2.1. Gas Phase
Gas-phase energy results are provided in
Table 1. Although some of the structures in
Scheme 1 and
Scheme 2 are rather peculiar, all of them have turned out to form local-minimum-energy tautomers/conformers on the basis of quantum chemical calculations.
The
s-trans/
anti form for 2-propene-1-imine (1) is more stable in free energy, ΔE
gint + ΔG
gth (see
Section 3), by 11.0 kJ/mol than the
s-cis/
anti (3) conformation, as calculated at the B97D level. Penn [
29] recorded the gas-phase microwave spectrum for this molecule and found only the
s-trans conformer. An
s-trans to
s-cis conformational change requires the rotation about the central C–C bond. The torsion barrier for (1) to (3) is 29–36 kJ/mol at the two theoretical levels (
Table 1) with very similar transition state CCCN torsion angles of 95.4° and 96.1°. Although the molecules could acquire the calculated activation energy upon collision for a gas-phase transformation, the experimental result for the lack of the
s-cis conformer is in good accord with the B97D relative free energy or the calculated CCSD(T)
CBS activation energy of 11.4 kJ/mol (the B97D and MP2 ΔG
gth values could be taken as similar, as revealed from the specifically calculated MP2 values for species (11), see below, thus an estimate for the CCSD(T)
CBS relative free energy is 9 kJ/mol, still too high).
Table 1.
Relative energy/free energy components for tautomeric/conformational isomers a.
Table 1.
Relative energy/free energy components for tautomeric/conformational isomers a.
| Structures in Schemes | Gas | Dichloromethane | Water |
|---|
| ΔEgint | ΔGgth | ΔEsint | ΔGsth | ΔGsolv | ΔEsint | ΔGsth | ΔGsolv |
|---|
| CH2=CH–CH=NH (1) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS (1 to 2) | 113.9 | −9.9 b | 111.5 | −10.1 b | 7.8 | 110.3 | −10.3 b | 10.3 |
| | 123.2 c | | | | | | | |
| CH2=CH–CH=NH (2) | 2.7 | −0.2 | 2.9 | −0.5 | −1.8 | 2.9 | −0.5 | −2.0 |
| | 3.1 c | | 3.8 c | | −2.1 d | 3.7 c | | −2.3 d |
| TS (1 to 3) | | | | | | | | |
| (95.4°, 96.0°, 96.3°) e | 35.5 | −3.5 b | 35.4 | −3.0 b | −0.6 | 35.5 | −3.0 b | −0.9 |
| (96.1°) | 29.3 c | | | | | | | |
| CH2=CH–CH=NH (3) | 13.4 | −2.4 | 14.3 | −4.0 b,f | 1.3 | 13.0 | −4.0 b,f | 2.2 |
| | 11.4 c | | 10.1 c | | 2.6 d | 9.7 c | | 3.2 d |
| CH2=CH–CH=NH (4) | 15.1 | −2.8 b | 16.1 | −2.3 b | −8.7 | 15.1 | −2.5 b | −1.4 |
| | | | | | | 13.1 c | | −0.9 d |
| O=CH–CH=NH (5) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| O=CH–CH=NH (6) | 2.9 | −0.5 | 3.5 | 0.2 | −1.3 | 3.8 | 0.0 | −2.0 |
| TS (6 to 7) | | | | | | | | |
| (87.3°, 88.2°, 88.4°) e | 29.1 | −3.4 b | 29.9 | −3.0 b | −3.3 | 30.4 | −3.0 b | −4.5 |
| (89.2°) | 30.8 | | | | | | | |
| O=CH–CH=NH (7) | 7.8 | −0.1 | 8.0 | 1.0 | 0.4 | 8.2 | 0.4 | 0.2 |
| | 7.8 c | | 9.6 c | | −1.9 d | 10.5 c | | −3.0 d |
| O=CH–CH=NH(8) | 23.1 | −5.4 d | 26.8 | −3.6 b | −10.6 | 29.0 | −3.3 b | −14.9 |
| | 24.4 | | 32.5 c | | −16.2 d | 36.1 c | | −22.3 d |
| O=C=CH–NH2 (9) | 20.8 | −1.0 | 19.7 | | 7.4 | 19.4 | −0.8 b | 8.2 |
| | 35.1 | | 32.0 c | | 9.2 d | 31.4 c | | 10.3 d |
| HN=CH–CH=NH (10) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HN=CH–CH=NH (11) | 4.6 | −2.3 b | 5.8 | −2.9 b | −3.4 | 6.5 | −3.0 b | −4.8 |
| | 6.2 c | | 8.7 c | −2.7 b | −5.0 d | 9.8 c | −2.9 b | −6.8 d |
| TS (11 to 13) | | | | | | | | |
| (92.7o, 93.2o, 93.4°) e | 32.6 | −3.6 b | 34.3 | −2.8 b | −5.2 | 35.2 | −3.0 b | −7.2 |
| (93.6°) | 31.3 | | | | | | | |
| HN=CH–CH=NH (13) | 11.8 | −2.8 b | 13.0 | −3.3 b | −1.5 | 13.6 | −4.1 b | −2.3 |
| | 10.6 c | | 13.9 c | | −4.5 d | 15.3 c | | −6.2 d |
| HN=CH–CH=NH (12) | 6.9 | −1.1 | 7.8 | −1.4 | −3.6 | 8.2 | −1.7 | −4.8 |
| | 9.4 c | | 11.6 c | | −4.9 d | 12.6 c | | −7.1 d |
| HN=CH–CH=NH (14) | 16.1 | | 16.7 | −0.9 b | −3.0 | 17.0 | −2.2 b | −4.0 |
| | | | 20.8 c | | −5.7 d | 21.7 c | | −7.2 d |
| TS (10 to 15) | | | | | | | | |
| (86.3°, -, 93.5°) e | 35.2 | −4.0 b | | | | 38.3 | −3.8 b | −9.8 |
| (83.6o) | 32.3 | | | | | | | |
| HN=CH–CH=NH (15) | 28.8 | | 33.3 | −4.7 b | −12.8 | 36.2 | −5.4 b | −18.5 |
| | | | 36.7 c | | −17.4 c | 40.8 c | | −24.4 d |
| HN=C=CH–NH2 (16) | 49.2 | | 49.8 | | 0.1 | 50.2 | −7.5 g | −0.8 |
| | 64.0 c | | | | | | | |
| H2N–C≡C–NH2 (17) | 98.6 | −7.4 g | 99.2 | | −2.6 | 99.2 | −5.0 g | −4.0 |
Penn found, however, two s-trans conformers with C–C=N–H anti (1) and syn (2) arrangements. The latter structure was predicted to be higher in energy by 3.8 ± 0.4 kJ/mol. The calculated relative energy is 2.7 and 3.1 kJ/mol at the DFT and the CCSD(T)CBS levels, respectively. But how can the anti and syn forms equilibrate in the gas phase?
The experiment was taken at T = 673 K, where a stably existing dimer is unlikely. Furthermore, as will be discussed below, a doubly hydrogen-bonded dimeric structure, preferable for a double proton-relay, is not favored even in dichloromethane solution, where a weak association trend has been still predicted. Then possible intramolecular routes for forming the s-trans/syn species are H rotation about the C=N double bond or the nitrogen inversion.
B97D transition state geometry optimization for the
s-trans/
anti to
s-trans/
syn CH
2CHCHNH, starting from a C–C=N–H torsion angle of 90° and C=N–H bond angle of about 110° as a guess for the potential energy maximum for the H rotation about the C=N bond, led quickly to a TS geometry with C=N–H bond angle of 179.3° and CCNH torsion angle of 124.9°. The torsion angle for the
s-
trans CCCN moiety was maintained at 180°. The TS structure was reoptimized at the MP level resulting in CNH angle of 179.2° and CCNH and CCCN torsion angles of 124.5° and 180.0°, respectively (
Supplementary Table S1). These results correspond to a combined mechanism for the
s-trans/
anti (1) to
s-trans/
syn (2) transformation along H rotation about the C=N bond and an increase in the CNH bond angle to almost linear, corresponding to N-inversion. At CNH >179° it is not important what the CCNH torsion angle is. B97D and MP2 predictions of the TS geometry through aug-cc-pvtz optimization are in agreement. The calculated energy barrier is, however, very high, 113.8 kJ/mol and the activation free energy is still about 105 kJ/mol at the B97D/aug-cc-pvqz level. The energy barrier is 123.2 kJ/mol at the CCSD(T)
CBS level (
Table 1).The average kinetic energy for a gas molecule is 1.5
RT/mol = 8.4 kJ/mol at T = 673 K, thus the necessary additional kinetic energy to distort the
s-trans/
anti structure to reach the transition state is at least 105.4 kJ/mol. From a Boltzmann distribution, the ratio of the molecules with the necessary additional energy and the average-energy molecules is about 7 × 10
−9. This is a small ratio, indeed, but even assuming only 1% for the average-energy particles, the number of the molecules with the required activation energy is 6 × 10
21 × 7 × 10
−9 ≈ 4 × 10
13/mol. Thus the fraction is small but the number itself is still large, which, on the basis of the experimental results, could trigger the
anti/
syn equilibration.
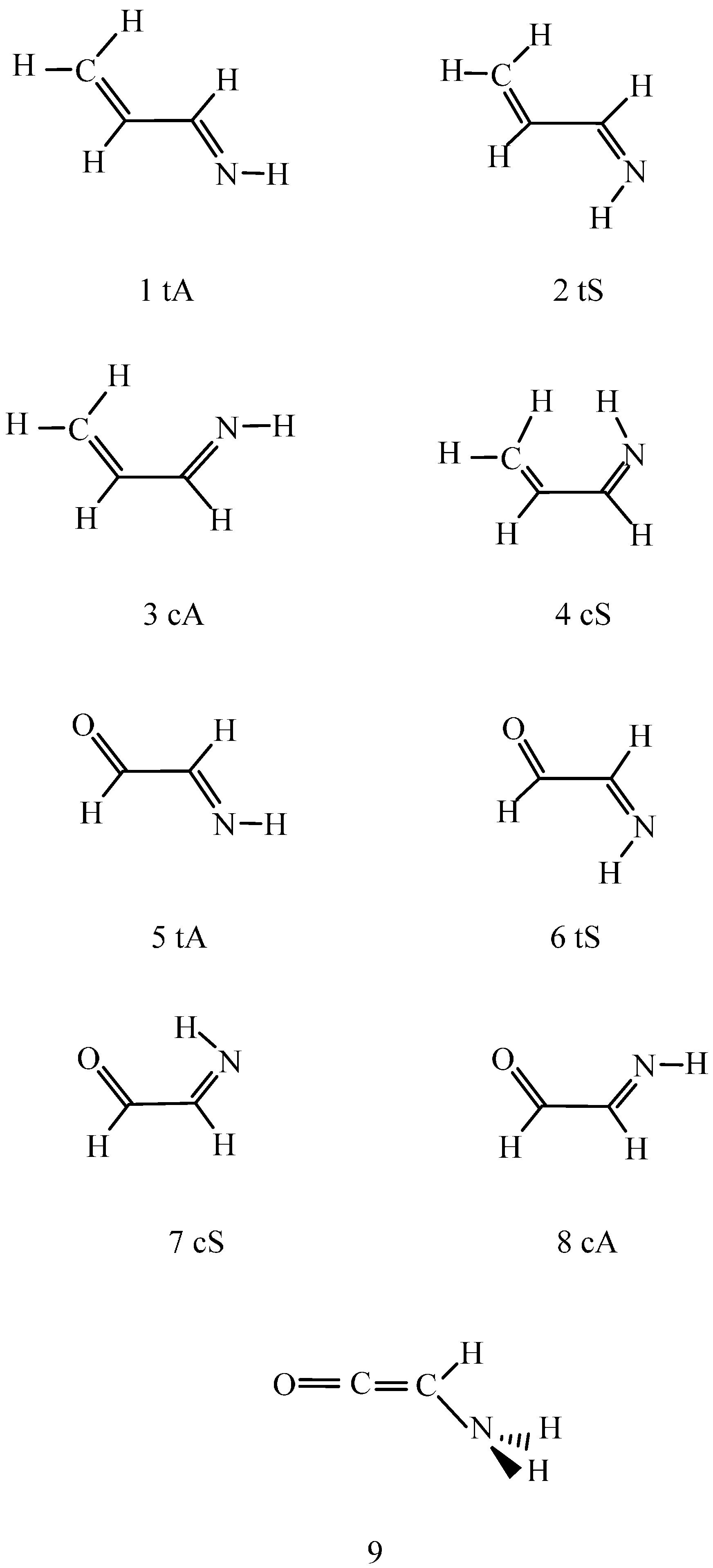
For the C2H3NO molecules, the two s-trans 2-imino-acetaldehyde structures with anti and syn positions for the imino hydrogen (5,6) differ by 2.9 kJ/mol in internal energy. The B97D calculations predict the existence of both conformers in the gas phase, although the required activation energy for the equilibration is supposed to be similar to that for the corresponding CH2=CH–CH=NH conformers. The relative energies of the s-cis conformers (7) and mainly that of (8) are considerable at both theoretical levels predicting nearly equal values. The relative energy of (7) is, however, lower by 3.6–5.6 kJ/mol than that for the structurally related (3) species. How can it be rationalized?
