
Pathological Bases for a Robust Application of Cancer Molecular Classification

Abstract
:1. Introduction
2. General Features of Tumor Classification: Current Status
3. Key Cellular and Molecular Processes to Incorporate in a Robust Classification
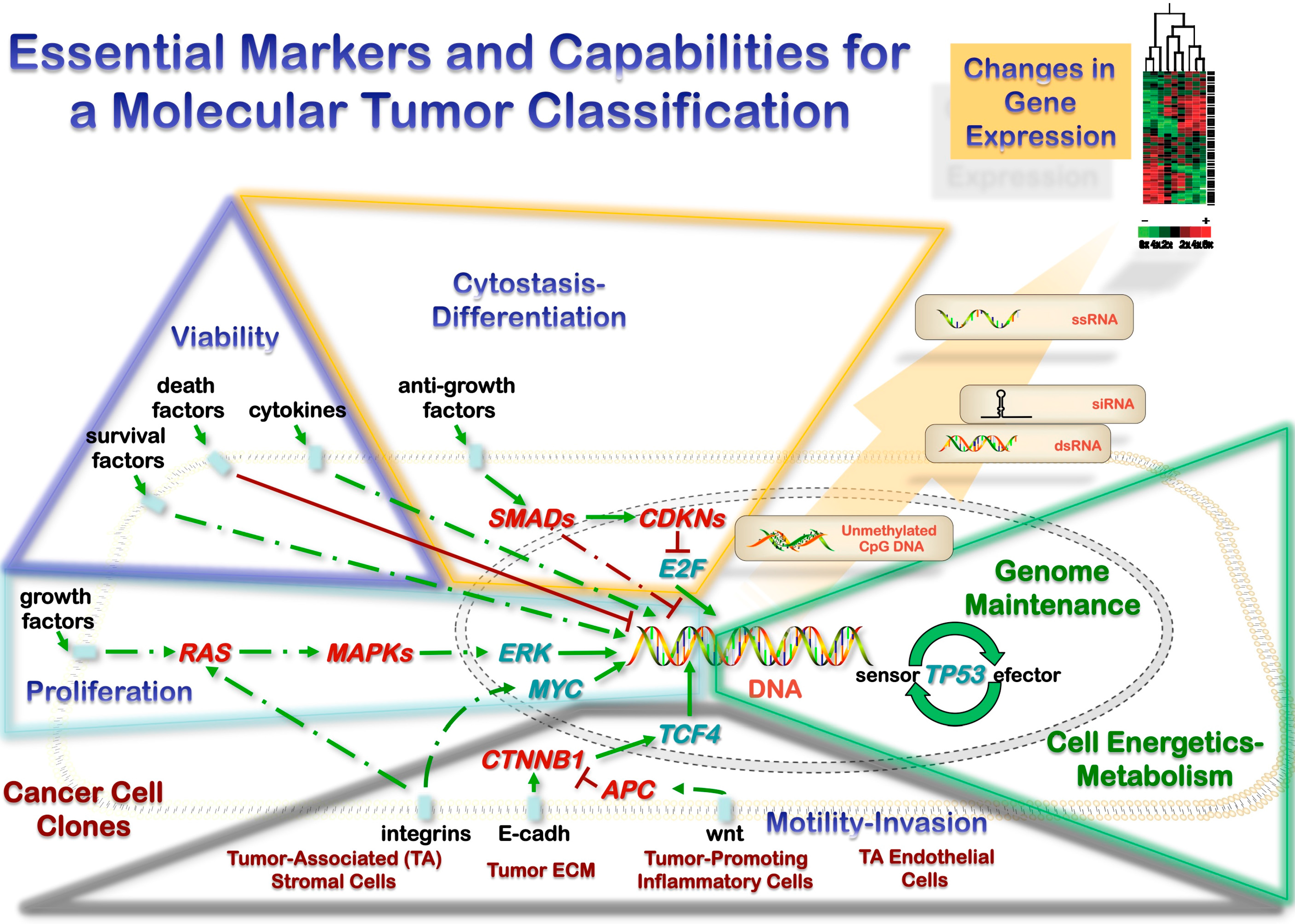
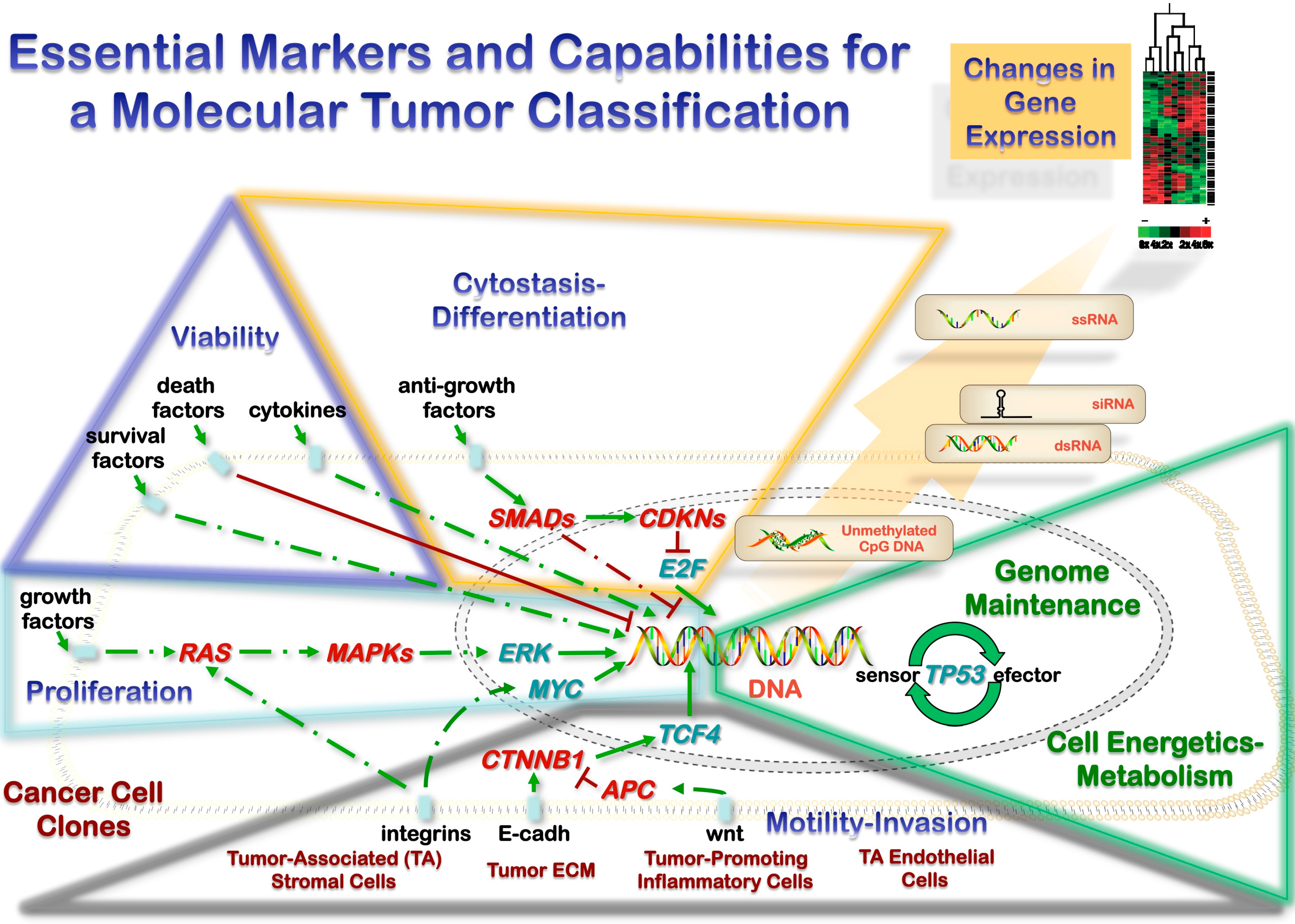
3.1. Heterotypic Tumor Biology: Tumor Cells-Microenvironment Interactions
3.2. Microenvironment and Metastasis
3.3. Gene Expression: Transfer of Genetic Material and Sequence-Independent Modifications
3.4. Molecular Test Scoring System for Practical Implementation
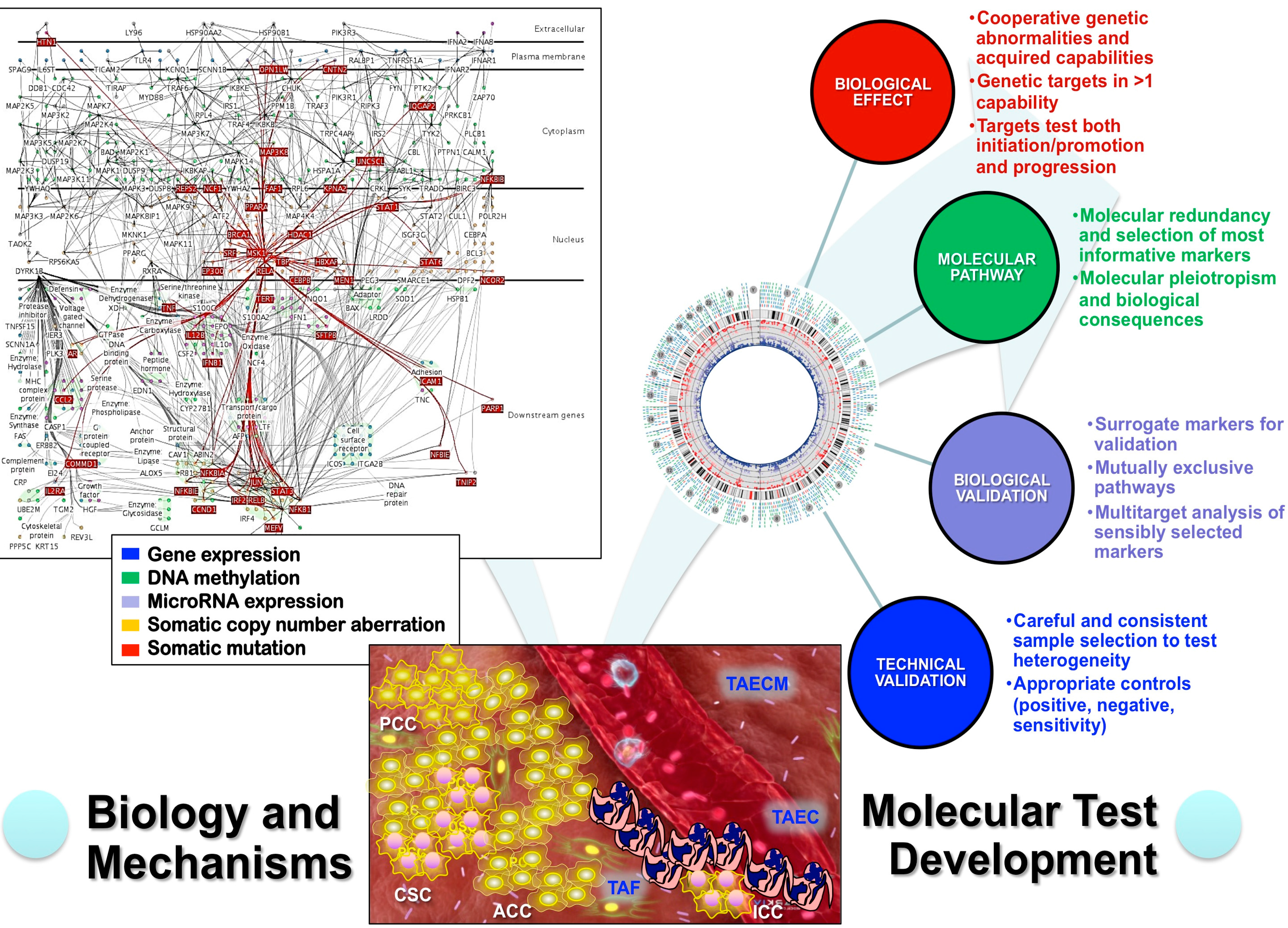
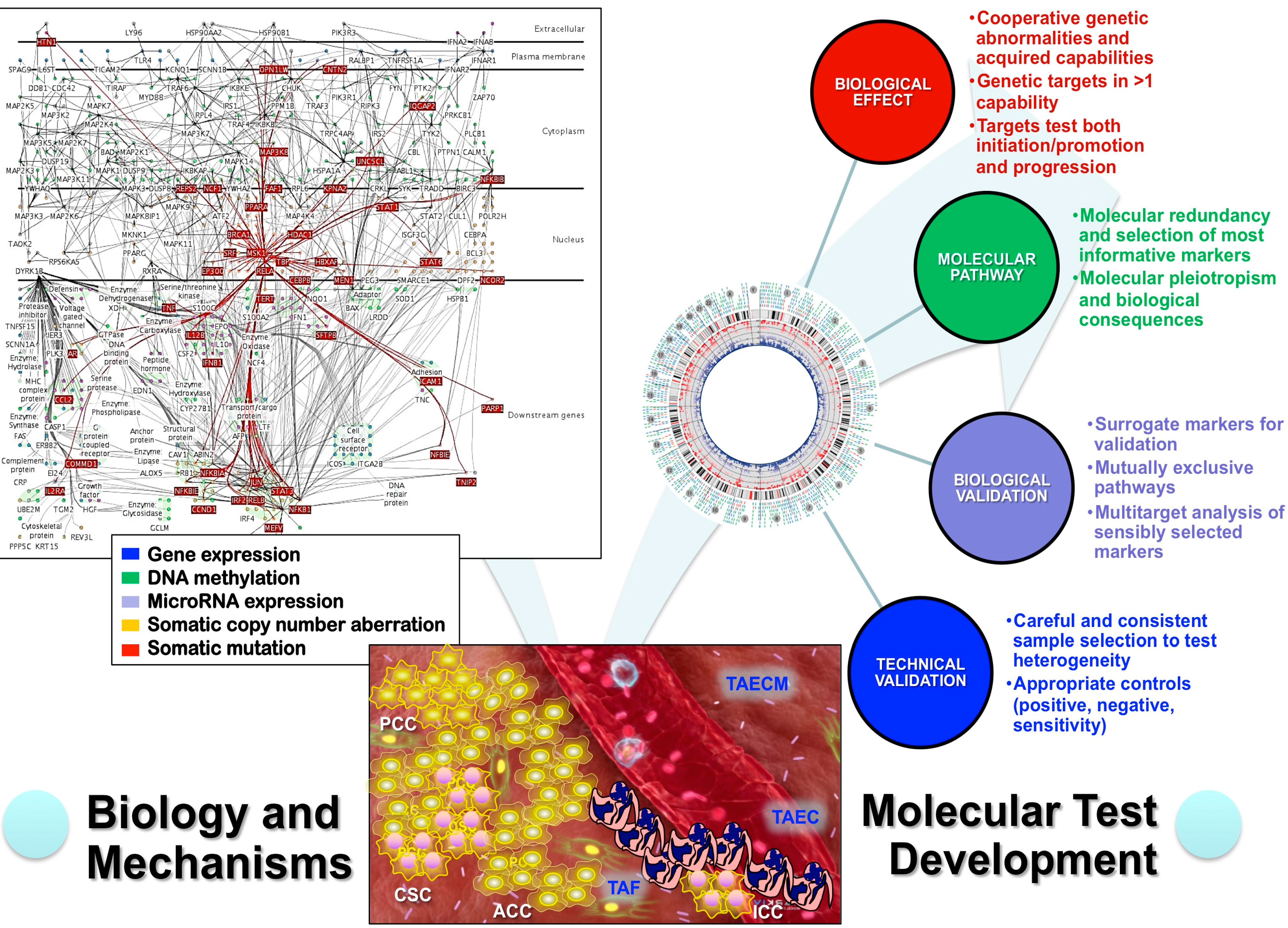
4. Analytical Genomic Classification of Tumors (AGCT)

{kind=link}
{kind=link}
| General Features | Specific Features |
|---|---|
| A classification is a hierarchical grouping | Each group is defined by the greatest number of informative features that can apply to every instance of the class. |
| Nomenclature should refer to differentiation/developmental terms internationally accepted. | |
| Every instance of the knowledge domain must fit the classification. | |
| Every instance and class must have exactly one slot in the classification. | |
| Instances of one class cannot migrate to a different class but must remain in the same class or a subclass of the same class. | |
| Instances and classes are separable from other instances and classes by informative features. | |
| All new findings of subpopulations of tumors can be considered candidate function to characterize a class and distinguish the class from other classes. | |
| Subclasses inherit the properties (shared informative features) of their ancestor classes. | |
| Familial syndromes and epidemiologic features | Prevalence of the disease should be significantly higher in those carrying the genetic abnormalities (if familial model exists). |
| Animal models of the genetic abnormality–Etiopathogenic features | Marker gene should be more commonly abnormally expressed in animals with the disease than in controls without the disease when all risk factors are held constant. |
| Incidence of the disease should be significantly higher in those animals with the abnormal gene than in those not exposed. | |
| A spectrum of preinvasive changes should follow the expression of the abnormal gene along a logical biologic gradient from mild to severe in the grading during neoplastic transformation (in particular for epithelial malignancies). | |
