biol. Chem. 2005, 280, 18229–18236. [Google Scholar] [CrossRef] [PubMed]
- King, F.W.; Shtivelman, E. Inhibition of nuclear import by the proapoptotic protein cc3. Mol. Cell. Biol. 2004, 24, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xiao, L.; Li, C.; Liu, X.; Liu, M.; Shao, Q.; Wang, D.; Huang, A.; He, C. Tip30 inhibits oligodendrocyte precursor cell differentiation via cytoplasmic sequestration of Olig1. Glia 2015, 63, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.A.; Tang, D.G.; Cheng, L.; Prochiantz, A.; Mudge, A.W.; Raff, M.C. Evidence that axon-derived neuregulin promotes oligodendrocyte survival in the developing rat optic nerve. Neuron 2000, 28, 81–90. [Google Scholar] [CrossRef]
- Lai, C.; Feng, L. Implication of γ-secretase in neuregulin-induced maturation of oligodendrocytes. Biochem. Biophys. Res. Commun. 2004, 314, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Raff, M.C. Axonal control of oligodendrocyte development. J. Cell Biol. 1999, 147, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.Y.; Murphy, M.P.; Golde, T.E.; Carpenter, G. Gamma-secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001, 294, 2179–2181. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, M.K.; Kosciuczyk, K.; Tapley, L.; Karimi-Abdolrezaee, S. Dysregulation of the neuregulin-1-ErbB network modulates endogenous oligodendrocyte differentiation and preservation after spinal cord injury. Eur. J. Neurosci. 2013, 38, 2693–2715. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, B.G.; Agarwal, A.; Sereda, M.W.; Garratt, A.N.; Muller, T.; Wende, H.; Stassart, R.M.; Nawaz, S.; Humml, C.; Velanac, V.; et al. Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 2008, 59, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Tynyakov-Samra, E.; Auriel, E.; Levy-Amir, Y.; Karni, A. Reduced ErbB4 expression in immune cells of patients with relapsing remitting multiple sclerosis. Mult. Scler. Int. 2011, 2011, 561262. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yamada, T.; Wang, F.; Vulin, A.I.; Samuels, H.H. Novel roles of retinoid X receptor (RXR) and RXR ligand in dynamically modulating the activity of the thyroid hormone receptor/RXR heterodimer. J. Biol. Chem. 2004, 279, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.G.; Hanover, J.A.; Hager, G.L.; Cheng, S.Y. Hormone-induced translocation of thyroid hormone receptors in living cells visualized using a receptor green fluorescent protein chimera. J. Biol. Chem. 1998, 273, 27058–27063. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer, J.H.; Schwartz, H.L. Molecular basis of thyroid hormone-dependent brain development. Endocr. Rev. 1997, 18, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A.; Lazar, M.A.; Raff, M.C. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development 1994, 120, 1097–1108. [Google Scholar] [PubMed]
- Lefebvre, P.; Benomar, Y.; Staels, B. Retinoid x receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol. Metab. 2010, 21, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Schrage, K.; Koopmans, G.; Joosten, E.A.; Mey, J. Macrophages and neurons are targets of retinoic acid signaling after spinal cord contusion injury. Eur. J. Neurosci. 2006, 23, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.K.; Jarjour, A.A.; Nait Oumesmar, B.; Kerninon, C.; Williams, A.; Krezel, W.; Kagechika, H.; Bauer, J.; Zhao, C.; Evercooren, A.B.; et al. Retinoid X receptor γ signaling accelerates CNS remyelination. Nat. Neurosci. 2011, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Saluja, I.; Granneman, J.G.; Skoff, R.P. PPAR δ agonists stimulate oligodendrocyte differentiation in tissue culture. Glia 2001, 33, 191–204. [Google Scholar] [CrossRef]
- Woods, J.W.; Tanen, M.; Figueroa, D.J.; Biswas, C.; Zycband, E.; Moller, D.E.; Austin, C.P.; Berger, J.P. Localization of ppardelta in murine central nervous system: Expression in oligodendrocytes and neurons. Brain Res. 2003, 975, 10–21. [Google Scholar] [CrossRef]
- Bernardo, A.; de Simone, R.; de Nuccio, C.; Visentin, S.; Minghetti, L. The nuclear receptor peroxisome proliferator-activated receptor-γ promotes oligodendrocyte differentiation through mechanisms involving mitochondria and oscillatory Ca2+ waves. Biol. Chem. 2013, 394, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Almad, A.; McTigue, D.M. Chronic expression of PPAR-δ by oligodendrocyte lineage cells in the injured rat spinal cord. J. Comp. Neurol. 2010, 518, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Unoda, K.; Doi, Y.; Nakajima, H.; Yamane, K.; Hosokawa, T.; Ishida, S.; Kimura, F.; Hanafusa, T. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013, 256, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Tanczos, E.; Simu, M.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Elevated levels of PPAR-γ in the cerebrospinal fluid of patients with multiple sclerosis. Neurosci. Lett. 2013, 554, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, S.; Mir, F.; Gould, R.M.; le Breton, G.C. Characterization of thromboxane a2 receptor signaling in developing rat oligodendrocytes: Nuclear receptor localization and stimulation of myelin basic protein expression. J. Neurosci. Res. 2006, 84, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Mir, F.; le Breton, G.C. A novel nuclear signaling pathway for thromboxane a2 receptors in oligodendrocytes: Evidence for signaling compartmentalization during differentiation. Mol. Cell. Biol. 2008, 28, 6329–6341. [Google Scholar] [CrossRef] [PubMed]