Scheme 1 shows that species (3) cannot have a strong intramolecular hydrogen bond, and could be subject only to a favorable C–H…N interaction with H…N distance of 267 pm. In a recent review [
30], problems related to intramolecular
vs intermolecular hydrogen bonds (mainly in solution for the latter) were surveyed. Upon the 2011 IUPAC (International Union of Pure and Applied Chemistry) recommendations, no clear-cut upper limit was defined for the atom separation in a X…H hydrogen bond, where X is generally an electronegative element. The former “golden rule” was that the X…H bond length must be shorter than the sum of the van der Waals radii of the two atoms [
31]. Then the above interaction could be qualified as a hydrogen bond since 267 pm is within this range. An important, but not mandatory feature of a H-bond by IUPAC is the existence of a (3, −1) bond criteria point (BCP). Furthermore, an important feature of a
red-shifting (classical) X–H…Y hydrogen bond is that the X–H bond length elongates with respect to the reference system without the H-bond and charge is transferred from Y to the X–H. The C–H distance from B97D in (1) is 109.0 pm, the molecular electrostatic potential fitted CHELPG [
32] C, H, N charges are −0.337, 0.138, and −0.682 atomic units, respectively. The C–H distance in (3) becomes shorter to 108.8 pm, and the corresponding atomic charges are −0.149, 0.109, and −0.638. Thus quite a remarkable charge was transferred from the nitrogen to the C–H proton. The important finding was that each of the symmetrical and asymmetrical C–H stretching frequencies increased by 10 cm
−1. Taking the structural information together, the system meets all requirements for a
blue-shifting hydrogen bonding, which could be, however, weak [
33]. From MP2 geometry optimizations, the C–H distance decreases from 108.3 to 108.2 pm and the above net atomic charges are −0.375, 0.155, −0.695
vs. −0.189, 0.123, and −0.647. The differences in the sets by the two methods are close.
For the pair of (6) and (7), the B97D/aug-cc-pvtz calculated N–H bond length increases from 102.5 in (6) to 103.1 in (7), the stretching frequency decreases from 3370 to 3296 cm−1, and the calculated atomic charges for O, H, N are −0.444, 0.358, and −0656, respectively, in (6) compared with −0.408, 0.317, and −0.562 in (7). The O…H separation is 245.0 pm in (7), the O…H–N bond angle is 105.4°. It is questionable whether such a bent bond still can be assigned to a hydrogen bond category, but the effect of the interactions to the important structural parameters for a red-shifting hydrogen bond formation has been clearly demonstrated. Even if the interaction is only pure electrostatic, the decrease of the relative energy from 11.4-13.4 kJ/mol for (3) to 7.8 for (7) is reasonable.
The transition state activation energy from (6) to (7) was calculated at 29.1–30.8 kJ/mol with OCCN torsion angles of 87.3°–89.2°. The barrier can be overridden upon collision in the gas-phase, and species (7) can be present with (5):(7) ratio of about 4:96 at room temperature. The large relative energy for (8) can be attributed to the repulsion of the nitrogen and oxygen lone pairs existing in the almost planar heavy atom arrangement with OCCN torsion angle of 0.4° (
Table S1).
The amino ketene tautomer (9) is higher in energy then species (5) by 20.8 kJ/mol and 35.1 kJ/mol at the DFT and the
ab initio level, respectively. The calculated values suggest that the predominant conformer/tautomer in the gas phase is structure (5). If the large, possibly about 100 kJ/mol, activation energy can be provided in the gas phase for the formation of (6) (in analogy to the formation of (2), as was proven by the experiment of Penn [
29], the calculated (5):(6) equilibrium ratio is 28:72 (the final equilibrium composition depends on all considered species existing in the mixture but the ratios for selected pairs will not change).
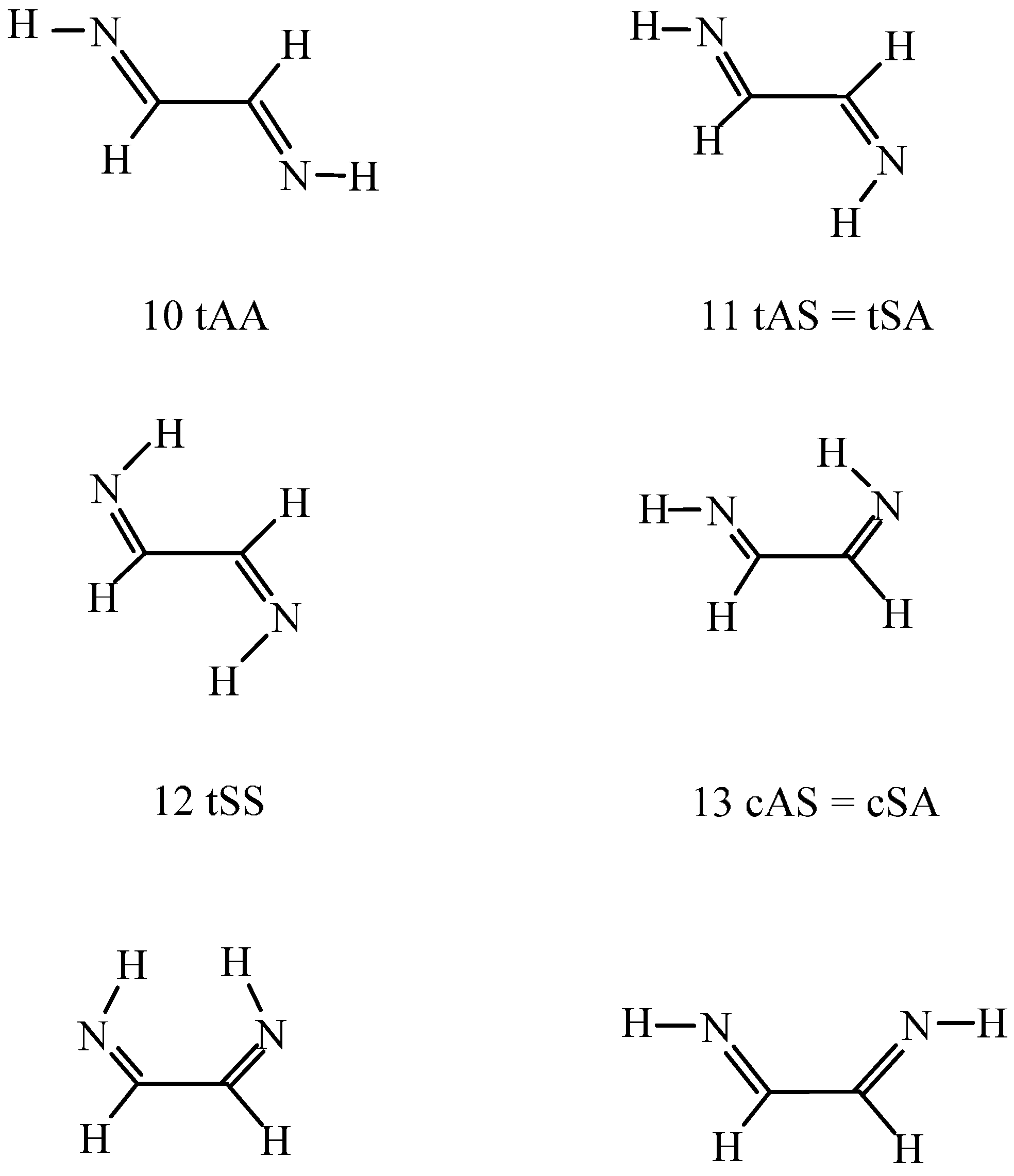
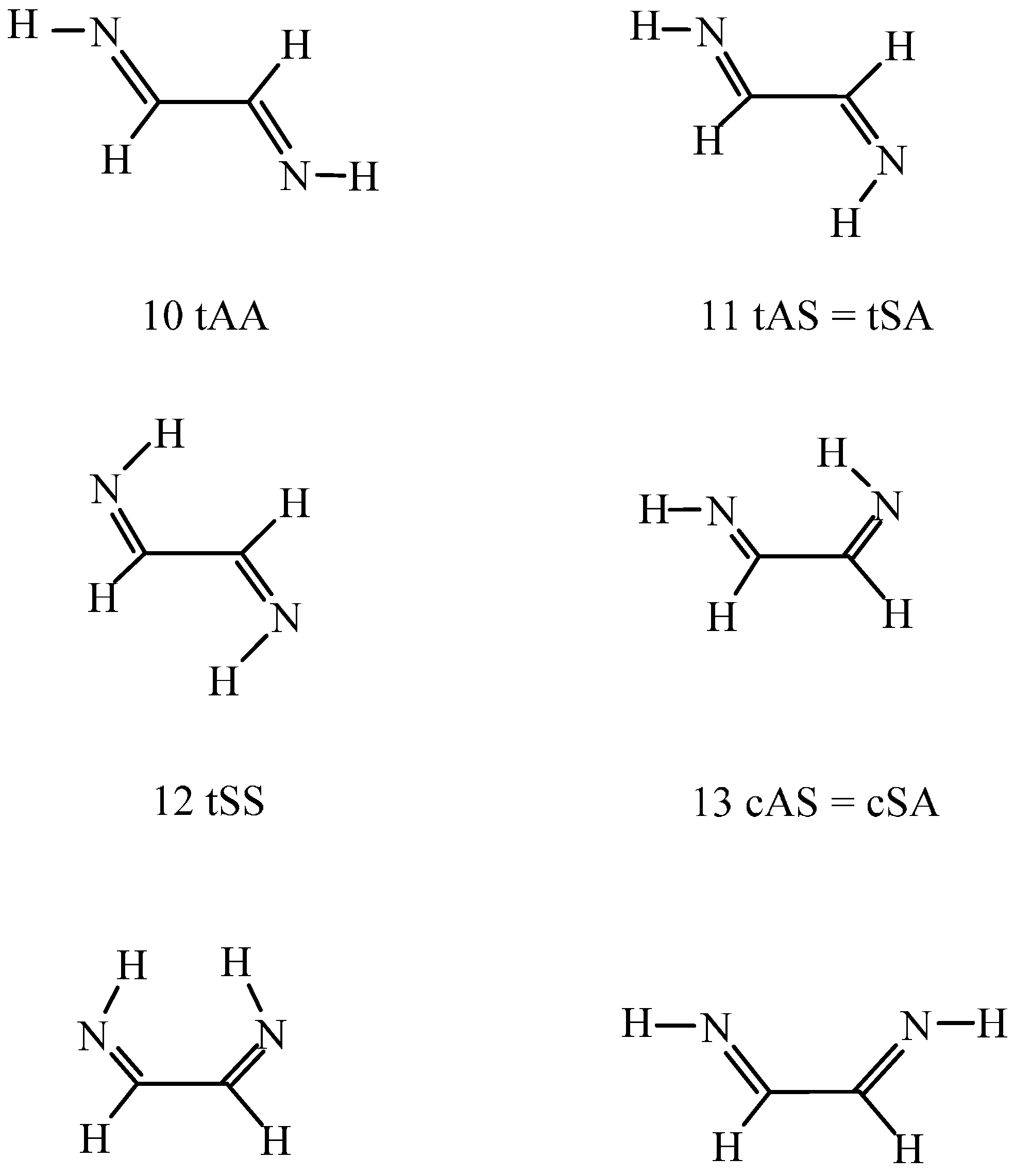
For the studied C
2H
4N
2 systems (the isomeric H
2N–CH
2–CN amino-acetonitrile was not investigated), the prevalent 1,2-ethanediimine species is the (10, tAA) (see
Scheme 2) in the gas phase. It is more stable in free energy by 2.3–5.8 kJ/mol than (11, tAS) and (12, tSS) at the DFT level and must be more stable by a further 2–3 kJ/mol than species (11) and (12) as calculated
ab initio. The relative energies of the
s-cis conformers (13–15) were calculated at 11–29 kJ/mol. Relative energies are even much higher for the amino imino-ketene (16) with a cumulated double-bonded structure and the 1,2-diamino acetylene (17). The values calculated at the DFT level are 49 and 99 kJ/mol, respectively. The CCSD(T)
CBS relative energy for structure (16) is 64 kJ/mol. The analysis of the energy components in Equation (2) (section 3) revealed that the ΔE
MP2CBS term is about 90% responsible for the indicated relative energy, whereas (ΔE
CCSD(T) − ΔE
MP2)
aug-cc-pvdz accounts for the about 10% remaining. This means that the post-MP2 correction is meaningful for tautomeric systems (5.33 kJ/mol in the present case). The relative post-MP2 correction accounted for an even larger share of the total relative internal energy for species (9). Its contribution to the final value of 35.1 kJ/mol was 10.1 kJ/mol. In contrast, the (ΔE
CCSD(T) − ΔE
MP2)
aug-cc-pvdz contributions to the relative conformational internal energies have been found to be small, amounting only to 1–2 kJ/mol in general.