| Elimination or modification of the putative gene or of the vector carrying it should decrease the incidence of the disease. |
5. Conclusions
Conflicts of Interest
References
- Blanes, A.; Sanchez-Carrillo, J.J.; Diaz-Cano, S.J. Topographic molecular profile of pheochromocytomas: Role of somatic down-regulation of mismatch repair. J. Clin. Endocrinol. Metab. 2006, 91, 1150–1158. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.; Blanes, A.; Diaz-Cano, S.J. Hashimoto’s thyroiditis shares features with early papillary thyroid carcinoma. Histopathology 2002, 41, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Baithun, S.I.; Naase, M.; Blanes, A.; Diaz-Cano, S.J. Molecular and kinetic features of transitional cell carcinomas of the bladder: Biological and clinical implications. Virchows Arch. Int. J. Pathol. 2001, 438, 289–297. [Google Scholar] [CrossRef]
- Diaz-Cano, S.J. Designing a molecular analysis of clonality in tumours. J. Pathol. 2000, 191, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Pozo-Garcia, L.; Diaz-Cano, S.J. Clonal origin and expansions in neoplasms: Biologic and technical aspects must be considered together. Am. J. Pathol. 2003, 162, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Clarke, B.A.; Hasselblatt, M.; Majewski, J.; Albrecht, S.; McCluggage, W.G. No small surprise–small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J. Pathol. 2014, 233, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, P.; Mueller, J.J.; Olvera, N.; Dao, F.; Scott, S.N.; Shah, R.; Gao, J.; Schultz, N.; Gonen, M.; Soslow, R.A.; et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat. Genet. 2014, 46, 424–426. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, L.; Carrot-Zhang, J.; Albrecht, S.; Fahiminiya, S.; Hamel, N.; Tomiak, E.; Grynspan, D.; Saloustros, E.; Nadaf, J.; Rivera, B.; et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat. Genet. 2014, 46, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J. General morphological and biological features of neoplasms: Integration of molecular findings. Histopathology 2008, 53, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J. Tumor heterogeneity: Mechanisms and bases for a reliable application of molecular marker design. Int. J. Mol. Sci. 2012, 13, 1951–2011. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.-Y.; Bonvalot, S.; Casali, P.; Choi, H.; Debiec-Richter, M.; Dei Tos, A.P.; Emile, J.-F.; Gronchi, A.; Hogendoorn, P.C.W.; Joensuu, H.; et al. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST consensus conference of 20–21 march 2004, under the auspices of ESMO. Ann. Oncol. 2005, 16, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.T.; Zhao, Q.; Cai, Z.; Butte, A.J.; Kim, J.Y.H.; Pomeroy, S.L.; Rowitch, D.H.; Kohane, I.S. Conserved mechanisms across development and tumorigenesis revealed by a mouse development perspective of human cancers. Genes Dev. 2004, 18, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.S. Causation and disease: The henle-koch postulates revisited. Yale J. Biol. Med. 1976, 49, 175–195. [Google Scholar] [PubMed]
- Berman, J.J. Tumor classification: Molecular analysis meets aristotle. BMC Cancer 2004, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Shiota, K.; Kogo, Y.; Ohgane, J.; Imamura, T.; Urano, A.; Nishino, K.; Tanaka, S.; Hattori, N. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes Cells 2002, 7, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Allegrucci, C.; Thurston, A.; Lucas, E.; Young, L. Epigenetics and the germline. Reproduction 2005, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br. J. Cancer 2007, 96, R26–R30. [Google Scholar] [PubMed]
- Ooi, S.L.; Henikoff, S. Germline histone dynamics and epigenetics. Curr. Opin. Cell Biol. 2007, 19, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Boorman, G.A.; Anderson, S.P.; Casey, W.M.; Brown, R.H.; Crosby, L.M.; Gottschalk, K.; Easton, M.; Ni, H.; Morgan, K.T. Toxicogenomics, drug discovery, and the pathologist. Toxicol. Pathol. 2002, 30, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Spradling, A.C. Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Reddig, P.J.; Juliano, R.L. Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005, 24, 425–439. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Durrett, R.; Foo, J.; Leder, K.; Mayberry, J.; Michor, F. Intratumor heterogeneity in evolutionary models of tumor progression. Genetics 2011, 188, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J. Clonality studies in the analysis of adrenal medullary proliferations: Application principles and limitations. Endocr. Pathol. 1998, 9, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J.; Blanes, A.; Wolfe, H.J. Pcr techniques for clonality assays. Diagn. Mol. Pathol. 2001, 10, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E. The evolving concept of tumor microenvironments. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 738–744. [Google Scholar] [CrossRef]