- Boccaccio, G.L.; Carminatti, H.; Colman, D.R. Subcellular fractionation and association with the cytoskeleton of messengers encoding myelin proteins. J. Neurosci. Res. 1999, 58, 480–491. [Google Scholar] [CrossRef]
- Barbarese, E.; Koppel, D.E.; Deutscher, M.P.; Smith, C.L.; Ainger, K.; Morgan, F.; Carson, J.H. Protein translation components are colocalized in granules in oligodendrocytes. J. Cell Sci. 1995, 108 Pt 8, 2781–2790. [Google Scholar] [PubMed]
- Carson, J.H.; Kwon, S.; Barbarese, E. RNA trafficking in myelinating cells. Curr. Opin. Neurobiol. 1998, 8, 607–612. [Google Scholar] [CrossRef]
- Czaplinski, K.; Singer, R.H. Pathways for mRNA localization in the cytoplasm. Trends Biochem. Sci. 2006, 31, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Kiebler, M.A.; Bassell, G.J. Neuronal RNA granules: Movers and makers. Neuron 2006, 51, 685–690. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Gonsior, C.; Bauer, N.M.; Kramer-Albers, E.M.; Luhmann, H.J.; Trotter, J. Heterogeneous nuclear ribonucleoprotein (hnRNP) F is a novel component of oligodendroglial rna transport granules contributing to regulation of myelin basic protein (MBP) synthesis. J. Biol. Chem. 2012, 287, 1742–1754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lacroix, G.; Haines, J.; Doukhanine, E.; Almazan, G.; Richard, S. The QKI-6 RNA binding protein localizes with the MBP mRNAs in stress granules of Glial cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Levin, M. Novel somatic single nucleotide variants within the RNA binding protein hnRNP A1 in multiple sclerosis patients. F1000Research 2014, 3, 132. [Google Scholar] [CrossRef] [PubMed]
- Han, S.P.; Friend, L.R.; Carson, J.H.; Korza, G.; Barbarese, E.; Maggipinto, M.; Hatfield, J.T.; Rothnagel, J.A.; Smith, R. Differential subcellular distributions and trafficking functions of hnRNP A2/B1 spliceoforms. Traffic 2010, 11, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mrnp remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Nadezhdina, E.S.; Lomakin, A.J.; Shpilman, A.A.; Chudinova, E.M.; Ivanov, P.A. Microtubules govern stress granule mobility and dynamics. Biochim. Biophys. Acta 2010, 1803, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Pilotte, J.; Larocque, D.; Richard, S. Nuclear translocation controlled by alternatively spliced isoforms inactivates the quaking apoptotic inducer. Genes Dev. 2001, 15, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ozgen, H.; Kahya, N.; de Jonge, J.C.; Smith, G.S.; Harauz, G.; Hoekstra, D.; Baron, W. Regulation of cell proliferation by nucleocytoplasmic dynamics of postnatal and embryonic exon-II-containing MBP isoforms. Biochim. Biophys. Acta 2014, 1843, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, L.; Fidler, L.; Staugaitis, S.M.; Colman, D.R. The active transport of myelin basic protein into the nucleus suggests a regulatory role in myelination. Neuron 1997, 18, 579–589. [Google Scholar] [CrossRef]
- Smith, G.S.; Paez, P.M.; Spreuer, V.; Campagnoni, C.W.; Boggs, J.M.; Campagnoni, A.T.; Harauz, G. Classical 18.5-and 21.5-kDa isoforms of myelin basic protein inhibit calcium influx into oligodendroglial cells, in contrast to golli isoforms. J. Neurosci. Res. 2011, 89, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.S.; Samborska, B.; Hawley, S.P.; Klaiman, J.M.; Gillis, T.E.; Jones, N.; Boggs, J.M.; Harauz, G. Nucleus-localized 21.5-kDa myelin basic protein promotes oligodendrocyte proliferation and enhances neurite outgrowth in coculture, unlike the plasma membrane-associated 18.5-kDa isoform. J. Neurosci. Res. 2013, 91, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.T.; Heng, M.Y.; Ptacek, L.J.; Fu, Y.H. Regulation of myelination in the central nervous system by nuclear lamin b1 and non-coding rnas. Transl. Neurodegener. 2014, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.T.; Fu, Y.H. MIR-23 regulation of lamin b1 is crucial for oligodendrocyte development and myelination. Dis. Models Mech. 2009, 2, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.D.; Herbin, O.; de la Hera, B.; Vidaurre, O.G.; Moy, G.A.; Sun, Q.; Fung, H.Y.; Albrecht, S.; Alexandropoulos, K.; McCauley, D.; et al. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat. Neurosci. 2015, 18, 511–520. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Göttle, P.; Küry, P. Intracellular Protein Shuttling: A Mechanism Relevant for Myelin Repair in Multiple Sclerosis? Int. J. Mol. Sci. 2015, 16, 15057-15085. https://doi.org/10.3390/ijms160715057
Göttle P, Küry P. Intracellular Protein Shuttling: A Mechanism Relevant for Myelin Repair in Multiple Sclerosis? International Journal of Molecular Sciences. 2015; 16(7):15057-15085. https://doi.org/10.3390/ijms160715057
Chicago/Turabian StyleGöttle, Peter, and Patrick Küry. 2015. "Intracellular Protein Shuttling: A Mechanism Relevant for Myelin Repair in Multiple Sclerosis?" International Journal of Molecular Sciences 16, no. 7: 15057-15085. https://doi.org/10.3390/ijms160715057