The (11) to (13) conformational change is possible along a rotation about the NCCN bond. The transition state torsion angle was predicted at 92.7°–93.6° by the two theoretical approaches with activation energy of 31–33 kJ/mol. The activation free energy must be smaller by 11% from B97D estimation. When (13) is formed the question can be raised whether there is an N–H…N hydrogen bond. Present computational results cannot give a unique answer. The right-hand side N–H bond length of 102.94 pm in 13 cAS (
Scheme 2) shows a very little increase from 102.88 pm in 11 tAS.
Scheme 2 clarifies that this N–H bond (CCNH
syn) can act as the hydrogen bond donor, whereas the left-hand side nitrogen (CCNH
anti) would be the acceptor atom. The small increase of the
syn N–H bond in combination with a remarkable charge transfer from the nitrogen in the CCNH
anti moiety, (atomic charges on this nitrogen are −0.670 and −0.637 in (11) and (13) respectively) suggest a red-shifting hydrogen bond. However, the vibrational frequency for the
syn N–H bond increases from 3306 cm
−1 in (11) to 3312 cm
−1 in (13). Since the bond length increases (even though very slightly), a small decrease instead of the noted small increase in the vibrational frequency would have been expected. For the other N–H bond in the CCNH
anti substructure, the bond length decreased from 102.37 pm in (11) to 102.21 pm in (13) and the concomitant frequency change was an increase from 3377 to 3398 cm
−1. The N…H distance was calculated at 245.9 pm at the B97D level with N–H…N bond angle of 105.6°. These geometric data would still comply with a strongly bent regular H-bond, although it was unable to identify the hydrogen bond character in the gas phase on the basis of the above contradictory data. In contrast, the stretching frequency of the donor N–H in the
s-cis/
syn conformation decreased by 10 and 20 cm
−1 in dichloromethane and water, respectively, completing the required conditions for a red-shifting intramolecular hydrogen bond.
Finally, the transformation of species (10) to (15) was studied through rotation about the C–C bond. The NCCN torsion angle is 83.6°–86.3° in the transition state with energy 32–35 kJ/mol above that for the reference (10) conformation. The two theoretical methods thus provide similar results. Conformer (15) corresponds to a NCCN
s-cis structure with two HNCC
anti moieties (cAA in
Scheme 2). Although the molecule is not planar, the NCCN torsion angle of 26.8° could only partially diminish the repulsion of the nitrogen lone pairs. Consequently, the relative energy is high, 28.8 kJ/mol at the B97D level and the conformer is not expected to appear in the gas-phase equilibrium mixture.
Whereas no comparison of the calculated relative energies for the studied C
2H
4N
2 systems is possible in the absence of experimental data for 1,2-ethanediimine itself, Hargittai and Seip [
34] investigated the molecular structure of its
N,
N'-
di-tertiary butyl derivative by gas electron diffraction. The predominant structure was found
gauche with N=C–C=N torsion angle of 65° by a rotation from the
syn form (corresponding to
s-cis in this paper), although a small fraction of the
anti (
s-
trans) conformer was also assigned. The present author attributes this experimental result mainly to the favorable dispersion interactions of the two bulky t-butyl groups in the
gauche rather than in the
s-trans form, and the decrease of the relative free energy by
RT ln2 = 2.03 kJ/mol at the temperature of the experiment (~353 K) due to the mixing of the two optical antipodes emerging in the case of a
gauche structure.
A structure, closely related to 1,2-diamino acetylene, μ
2,η
2-1,2-diaminoethylene was characterized by using Fourier transform-reflection absorption infrared spectroscopy (FT-RAIRS) as a surface species [
35]. The C atoms in this structure are bound to a Pt surface and the spectra predicted a H
2N–C–C–NH
2 moiety, with delocalized π electrons along the NCCN path. Two different amino N–H stretching frequencies were assigned according to the C
2v symmetry. The relevance of this result with respect to the present study is that no imine frequencies were assigned. Thus the H
2N–C≡C–NH
2 tautomer could be also stable in the gas phase in spite of its about 99 kJ/mol relative energy.
2.2. In-Solution Results
Table 1 summarizes the results of the quantum mechanical calculations for solutes in dichloromethane and water solvents, as well. Internal energy differences and IEF-PCM/ΔG
solv values were obtained from B97D/aug-cc-pvqz//B97D/aug-cc-pvtz single point calculations in every case. In a number of cases, relative internal energies were calculated from CCSD(T)
CBS values following the IEF-PCM/MP2/aug-cc-pvtz geometry optimization. The related ΔG
solv values were obtained at this optimization level.
The energy of a structure, E
gint, is lowest at a theoretical level if the geometry is optimized in the gas phase. If the molecule is imbedded in a solvent environment, its internal energy will increase: E
sint becomes less negative than E
gint in order to make G
solv/PCM optimally negative. The SCF (self-consistent field) procedure through the IEF-PCM in-solution geometry optimization stops where the (E
sint + G
solv) term reaches a local minimum. Increase of the internal energy has two sources: the geometry distortion as compared to the gas-phase structure and the solute polarization.
Table S1 shows that the main geometric parameters change only slightly due to solvation, thus the increase of E
int in solution must be attributed mainly to polarization effects. In a former study, Alagona
et al. [
36] calculated the separate effects of the two contributions for small molecules, and the polarization effect was found to be of significantly larger importance in DFT calculations.
ΔEsint from IEF-PCM/B97D calculations are both larger and smaller than the corresponding gas-phase values by 1–2 kJ/mol in general. It is worth mentioning that this feature of the relative internal energy is allowed theoretically upon solvation, whereas the individual Eint values always increase. The variation of the in-solution ΔEsint value in comparison with its gas-phase counterpart, ΔEgint, depends on which species undergoes smaller energy increase upon solvation. The smaller increase in Eint for the more stable form leads to an increased ΔEsint and vice versa.
Remarkable increases in ΔEint were obtained for structures (8) and (15) with outstandingly large solvent effects in water. Since ΔGsolv/PCM is conspicuously negative, the concomitant ΔEsint values increased by 5.9 and 7.4 kJ/mol as compared with ΔEgint of 23.1 and 28.8 kJ/mol, respectively, suggesting considerable internal energy increases for structures (8) and (15) in aqueous solution.
Bond lengths and bond angles change negligibly upon solvation. In contrast, the XCCY torsion angles for heavy atoms significantly deviate from 0° or 180° in solution for species (4), (8), and (15). The heavy atoms in tautomers (9) and (16) are far from coplanarity even in the gas phase. The solvation changes the XCCY torsion angles only slightly. Indicated HNNH torsion angles for (17) in Supplementary
Table S1 also do not change remarkably leaving the structure without any symmetry.
The in-solution ΔGsth values are generally moderate in comparison with the corresponding ΔEsint values. The gas-phase molecular symmetries were preserved in solution, thus the related entropy contributions to the corresponding Gsth values still hold. The sign of the ΔEsint + ΔGsth + ΔGsolv/PCM sum was always dominated by the sign of the ΔEsint term. Accordingly, the sign of the total relative free energy, ΔGstot is equal to that for ΔEsint in every case.
IEF-PCM/CCSD(T)CBS calculations predicted that ΔEsint would increase for the conformers by up to 7.1 kJ/mol (except (3) and (4) with a decrease up to 4.2 kJ/mol) as compared with the B97D/aug-cc-pvqz result for structures in dichloromethane and water, and the increase by about 12.0 kJ/mol for species (9) is similar to that found for this latter tautomer in the gas-phase. The most surprising result was, however, that the IEF-PCM/MP2/aug-cc-pvtz ΔGsolv for (7) was significantly negative in comparison with the small positive value by the DFT calculations. In other cases, the MP2/aug-cc-pvtz relative solvation free energies preserved at least the sign of ΔGsolv/PCM as calculated by B97D. In general, if the ab initio ΔEsint increases, the related MP2/ΔGsolv becomes more negative for the conformers and vice versa. The deviations call attention to the probably different account for the solute polarization by the two theoretical methods. For tautomer (9), however, both ΔEsint and ΔGsolv/PCM increase.