- Laconi, E.; Doratiotto, S.; Vineis, P. The microenvironments of multistage carcinogenesis. Semin. Cancer Biol. 2008, 18, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E.; Sonnenschein, C. Cancer development at tissue level. Semin. Cancer Biol. 2008, 18, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Bindra, R.S.; Glazer, P.M.; Harris, A.L. Hypoxia-induced genetic instability—A calculated mechanism underlying tumor progression. J. Mol. Med. 2007, 85, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Oncogenic resistance to growth-limiting conditions. Nat. Rev. 2002, 2, 221–225. [Google Scholar] [CrossRef]
- Breivik, J. The evolutionary origin of genetic instability in cancer development. Semin. Cancer Biol. 2005, 15, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bielas, J.H.; Loeb, L.A. Mutator phenotype in cancer: Timing and perspectives. Environ. Mol. Mutagen. 2005, 45, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Blanes, A.; Diaz-Cano, S.J. Complementary analysis of microsatellite tumor profile and mismatch repair defects in colorectal carcinomas. World J. Gastroenterol. WJG 2006, 12, 5932–5940. [Google Scholar]
- Rubio, J.; Blanes, A.; Sanchez-Carrillo, J.J.; Diaz-Cano, S.J. Microsatellite abnormalities and somatic down-regulation of mismatch repair characterize nodular-trabecular muscle-invasive urothelial carcinoma of the bladder. Histopathology 2007, 51, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Cahill, D.P.; Lederer, G.; Speicher, M.R.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Carcinogen-specific induction of genetic instability. Proc. Natl. Acad. Sci. USA 2001, 98, 5770–5775. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Vegf-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGFβ signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Blanes, A.; Diaz-Cano, S.J. DNA and kinetic heterogeneity during the clonal evolution of adrenocortical proliferative lesions. Hum. Pathol. 2006, 37, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Blanes, A.; Rubio, J.; Martinez, A.; Wolfe, H.J.; Diaz-Cano, S.J. Kinetic profiles by topographic compartments in muscle-invasive transitional cell carcinomas of the bladder: Role of TP53 and NF1 genes. Am. J. Clin. Pathol. 2002, 118, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J.; Blanes, A.; Rubio, J.; Matilla, A.; Wolfe, H.J. Molecular evolution and intratumor heterogeneity by topographic compartments in muscle-invasive transitional cell carcinoma of the urinary bladder. Lab. Investig. J. Tech. Methods Pathol. 2000, 80, 279–289. [Google Scholar] [CrossRef]