IEF-PCM relative solvation free energies for conformational local energy minima scatter in a wide range of 3.2 and –24.4 kJ/mol, whereas those for tautomers (9,16,17) vary between 10.3 and −4.0 kJ/mol. The remarkable cases from the point of view of the estimation of the equilibrium composition are when the ΔE
sint and ΔG
solv values are of opposite signs, thus when ΔG
solv is negative in
Table 1 (with positive ΔG
solv/PCM, the species is even less preferable than calculated on the basis of the ΔE
sint + ΔG
sth term). For CH
2=CH–CH=NH (2), ΔG
stot from B97D is only 0.4–0.6 kJ/mol, considerably smaller than the gas-phase value of 2.5 kJ/mol. The reduction means that the (2), the
s-trans/syn fraction is 44%–46% of the total
s-trans form in solution at room temperature. For O=CH–CH=NH (6), also at T = 298 K, the B97D ΔG
stot values are 2.4 and 1.8 kJ/mol in dichloromethane and water, respectively, corresponding to 28:72 and 33:67 molar fractions in the respective solvents for the less and more stable
s-trans conformers with the H–N bond pointing in different directions. The ratio is about 24:76 in the gas phase as calculated from energies, thus the solvent effects are small for the
s-trans 2-imino-acetaldehyde.
The most stable conformation of 1,2-ethanediimine seems to be a delicate question at the IEF-PCM level. Whereas (10), the s-trans/anti/anti conformation is clearly the most stable in the gas phase, ΔGstot for the (11) s-trans/anti/syn conformation is −0.5 and −1.3 kJ/mol in dichloromethane and water, respectively, at the B97D level. For a comparable estimation, the time-consuming MP2/aug-cc-pvtz frequencies were calculated for the corresponding pair, showing that the derived ΔGsth values hardly deviated from the B97D values in the four cases studied. This finding supports that one may use the B97D values in other cases, as well, when the relative CCSD(T)CBS free energies are to be calculated. For the present case, (10) is just slightly more stable than (11) at the ab initio level even in aqueous solution. Nonetheless, the problem will be studied using the FEP/MC method and discussed below.
In the s-cis/anti/anti conformation of HN=CH–CH=NH (15), when the two lone pairs of the nitrogen atoms point mostly toward each other, the relative internal energy largely increases compared with (13) and (14). Although the considerably more negative ΔΔGsolv/PCM values in water indicates that the solvation strongly supports the formation of this conformer, ΔGstot remains too positive preventing the observable appearance of this conformer in the equilibrium mixture.
ΔGsolv/PCM values are negative for the TS structures regarding rotations about the central C–C bond. All calculated XCCN torsion angles for such conformer transformations are about 90°. Apparently, solvation favors the nearly perpendicular rather than coplanar arrangements for the XCC and CCY planes of the heavy atoms. In contrast, ΔGsolv/PCM for the TS corresponding to the combined H rotation about the C=N bond/N-inversion for the s-trans CH2=CH–CH=NH is strongly positive, with coplanar CCCN skeleton.
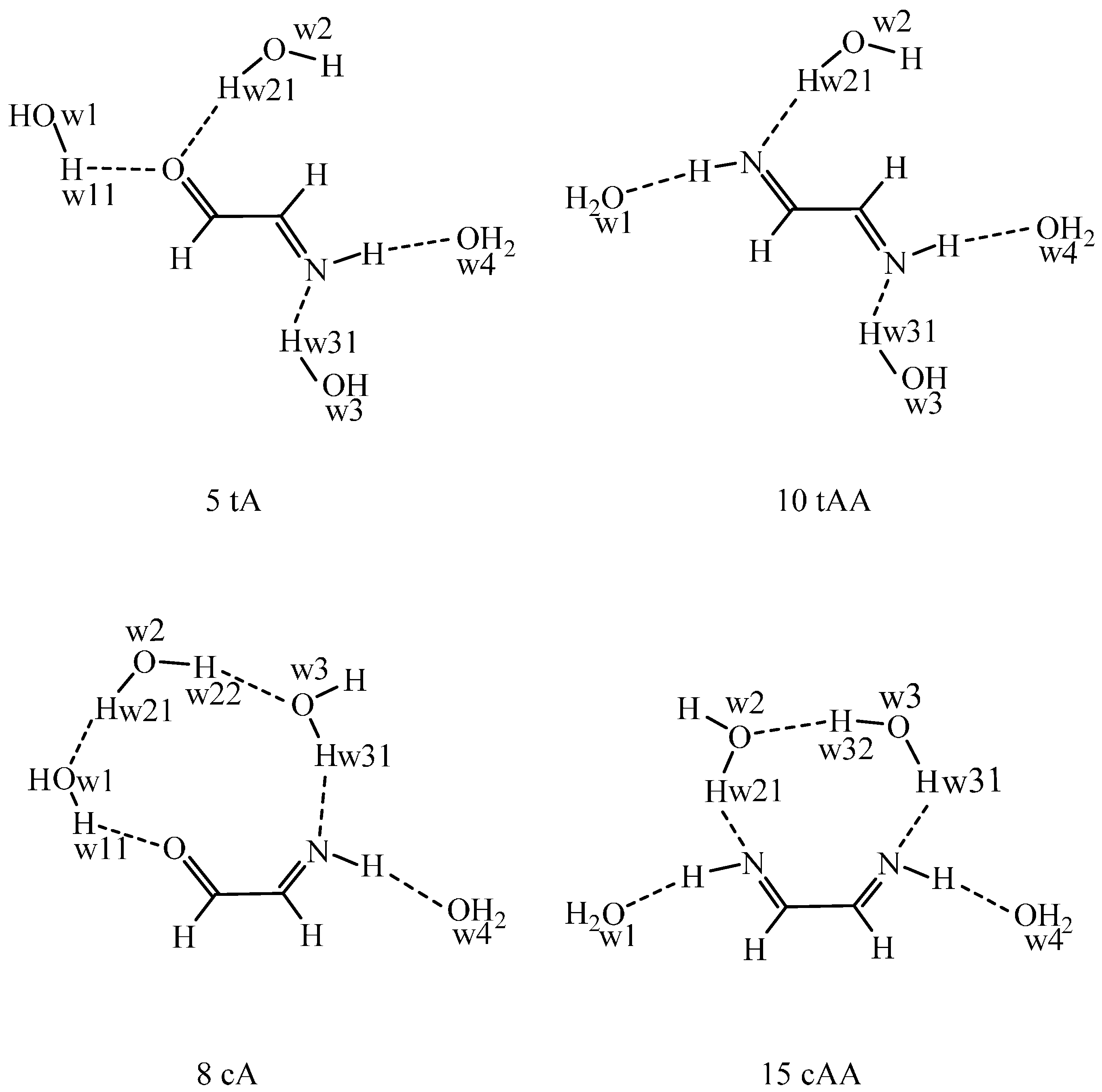
A well-known shortcoming of the continuum dielectric solvent model is that it may underestimate the solvation effects on the possible solute-solvent hydrogen bonds, when the pure solute is placed into the cavity carved within the solvent. For improving the performance of the IEF-PCM, a supermolecule may be considered, where the solute is surrounded by a number of water molecules and this supermolecule is embedded into the cavity within the continuum solvent. To this aim, tetrahydrate models have been studied for two solutes in two conformations for each (
Figure 1). By an explicit solvent Monte Carlo method, the thermally averaged solute-solvent hydrogen bond interactions can be considered. In the present, classical MC, the solute has to be characterized by preset net atomic charges, which can be derived by a fit to the in-solution molecular electrostatic potential (
Table 2).
In the optimized tetrahydrate structures, the OCCN and NCCN torsion angles are about 180° for 5 tA and 10 tAA in accord with their values for the pure solute in the water cavity, whereas the torsion angles for 8 cA and 15 cAA increase by 12°–17° as compared with their B97D values in
Table S1. There are four solute-solvent hydrogen bonds to 5 tA, 10 tAA, and 15 cAA. In 8 cA a water-water hydrogen bond (w1…w2) comes into existence instead of a =O…H
w21O
w2 bond. The hydrogen bonds are shorter when the water hydrogen is the donor than when a N–H…O
w bond is formed.
The problems related to the supermolecule approach when considering a limited number of water molecules becomes evident by the structures in
Figure 1. The four hydrogen-bonding sites are far from each other in 5 tA and 10 tAA and the connecting water molecules cannot form a water-water hydrogen bond in parallel with the formation of solute-water hydrogen bonds. In the
s-cis conformations, a three-member water chain is formed for 8 cA and a water-water hydrogen bond is stable in 15 tAA. As a result, the
s-cis imino-aldehyde tetrahydrate (8 cA) is more stable than the
s-trans (5 tA) tetrahydrate by 2.7 kJ/mol, in comparison with the relative ΔE
sint + ΔG
solv/PCM energies of 29.1–14.9 = 14.2 kJ/mol for the pure 8 cA solute at the IEF-PCM/B97D/aug-cc-pvtz level (
Table 1 contains aug-cc-pvqz values). The tetrahydrate result is a consequence of the two water-water hydrogen bonds along the three-water chain. Their BSSE-corrected interaction energy in the gas phase (no BSSE available in IEF-PCM) is 25.4 kJ/mol. If the relative tetrahydrate energy is corrected by this value, the
s-cis 8 cA system becomes 22.7 kJ/mol less stable than the 5 tA tetrahydrate vs 14.2 kJ/mol for the pure solutes. This correction is justified, because the water bridges, affecting the total energy only for 8 cA, specifically emerge for this structure due to considering only a limited number for explicit water molecules.
In a real system, each water molecule in the first hydration shell is in hydrogen bond(s) with waters in the second shell or with those around the CH sites, thus the discussed peculiarity will not be relevant. In an aqueous solution, w1, w2, and w3 of 5 tA are also in hydrogen bonds with its neighbors, and w4 for both conformers, too. In conclusion, the reversal of the relative energy is the consequence of considering only four water molecules in the supermolecule.
Figure 1.
The structures of the tetrahydrates of the
s-trans species 5 and 10 and the
s-cis species 8 and 15 as optimized in continuum water solvent at the IEF-PCM/B97D/aug-cc-pvtz level. The hydrogen bond parameters (from left to right) in
Figure 1 are as follow. 5 tA: OCCN = 180°, the O=C, O…H
w11 (198 pm), and O…H
w21 (201 pm) bonds are coplanar, bond angles: O
w1H
w11…O = 177°, O
w2H
w21…O = 178°. The C=N, N–H and the N…H
w31 (187 pm) bonds are coplanar, H…O
w4 = 208 pm. The N…H
w31O
w3 bond angle is 177°, N–H…O
w4 = 173°. 10 tAA: NCCN = 179.9°, the N=C, N–H, and N…H
w21 (187 pm) bonds are coplanar, H…O
w1 = 209 pm. Bond angles: O
w1…H–N = 173°, O
w2H
w21…N = 177°. The C=N, N–H and the N…H
w31 (187 pm) bonds are coplanar, H…O
w4 = 208 pm. The N…H
w31O
w3 bond angle is 177°, N–H…O
w4 = 173°. 8 cA: OCCN = 23.7°, only one water—carbonyl hydrogen bond: O…H
w11 (194 pm), O
w1H
w11…O = 161°. Waters 1 and 2 form a solvent-solvent hydrogen bond with O
w1…H
w21 distance of 194 pm and O
w1…H
w21O
w2 angle of 177°. Water 2 and water 3 are also linked by a solvent-solvent hydrogen bond: H
w22…O
w3 = 187 pm and O
w2H
w22…O
w3 = 177°. The C=N, N–H and the N…H
w31 (186 pm) bonds are almost coplanar (the sum of the bond angles is 359°), H…O
w4 = 201 pm. The N…H
w31O
w3 bond angle is 178°, N–H…O
w4 = 176°. 15 cAA: NCCN = 30.5°, the N=C, N–H, and N…H
w21 (182 pm) bonds are coplanar, H…O
w1 = 207 pm. Bond angles: O
w1…H–N = 176°, O
w2H
w21…N = 177°. Water 2 and water 3 form a solvent–solvent hydrogen bond: O
w2…H
w32 = 193 pm and O
w2…H
w32O
w3 = 161°. The C=N, N–H and the N…H
w31 (190 pm) bonds are coplanar, H…O
w4 = 210 pm. The N…H
w31O
w3 bond angle is 175°, N–H…O
w4 = 172°.
Figure 1.
The structures of the tetrahydrates of the
s-trans species 5 and 10 and the
s-cis species 8 and 15 as optimized in continuum water solvent at the IEF-PCM/B97D/aug-cc-pvtz level. The hydrogen bond parameters (from left to right) in
Figure 1 are as follow. 5 tA: OCCN = 180°, the O=C, O…H
w11 (198 pm), and O…H
w21 (201 pm) bonds are coplanar, bond angles: O
w1H
w11…O = 177°, O
w2H
w21…O = 178°. The C=N, N–H and the N…H
w31 (187 pm) bonds are coplanar, H…O
w4 = 208 pm. The N…H
w31O
w3 bond angle is 177°, N–H…O
w4 = 173°. 10 tAA: NCCN = 179.9°, the N=C, N–H, and N…H
w21 (187 pm) bonds are coplanar, H…O
w1 = 209 pm. Bond angles: O
w1…H–N = 173°, O
w2H
w21…N = 177°. The C=N, N–H and the N…H
w31 (187 pm) bonds are coplanar, H…O
w4 = 208 pm. The N…H
w31O
w3 bond angle is 177°, N–H…O
w4 = 173°. 8 cA: OCCN = 23.7°, only one water—carbonyl hydrogen bond: O…H
w11 (194 pm), O
w1H
w11…O = 161°. Waters 1 and 2 form a solvent-solvent hydrogen bond with O
w1…H
w21 distance of 194 pm and O
w1…H
w21O
w2 angle of 177°. Water 2 and water 3 are also linked by a solvent-solvent hydrogen bond: H
w22…O
w3 = 187 pm and O
w2H
w22…O
w3 = 177°. The C=N, N–H and the N…H
w31 (186 pm) bonds are almost coplanar (the sum of the bond angles is 359°), H…O
w4 = 201 pm. The N…H
w31O
w3 bond angle is 178°, N–H…O
w4 = 176°. 15 cAA: NCCN = 30.5°, the N=C, N–H, and N…H
w21 (182 pm) bonds are coplanar, H…O
w1 = 207 pm. Bond angles: O
w1…H–N = 176°, O
w2H
w21…N = 177°. Water 2 and water 3 form a solvent–solvent hydrogen bond: O
w2…H
w32 = 193 pm and O
w2…H
w32O
w3 = 161°. The C=N, N–H and the N…H
w31 (190 pm) bonds are coplanar, H…O
w4 = 210 pm. The N…H
w31O
w3 bond angle is 175°, N–H…O
w4 = 172°.
![Ijms 16 10767 g001]()
Table 2.
B97D/aug-cc-pvtz polar atomic and net solute charges from IEF-PCM calculations a.
Table 2.
B97D/aug-cc-pvtz polar atomic and net solute charges from IEF-PCM calculations a.
| Structures in Schemes | Polar Atoms | Pure Solute | Solute + 4H2O | Net Solute Charges |
|---|
| Pure Solute | in Supermolecule |
|---|
| O=CH–CH=NH (5) | O | −0.510 | −0.340 | 0.000 | 0.268 |
| | N | −0.754 | −0.495 | | |
| | H | 0.419 | 0.406 | | |
| O=CH–CH=NH (8) | O | −0.488 | −0.389 | 0.000 | 0.184 |
| | N | −0.726 | −0.489 | | |
| | H | 0.404 | 0.368 | | |
| HN=CH–CH=NH (10) | H | 0.404 | 0.460 | 0.000 | 0.186 |
| | N | −0.800 | −0.666 | | |
| | N | −0.800 | −0.641 | | |
| | H | 0.404 | 0.439 | | |
| HN=CH–CH=NH (15) | H | 0.388 | 0.354 | 0.000 | 0.208 |
| | N | −0.757 | −0.531 | | |
| | N | −0.757 | −0.479 | | |
| | H | 0.388 | 0.325 | | |
The same analysis can be performed for the 1,2-ethanediimine conformers. In this case, 10 tAA is still more stable than 15 cAA by 9.8 kJ/mol in comparison with ΔEsint + ΔGsolv/PCM of 36.3–18.6 = 17.7 kJ/mol for the pure solutes. The correct preference was still maintained because there is only one water-water hydrogen bond in 15 cAA, for which the BSSE corrected interaction energy is –11.0 kJ/mol. Considering this term, 10 tAA becomes more stable than 15 cAA by 20.8 kJ/mol, near the relative in-solution energy for the pure solutes. In the transition state, no water-water hydrogen bond formation was noticed. The NCCN torsion angle was found as 95.1° in comparison with 93.5° for the pure solute. The tetrahydrate transition state is higher in energy by 27 kJ/mol compared with the calculated value of 38.3 − 9.8 = 28.5 kJ/mol for the pure solute. The agreement is very good; consideration of explicit water molecules for the transition state along the s-trans to s-cis transformation results in only a small energy decrease.
As a partial summary, supermolecule models in a continuum dielectric solvent may distort the relative energies if only a limited number of water molecules are considered. In some solute conformations the water molecules may form water-water hydrogen bonds, whereas such bonds do not appear in the case of other solute structures. The water-water hydrogen bond interactions make the related supermolecule energy artificially too negative. This phenomenon typically emerges for conformational equilibrium calculations of small molecules with two, near polar sites open to form solute-water hydrogen bonds. If the relative energies are approximately corrected by the water-water hydrogen-bond interactions existing in one but not in another conformation, the calculated relative energies differ by less than 8 kJ/mol in comparison with the pure solute in-solution energies in the cases studied here at the B97D/aug-cc-pvtz level.
Nonetheless, the optimized tetrahydrate structures can well be compared with the derived first hydration shell structure of different conformers to be discussed below. Those results stem from Monte Carlo simulations, for which IEF-PCM derived net atomic charges have been utilized (
Table 2).
In a hydrogen bond, charges are always transferred from the acceptor to the donor [
33]. Since the X…H distances are shorter in the X…HOH hydrogen bonds (X = N, O) than in the N–H…O (water) bonds, the former bonds must be stronger and consequently larger amount of charges are to be transferred from the solute to the water molecules than in the opposite direction through the formation of the N–H…O (water) bonds. The total solute charge is zero for a pure solute in the continuum solvent. The total supermolecule charge is also zero but the net solute charge, due to charge transfers, is generally not zero. Depending on the direction of the net charge transfer, the solute can be both positive and negative, As discussed above, more charge are expected to leave the present solutes than being received from the surrounding water molecules, thus the net solute charge should be positive. Numerical results in
Table 2 prove it.
Based on the above numerical values, the tetrahydrate solute charges cannot be utilized in atomic charge parameterization for MC because an essential requirement is the total zero charge for an explicit-solvent solution model.
Table 2 shows that the net solute charge from the tetrahydrate is strongly positive. Since the atomic charges for the applied TIP4P model are fixed and sum up strictly to zero (although the molecule is polarized in comparison with the gas-phase water for producing good density and heat of vaporization for the liquid water [
37]), only the charges derived for the pure solute could be used in the present calculations.
By the FEP/MC procedure [
38,
39], the relative solvation free energy was calculated for different conformational/tautomeric transformations. The results are compared with the corresponding IEF-PCM/B97D (upper row) and IEF-PCM/MP2 (lower row) values in
Table 3. The goal of the presented MC calculations is not to completely repeat the corresponding IEF-PCM solvation free energy calculations. Results in
Table 3 were intended to point out that consideration of explicit solvent molecules, primarily water molecules would affect the derived ΔG
solv due to supposedly account for the thermally averaged solute-water hydrogen bond interactions in the first hydration shell.
Regarding the B97D results in the table, the ΔGsolv/PCM values as calculated by using the aug-cc-pvtz basis set are also provided in parentheses. These values deviate from the corresponding aug-cc-pvqz values within the rounding error. Thus the difference of the MP2/aug-cc-pvtz and B97D/aug-cc-pvtz ΔGsolv/PCM values should be attributed to the applied method, just like for ΔEsint as discussed above.
The FEP/MC simulations utilized the IEF-PCM/B97D/aug-cc-pvqz molecular electrostatic potential fitted charges. The calculated ΔG
solv values for the
s-trans/anti to
syn conformational change (1 to 2) by the IEF-PCM and MC methods are close in aqueous solution, suggesting that the solute is similarly well exposed to hydration, and formation of solute-water hydrogen bonds are favored in both conformations (
Table 4). For other conformer pairs, however, the situation is apparently largely different. For (7)
vs. (5) and (13)
vs. (12), the possible intramolecular hydrogen bond (see for the discussions above) must strongly reduce the solute’s capacity for forming solute-solvent hydrogen bonds and the MC value is then largely increased. Tautomeric change to structure (9) is not supported in any solvent by any method. The MC result is outstandingly unfavorable in water. In contrast, aqueous solvations of (8) and (15) are highly favored at any level, even though the MC values are remarkably less negative than those from IEF-PCM. The structural basis for the favorable solvation is that the
anti HNCC arrangement allows favorable hydration of the N–H bonds, whereas the nitrogen lone pairs are also open to accept a hydrogen bond with a nearby water molecule. The O…N and N…N distances are 285 and 290 pm in (8) and (15), respectively. There is room enough for locating 1-2 hydrogen bond donor water molecules. Nonetheless, ΔE
sint is too high for each of these species and prevents their appearance in an aqueous solution
Table 3.
IEF-PCM and FEP/Monte Carlo relative solvation free energies, ΔGsolv a.
Table 3.