- Jimenez, J.J.; Blanes, A.; Diaz-Cano, S.J. Microsatellite instability in colon cancer. N. Engl. J. Med. 2003, 349, 1774–1776. [Google Scholar] [CrossRef] [PubMed]
- Bissig, H.; Richter, J.; Desper, R.; Meier, V.; Schraml, P.; Schaffer, A.A.; Sauter, G.; Mihatsch, M.J.; Moch, H. Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am. J. Pathol. 1999, 155, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Bostwick, D.G.; Li, G.; Wang, Q.; Hu, N.; Vortmeyer, A.O.; Zhuang, Z. Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer 1999, 85, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Kuukasjarvi, T.; Karhu, R.; Tanner, M.; Kahkonen, M.; Schaffer, A.; Nupponen, N.; Pennanen, S.; Kallioniemi, A.; Kallioniemi, O.P.; Isola, J. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997, 57, 1597–1604. [Google Scholar] [PubMed]
- Ramaswamy, S.; Ross, K.N.; Lander, E.S.; Golub, T.R. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003, 33, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Glas, A.M.; Wessels, L.F.; Witteveen, A.T.; Peterse, J.L.; van’t Veer, L.J. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc. Natl. Acad. Sci. USA 2003, 100, 15901–15905. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Hu, Z.; He, X.; Livasy, C.; Carey, L.A.; Ewend, M.G.; Glas, A.M.; Perou, C.M.; van’t Veer, L.J. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005, 65, 9155–9158. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Laitinen, S.; Khan, S.; Vihinen, M.; Kowalski, J.; Yu, G.; Chen, L.; Ewing, C.M.; Eisenberger, M.A.; Carducci, M.A.; et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat. Med. 2009, 15, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.T.; van de Kaa, C.A.; Schalken, J.A.; Debruyne, F.M.; Ruiter, D.J. Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J. Pathol. 1996, 180, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, C.; Beitel, L.K.; Sircar, K.; Aprikian, A.; Trifiro, M.; Gottlieb, B. Somatic mosaicism and cancer: A micro-genetic examination into the role of the androgen receptor gene in prostate cancer. Cancer Res. 2005, 65, 8514–8518. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Hornsby, P.J. What can we learn from gene expression profiling for adrenal tumor management? J. Clin. Endocrinol. Metab. 2005, 90, 1900–1902. [Google Scholar] [CrossRef] [PubMed]