IEF-PCM and FEP/Monte Carlo relative solvation free energies, ΔGsolv a.
| For Transformation | Dichloromethane | Water |
|---|
| IEF-PCM | MC | IEF-PCM | MC |
|---|
| CH2=CH–CH=NH | | | | |
| 1 to TS (toward 2) | 7.8 (7.8) | 5.1 ± 0.1 | | |
| 1 to 2 | −1.8 (−1.8) | −1.2 ± 0.2 | −2.0 (−2.1) | −1.6 ± 0.3 |
| | −2.1 | | −2.3 | |
| O=CH–CH=NH | | | | |
| 5 to 7 | 0.4 (0.4) | 1.4 ± 0.5 | 0.2 (0.3) | 9.5 ± 0.9 |
| | −1.9 | | −3.0 | |
| 5 to 8 | | | −14.9 (−14.8) | −9.0 ± 0.3 |
| | | | −22.3 | |
| 5 to 9 | 7.4 (7.4) | 5.7 ± 0.2 | 8.2 (8.2) | 14.1 ± 0.4 |
| | 9.2 | | 10.3 | |
| HN=CH–CH=NH | | | | |
| 10 to 11 | −3.4 (−3.3) | −0.8 ± 0.3 | −4.8 (−4.8) | −0.1 ± 0.4 |
| | −5.0 | | −6.8 | |
| 10 to 12 | | | −4.8 (−4.8) | 2.6 ± 0.4 |
| | | | −7.1 | |
| 10 to TS (toward 15) | | | −9.8 (−9.8) | −3.1 ± 0.2 |
| 10 to 15 | | | −18.5 (−18.6) | −9.4 ± 0.4 |
| | | | −24.4 | |
| 12 to 13 | 2.1 (2.1) | 4.5 ± 0.3 | 2.5 (2.5) | 10.2 ± 0.5 |
| | 0.4 | | 0.9 | |
Table 3 shows that the calculated B97D and the average MC relative solvation free energies differ by 0.6–2.7 kJ/mol in dichloromethane and the ΔG
solv signs always agree. The difference scatters between 0.4 and 9.3 kJ/mol for aqueous solutions and the signs calculated by the two methods agree in all cases but for ΔG
solv (10) to (12). This result, 2.6 ± 0.4 makes the relative free energy of the
s-trans/
syn/
syn conformer much less stable than calculated for the pure solute; ΔG
tot of 1.7 kJ/mol from
Table 1 increases to 9.1 kJ/mol. The result predicts only a small fraction for the
s-trans/
syn/
syn 1,2-diiminoathane conformer (12) in aqueous solution. The FEP/MC value is −0.1 ± 0.4 for the (10) to (11) transformation and would be −1.3 kJ/mol at the lower limit at the 3SD (99%) level, which is not enough to stabilize (11) relative to (10). ΔG
tot of −1.3 kcal/mol from
Table 1 increases to 2.2 kJ/mol on the basis of the MC calculations. A relative free energy of 2.2 kJ/mol corresponds to (11): (10) ratio of 29:71 at room temperature.
Table 4.
Coordination numbers (CN) and number of hydrogen bonds (nHB) in aqueous solution a.
Table 4.
Coordination numbers (CN) and number of hydrogen bonds (nHB) in aqueous solution a.
| Structures in Schemes | O/Ow | O/Hw | Nt/Ow | Nt/Hw | Nc/Ow | Nc/Hw | (N)Ht/Ow | (N)Hc/Ow | nHB b |
|---|
| CH2=CH–CH–NH (1) | | | 3.4 | 2.0 | | | 0.9 | | 2.3 (−3.0) c |
| CH2=CH–CH–NH (2) | | | 3.1 | 2.1 | | | 0.8 | | 2.4 (−3.0) c |
| O=CH–CH=NH (5) | 1.4 | 1.3 | 2.5 | 1.3 | | | 1.0 | | 2.9 (−3.0) |
| O=CH–CH=NH (7) | - d | 1.1 | | | 2.0 | 1.0 | | 0.8 | 1.4 (−3.5) c |
| O=CH–CH=NH (8) | 1.9 | 1.5 | | | 2.8 | 1.3 | | 1.0 | 3.0 (−3.5) |
| O=C=CH–NH2 (9) | - d | 0.6 e | - d | 1.0 | | | 0.7 | | 2.3 (−2.5) c |
| HN=CH–CH=NH (10) | | | 2.8 | 1.5 | | | 0.8 | | 3.9 (−3.0) |
| HN=CH–CH=NH (11) | | | 2.9, 3.1 | 1.3, 1.7 | | | 1.0, 1.0 | | 4.4 (−3.0) |
| HN=CH–CH=NH (12) | | | 3.0 | 1.5 | | | 0.95 | | 4.1 (−3.0) |
| HN=CH–CH=NH (13) f | | | | | 2.1, 2.7 | 1.3, 1.0 | | 0.4, 0.95 | 2.6 (−3.0) |
| TS (from
10 toward 15)f | | | 3.2 | 1.6 | | | | 1.0 | 4.3 (−3.0) |
| HN=CH–CH=NH (15) g | | | | | 2.9 | 1.6 | | 1.0 | 4.7 (−2.5) |
Since in any case but one the signs for ΔG
solv have been preserved when the IEF-PCM/B97D and the MC values are compared, consideration of the ΔG
solv/MC values would not modify the ΔG
tot results qualitatively as obtained by IEF-PCM (not even for (12)). In other words, the MC studies do not lead to the reversal of the preferred structure for the studied pairs. For the
s-cis O=CH-CH=NH conformer (7), even the IEF-PCM study predicted a fraction of only about 3% in the equilibrium composition in aqueous solution (whereas there is about 32% (6) also present as calculated from IEF-PCM values). On the basis of the MC results, the fraction must be further reduced. Each of the relative ΔE
sint and ΔG
solv is so much positive for O=C=CH–NH
2 (9) in both solvents (
Table 1) that this tautomer would not appear in the solution except if the kinetic control is in effect (see next section). For conformer (13), its fraction relative to the most stable (10)
s-trans form is about 3%–5% in both solvents upon the IEF-PCM calculations. On the basis of the FEP/MC simulations, this fraction practically disappears in solution. Thus each of the two models predicts small, possibly negligible fractions even for the most stable
s-cis conformations in the studied solutions.
2.3. Solution Structures
By utilization of the MC simulation results, some structural characteristics of the considered species in aqueous solution are summarized in
Table 4. The coordination numbers (CN) were calculated by integration of the radial distribution functions (rdf) [
40] up to their first minima, as indicated in the footnote of the table. The sites of the first minima scatter within a few tens of a pm. The
nHB values were obtained by integration of the solute-solvent pair-energy distribution functions (pedf) up to their first minima or until the middle of a plateau. According to Jorgensen
et al. [
37],
nHB may be considered as the number of the solute-solvent intermolecular hydrogen bonds in a protic solvent.
The CN and
nHB values are very similar for the
anti (1) and
syn (2)
s-trans CH
2=CH–CH–NH conformers. The slightly larger
nHB number for the
syn form and its dipole moment of 3.5 D
vs 2.9 D for the
anti form, calculated from point charges, are in accord with the −1.5 kJ/mol for ΔG
solv by MC. The B97D exact dipole moments are 3.6 and 3.0 D, respectively. In general, the point-charge-based dipole moments reproduced the exact values (
Table S1) generally within 0.2 D for the studied species.
While hydration characteristics of the
s-trans/anti (5) and
s-cis/
anti (8) (for which conformers tetrahydrates were also investigated in
Figure 1) are rather similar, coordination numbers for
s-cis/syn (7) 2-imino-acetaladehyde and for the amino-ketene tautomer (9) are rather different. The first solvation sphere around the carbonyl oxygen is well defined for (5) and (8) with O/O
w coordination numbers of 1.4–1.9. Comparing the O/O
w and O/H
w CN values for (5), the CN’s are nearly equal suggesting that every solvating water molecule forms one hydrogen bond pointing toward the carbonyl oxygen. O/H
w is less by 0.4 units than O/O
w for (8), suggesting that not all surrounding waters form hydrogen bonds with the carbonyl oxygen. This is in good accord with the 8 cA tetrahydrate structure prediction in
Figure 1, where water 2 is bound to water 1 instead of forming a =O…H
w21O
w2 hydrogen bond. No resolved O/O
w peaks were noted, however, for the other two C
2H
3NO species. There is an intramolecular hydrogen bond in the
s-cis conformer (7) and whereas the nitrogen can act as a full hydrogen-bond acceptor (N
c/H
w = 1.0), the (N)
cH proton can only partially act as a hydrogen-bond donor to a water oxygen as concluded from (N)
cH/O
w coordination number of 0.8, thus less than 1 for a full hydration. As a consequence, the
nHB value decreased from 2.9 for (5) to 1.4 for (7). Although the dipole moment is slightly larger for the latter (
Supplementary Table S1), the relative solvation free energy is remarkably positive, 9.5 ± 0.9 kJ/mol from MC simulations, probably due to the smaller number of the hydrogen bonds. The O/H
w CN for the ketene oxygen (9) is considerably smaller, 0.6 compared with 1.3 for (5), indicating a less strictly localized solvent sphere at this site. In contrast, the N
t/H
w as well as the (N)
tH/O
w coordination numbers for the two tautomers are much closer to each other, suggesting no basic hydration difference for the =NH and the –NH
2 groups. There are 1.0–1.3 water hydrogens around the nitrogen atoms for (5) and (9), which can form strong N…H
wO
w hydrogen bonds. Overall, the reduced
nHB number relative to that for (5) and the remarkably smaller dipole moment for (9) support the calculated ΔG
solv of 14.1 ± 0.4 kJ/mol.
The HN=CH–CH=NH
s-trans (10) and (12) conformers have a C
2h symmetry, whereas conformers (11) and (13) have only a C
s symmetry. The CN values (subscript “t” for (10–12) and “c” for (13)) must be very close for the equivalent nitrogens and hydrogens in structure (10) if statistical fluctuation is accepted (only the average of the almost equal individual values are presented). The same expectations apply for (12). The N–H groups are not identical in (13), for this species two values are presented. For conformers (10–12), the nitrogen atoms are open to hydration by water at their lone pair regions, and the imine hydrogens are also easily reachable by water oxygens. All these hydration patterns can assure formations of N…H
wO
w and N–H…O
w intermolecular hydrogen bonds. For conformer (13), the N
c/O
w and the (N)
cH/O
w values differ considerably from the corresponding N
t CNs. The differences stem from the deviations in the hydration abilities of the two imine groups. The right-hand side,
syn NH group of (13) can form a strongly bent intramolecular hydrogen bond with the left-hand side (
anti HN) nitrogen lone pair (
Scheme 2). This bond reduces the hydration capacity of the molecule in the top region, but allows for favorable N…H
wO
w bond formation at the right-hand side (CN = 1.3) and the (N)H…O
w hydration on the left-hand side (CN = 0.95). The reduced exposure of (13) to hydration, as revealed from the sum of the N
c/O
w values in comparison with the double of the N
t/O
w coordination number for structures (10) and (12) and with the sum of the N
t/O
w values for (11) necessarily leads to the decrease of the
nHB value by 1.3–1.8 units for the
s-cis conformer compared to that for the (10–12)
s-trans forms.
2.4. Equilibration Mechanism
Equilibration of some conformers is possible both in the gas phase and in the studied solvents by rotation about the central C–C bond because the calculated barrier heights are moderate. The studied pairs meeting the conditions are (1,3), (2,4), (5,8), (6,7), (10,15), (11,13) and (12,14). The calculated ΔG
stot values are, however, so large for (3, 4, 7, 8, 13–15) that the
s-cis conformers could be hardly observed experimentally. The intramolecular transformation for the pair (1,2) was found as proceeding along H rotation about the C=N bond followed by N-inversion, for which the activation energy was calculated at about 111 kJ/mol in solution for (2) (
Table 1).