- Garber, M.E.; Troyanskaya, O.G.; Schluens, K.; Petersen, S.; Thaesler, Z.; Pacyna-Gengelbach, M.; van de Rijn, M.; Rosen, G.D.; Perou, C.M.; Whyte, R.I.; et al. Diversity of gene expression in adenocarcinoma of the lung. Proc. Natl. Acad. Sci. USA 2001, 98, 13784–13789. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. New insights into the tumor metastatic process revealed by gene expression profiling. Am. J. Pathol. 2005, 166, 1291–1294. [Google Scholar] [CrossRef] [PubMed]
- Van de Rijn, M.; Rubin, B.P. Gene expression studies on soft tissue tumors. Am. J. Pathol. 2002, 161, 1531–1534. [Google Scholar]
- Van’t Veer, L.J.; Paik, S.; Hayes, D.F. Gene expression profiling of breast cancer: A new tumor marker. J. Clin. Oncol. 2005, 23, 1631–1635. [Google Scholar]
- Ouatas, T.; Salerno, M.; Palmieri, D.; Steeg, P.S. Basic and translational advances in cancer metastasis: Nm23. J. Bioenerg. Biomembr. 2003, 35, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Salerno, M.; Ouatas, T.; Palmieri, D.; Steeg, P.S. Inhibition of signal transduction by the nm23 metastasis suppressor: Possible mechanisms. Clin. Exp. Metastasis 2003, 20, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. Metastasis suppressors alter the signal transduction of cancer cells. Nat. Rev. 2003, 3, 55–63. [Google Scholar] [CrossRef]
- Steeg, P.S.; Ouatas, T.; Halverson, D.; Palmieri, D.; Salerno, M. Metastasis suppressor genes: Basic biology and potential clinical use. Clin. Breast Cancer 2003, 4, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Wulfkuhle, J.D.; Paweletz, C.P.; Steeg, P.S.; Petricoin, E.F., III; Liotta, L. Proteomic approaches to the diagnosis, treatment, and monitoring of cancer. Adv. Exp. Med. Biol. 2003, 532, 59–68. [Google Scholar] [PubMed]
- Diaz-Cano, S.J. Paratumoral gene expression profiles: Promising markers of malignancy in melanocytic lesions. Br. J. Dermatol. 2011, 165, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Lavotshkin, S.; Lyden, D. The secreted factors responsible for pre-metastatic niche formation: Old sayings and new thoughts. Semin. Cancer Biol. 2011, 21, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Szabo, T.G.; Pasztoi, M.; Pal, Z.; Misjak, P.; Aradi, B.; Laszlo, V.; Pallinger, E.; Pap, E.; Kittel, A.; et al. Membrane vesicles, current state-of-the-art: Emerging role of extracellular vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Feng, Y.; Trivedi, N.S.; Huang, S. Medusa structure of the gene regulatory network: Dominance of transcription factors in cancer subtype classification. Exp. Biol. Med. 2011, 236, 628–636. [Google Scholar] [CrossRef]
- Guo, Z.; Wu, F.; Asplund, A.; Hu, X.; Mazurenko, N.; Kisseljov, F.; Ponten, J.; Wilander, E. Analysis of intratumoral heterogeneity of chromosome 3p deletions and genetic evidence of polyclonal origin of cervical squamous carcinoma. Mod. Pathol. 2001, 14, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. The epigenetics of cancer etiology. Semin. Cancer Biol. 2004, 14, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cano, S.J. Molecular mechanisms in melanoma. N. Engl. J. Med. 2006, 355, 1395–1396. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum. Pathol. 2002, 33, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, F.; Corless, C.L.; Duensing, A.; Hornick, J.L.; Oliveira, A.M.; Heinrich, M.C.; Fletcher, J.A.; Fletcher, C.D.M. Kit-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. Am. J. Surg. Pathol. 2004, 28, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, S.; Hasegawa, T.; Sakuma, Y.; Takazawa, Y.; Motegi, A.; Nakajima, T.; Saito, K.; Fukayama, M.; Shimoda, T. Myxoid epithelioid gastrointestinal stromal tumor (gist) with mast cell infiltrations: A subtype of gist with mutations of platelet-derived growth factor receptor alpha gene. Hum. Pathol. 2004, 35, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Laé, M.; Fréneaux, P.; Sastre-Garau, X.; Chouchane, O.; Sigal-Zafrani, B.; Vincent-Salomon, A. Secretory breast carcinomas with ETV6-NTRK3 fusion gene belong to the basal-like carcinoma spectrum. Mod. Pathol. 2009, 22, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Makretsov, N.; He, M.; Hayes, M.; Chia, S.; Horsman, D.E.; Sorensen, P.H.B.; Huntsman, D.G. A fluorescence in situ hybridization study of ETV6-NTRK3 fusion gene in secretory breast carcinoma. Genes Chromosomes Cancer 2004, 40, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Leeman-Neill, R.J.; Kelly, L.M.; Liu, P.; Brenner, A.V.; Little, M.P.; Bogdanova, T.I.; Evdokimova, V.N.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 2014, 120, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Li, Y.; Liu, J.; Yang, X.; Wang, L.; Zhou, Z.; Han, B. Morphologic features of carcinomas with recurrent gene fusions. Adv. Anat. Pathol. 2012, 19, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Skálová, A.; Vanecek, T.; Majewska, H.; Laco, J.; Grossmann, P.; Simpson, R.H.W.; Hauer, L.; Andrle, P.; Hosticka, L.; Branžovský, J.; et al. Mammary analogue secretory carcinoma of salivary glands with high-grade transformation: Report of 3 cases with the ETV6-NTRK3 gene fusion and analysis of TP53, β-catenin, EGFR, and CCND1 genes. Am. J. Surg. Pathol. 