In a dilute aqueous solution, small polar molecules could be dissolved in monomeric form at least at a large fraction. For monomers, the CC=NH
anti to
syn transformation must be feasible by water catalysis if the favorable hydration of structures of (1), (5), and (10–12) are considered (
Table 4). More directly, conclusions from the tetrahydrate structures could be drawn (
Figure 1). The water molecule w3 can donate a hydrogen and forms an N…H
w31–O
w3 hydrogen bond, whereas the w4 water molecule forms an N–H…O
w4 intermolecular hydrogen bond. The
anti/
syn transformation would be carried out by the H
w31 jump over to the nitrogen, whereas it releases its covalently bound proton toward O
w4. Along a sequential mechanism, a solute-solvent ion-pair is formed, irrespective of the actual proton involved in the first proton jump. The system’s status after the first proton jump will be, however, different. If H
w32 jumps first, the solute will be protonated facing an OH
− anion. If the N–H proton leaves first (less likely, since imines are not strong acids), a negatively charged solute anion and a hydroxonium cation form an ion-pair. In any case, the ion-pair could be stabilized in water for a short time until the second proton jump takes place. Now the solute is neutral, whereas there is a O
w3H
w32 hydroxyl anion and a HO
w4H
2 hydroxonium cation. The water molecules are members of a water network. Either in a concerted mechanism or through a sequential process, a double proton-relay results in the
anti to
syn transformation [
10] and the neutralization of the hydroxyl-hydroxonium ion-pair will be reached by proton jumps along the water chain connecting O
w3 and O
w4. Calculated coordination and hydrogen-bond numbers are in conformity with this supposed mechanism. The outlined equilibration mechanism for the (1,2) and (5,6) pairs is a reasonable alternative of the H rotation about the C=N bond followed by an N-inversion in aqueous solution. This mechanism must fail, however, in a non-protic solvent like dichloromethane. In that solvent, equilibration may proceed along an alternative route, namely through solute association.
N–H containing molecules could produce (N–H)
n rings with Nlp…H–N bonds, where lp stands for the nitrogen lone pair. Formation of a three-member ring was found experimentally even in the gas phase [
41]. Through concerted proton jumps in a dimer, the
syn conformer could come into existence. Polar solute association may be favored in a non-protic solvent, which supports the formation of intermolecular hydrogen bonds for the polar sites. Strong dimerization was found for acetic acid in chloroform [
10] and for pyruvic acid in dichloromethane [
1].
IEF-PCM/B97D/aug-cc-pvtz geometry optimization for an
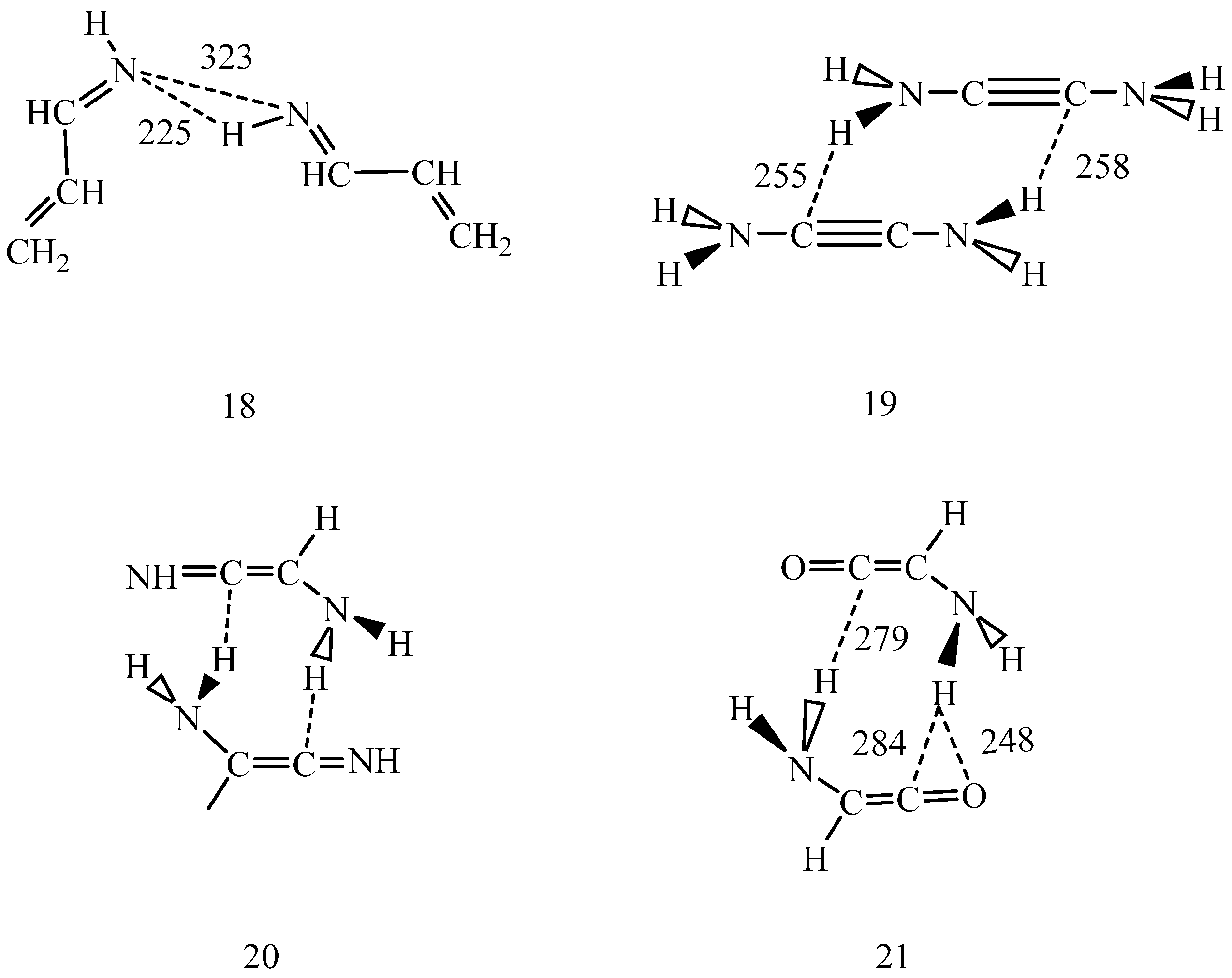
s-trans CH
2=CH–CH=NH dimer (18) (
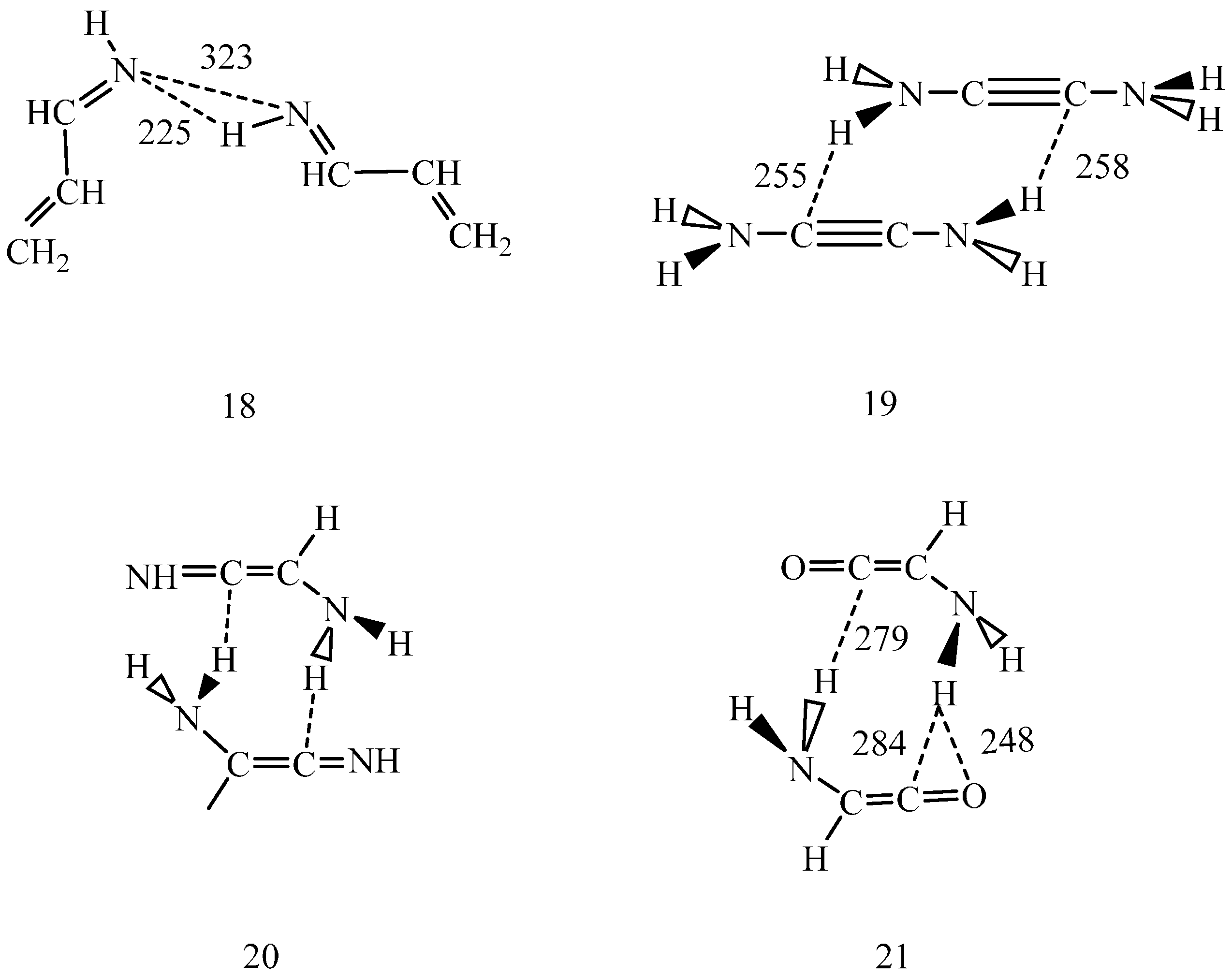
Scheme 3) in dichloromethane predicted individually planar monomers with C=N…N=C torsion angle of 42° at 323 pm N…N distance. One intermolecular hydrogen bond was noticed with N…H distance of 225 pm and N–H…N bond angle of 157°. The other N…H distance was determined as 391 pm.
Scheme 3.
IEF-PCM/B97D/aug-cc-pvtz optimized s-trans 2-propene-imine dimer (18) in dichloromethane. N–H…N angle 157°, C=N…N=C torsion angle 41.2°. 2-diamino acetylene dimer, optimized in the gas phase (19). Schematic structure of the 2-amino imino-ketene dimer (20). 2-amino ketene dimer, optimized in dichloromethane (21). Dashed lines indicate selected interatomic distances in pm.
Scheme 3.
IEF-PCM/B97D/aug-cc-pvtz optimized s-trans 2-propene-imine dimer (18) in dichloromethane. N–H…N angle 157°, C=N…N=C torsion angle 41.2°. 2-diamino acetylene dimer, optimized in the gas phase (19). Schematic structure of the 2-amino imino-ketene dimer (20). 2-amino ketene dimer, optimized in dichloromethane (21). Dashed lines indicate selected interatomic distances in pm.
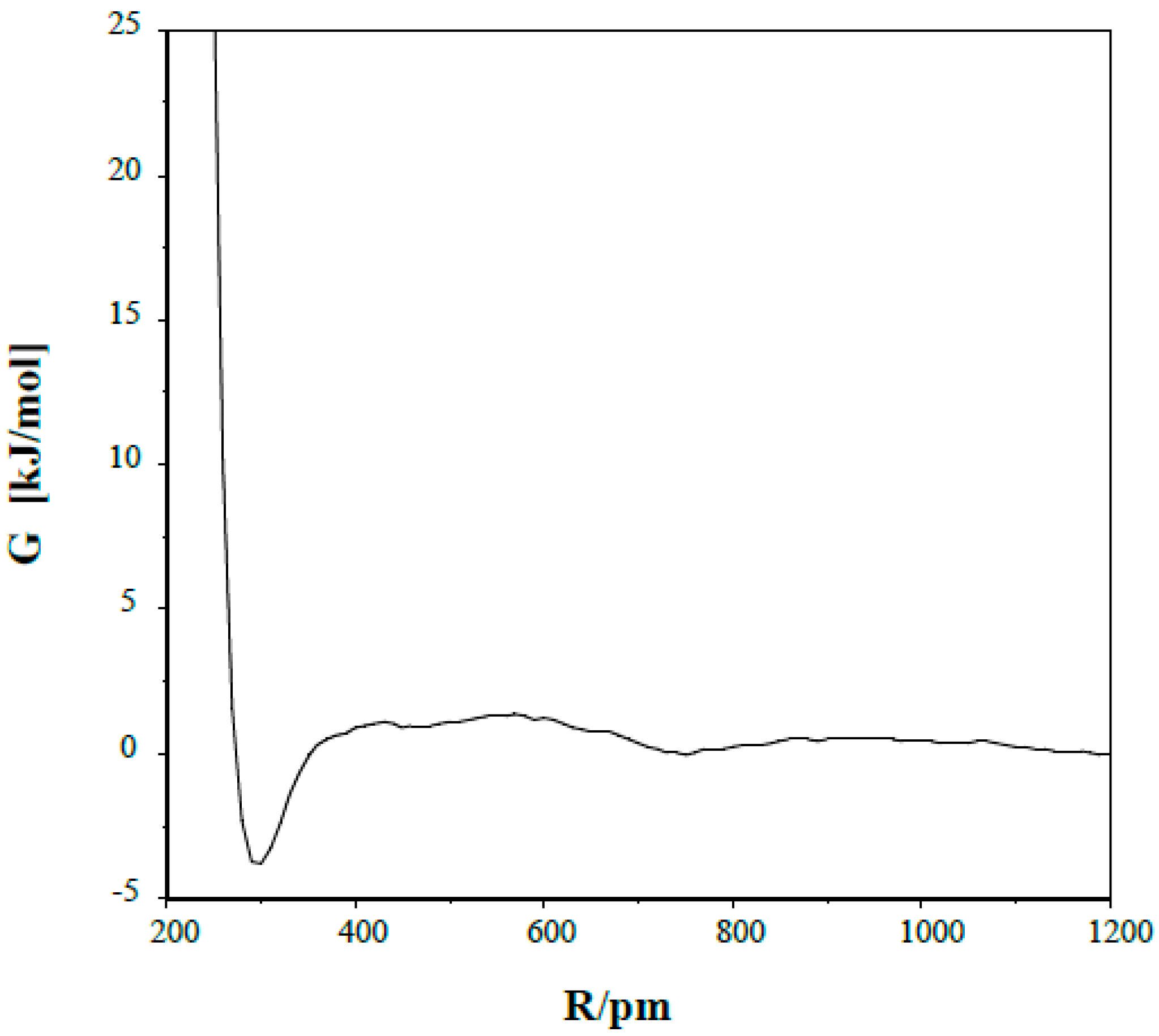
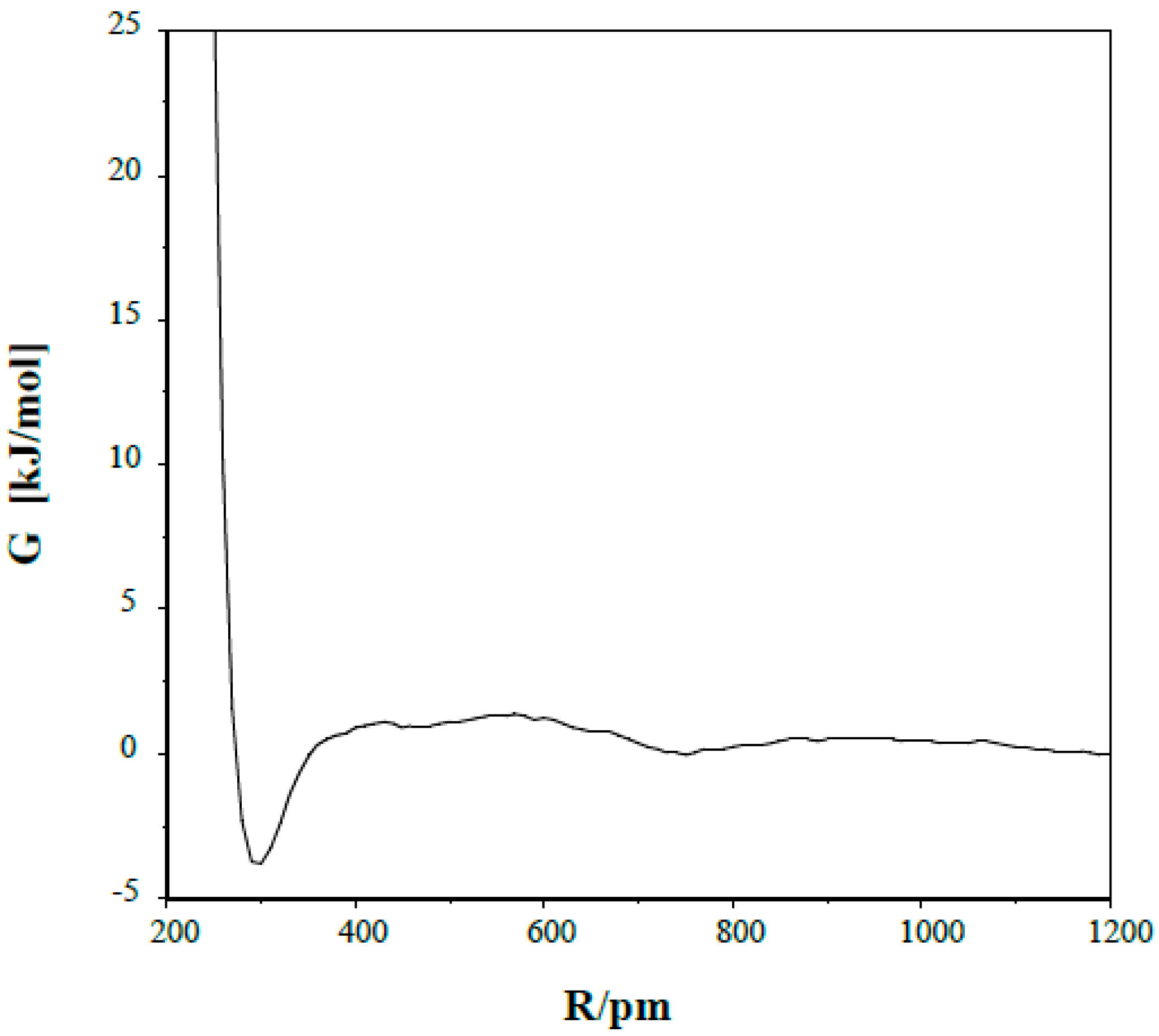
The potential of mean force curve (pmf) is shown in
Figure 2 for a pair of the (1) species in dichloromethane. The free energy of the system (with G = 0 at N…N separation of 1200 pm) has a maximum of 1.4 ± 0.3 kJ/mol at R(N…N) = 570 pm, becomes negative below R = 350 pm and reaches its minimum of −3.8 ± 0.4 kJ/mol with N…N separation of 300 pm.
The solution concentration is about 0.12 molar corresponding to two moles of solutes in a total volume of 17 dm
3. Assuming a small cube as its own volume for a solute, in the case of a uniform local solute density when the reference solute atoms reside at the centers of the cubes, their distances to the nearest neighbors are at about 2400 pm in a 0.12 molar solution. Thermal effects destroy this ordered arrangement, and as the calculated pmf shows, a fraction of the molecules tends to associate. The ratio of the integrals of the R
2exp(−ΔG/
RT) curve in the ranges of R(0, R(spec)) and R(0–2400) pm provides the fraction of the associated dimer relative to all other dimer arrangements [
42]. Here, R(spec) is an appropriately chosen N…N separation, which was accepted as 570 pm in the present calculation, corresponding to the site of the top of the association barrier. G was accepted as maintaining the reference value (G = 0) at R > 1200 pm. The calculated total associated fraction was slightly more than 1%, but is much less than that in the N…N separation range of 300–320 pm required for the formation of a hydrogen-bond within the dimer.
Figure 2.
Potential of mean force for the s-trans/anti dimer in dichloromethane.
Figure 2.
Potential of mean force for the s-trans/anti dimer in dichloromethane.
Upon inspection of some snapshots through MC simulations, arrangements with one, primarily linear N–H…N hydrogen bond was found for the dimer at the minimum (R = 300 pm) of the pmf. This orientation is not far from the calculated optimal dimer structure and is not favorable for a double proton-relay. Although the thermal motion may lead to some bent N–H…N structure, a prerequisite for forming another hydrogen bond, the pmf does not support the concerted double proton-relay. Jump over of a single proton would result in formation of an ion-pair temporarily, whose stabilization by a moderately polar solvent like CH2Cl2 is questionable. Thus the idea of a collision-based H rotation about the C=N bond/N-inversion is left in the dicholoromethane solvent.
Exploration of the tautomerization mechanism for H
2N–C≡C–NH
2 (17) is also a complicated problem. Geometry optimization in the gas phase predicts linear arrangement for the heavy atoms, whereas the rotational positions of the two amino groups break any symmetry for the molecule. In fact, two optical antipodes are possible, and no easily reachable path is available for an intramolecular 1,3 hydrogen relocation, which would lead to the formation of the HN=C=CH–NH
2 tautomer (
Scheme 2).
By studying dimeric structures (19–21,
Scheme 3), possibilities for their tautomerizations were investigated. As calculated at the B97D/aug-cc-pvtz level in the gas phase (for calculation details, see [
10]), only about 1% of the (19) monomers form a dimer. Concerted double proton-relay is unlikely along the two weak N–H…C hydrogen bonds. A sequential mechanism may be even less likely, because the formed ion-pair could be hardly stabilized in the gas phase. Considering the above experience with the pmf calculations, even dichloromethane may not have the satisfactory stabilizing effect to facilitate either the tautomeric transformation of (19) to (20) or the transformation of the (20) dimer to yield the dimer of some of (10–12).
Structure (21) shows the intermolecular hydrogen bond pattern calculated for the optimized amino ketene dimer in dichloromethane. The structure suggests that the N–H…O hydrogen bond must be stronger than any of the N–H…C hydrogen bonds in the dimer. Accordingly, if a proton jumps to the carbonyl oxygen, then the formed structure with an OH group will definitely differ from structures (5–8). This structure may later reorganize, but this reaction path strongly suggests that tautomerization of the studied systems in the gas phase and in non-polar solvents may not proceed via one-step double proton-relay mechanism.
An alternative is that the kinetic control is in effect regarding the tautomerization of the present systems. The ketenes (and possibly imino-ketenes) are stable in a non-aqueous solution [
12]. The calculations have found that the (9) and (16) structures correspond to local minima on the potential energy hypersurface in solution. If they were prepared in some non-aqueous solvent they may not convert to the corresponding most stable (5) and (10) structures according to the above discussion. Adding, however, even a catalytic amount of water to the solution, a provision of a proton to the carbon involved in cumulated double bonds and a drop-off of a proton at the NH
2 site could be a feasible process. The waters are in a water network again, and the neutralization of the formed ion-pair would easily proceed through a number of proton jumps along the water bridge.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}