2014, 38, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Versteege, I.; Sévenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Becker, A.J.; LoTurco, J.J. Contribution of tumor heterogeneity in a new animal model of cns tumors. Mol. Cancer Res. 2014, 12, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Le Loarer, F.; Zhang, L.; Fletcher, C.D.; Ribeiro, A.; Singer, S.; Italiano, A.; Neuville, A.; Houlier, A.; Chibon, F.; Coindre, J.-M.; et al. Consistent smarcb1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by fish in archival material. Genes Chromosomes Cancer 2014, 53, 475–486. [Google Scholar]
- Vu-Han, T.-L.; Frühwald, M.C.; Hasselblatt, M.; Kerl, K.; Nagel, I.; Obser, T.; Oyen, F.; Siebert, R.; Schneppenheim, R. Identifying molecular markers for the sensitive detection of residual atypical teratoid rhabdoid tumor cells. Cancer Genet. 2014, 207, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Vu-Phan, D.; Grachtchouk, V.; Yu, J.; Colby, L.A.; Wicha, M.S.; Koenig, R.J. The thyroid cancer PAX8-PPARG fusion protein activates Wnt/TCF-responsive cells that have a transformed phenotype. Endocr. Relat. Cancer 2013, 20, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Li, S.; Garcia-Rendueles, M.E.R.; Montero-Conde, C.; Voza, F.; Knauf, J.A.; Heguy, A.; Viale, A.; Bogdanova, T.; Thomas, G.A.; et al. Identification of kinase fusion oncogenes in post-chernobyl radiation-induced thyroid cancers. J. Clin. Investig. 2013, 123, 4935–4944. [Google Scholar] [CrossRef] [PubMed]
- Mackinnon, A.C.; Yan, B.C.; Joseph, L.J.; Al-Ahmadie, H.A. Molecular biology underlying the clinical heterogeneity of prostate cancer: An update. Arch. Pathol. Lab. Med. 2009, 133, 1033–1040. [Google Scholar] [PubMed]
- Mehra, R.; Han, B.; Tomlins, S.A.; Wang, L.; Menon, A.; Wasco, M.J.; Shen, R.; Montie, J.E.; Chinnaiyan, A.M.; Shah, R.B. Heterogeneity of TMPRSS2 gene rearrangements in multifocal prostate adenocarcinoma: Molecular evidence for an independent group of diseases. Cancer Res. 2007, 67, 7991–7995. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.; Valeri, A.; Mangin, P.; Cussenot, O. Prostate cancer. Epidemiology. Risk factors. Pathology. Ann. Urol. 2004, 38, 187–206. [Google Scholar] [CrossRef]
- Hodge, J.C.; Pearce, K.E.; Wang, X.; Wiktor, A.E.; Oliveira, A.M.; Greipp, P.T. Molecular cytogenetic analysis for TFE3 rearrangement in Xp11.2 renal cell carcinoma and alveolar soft part sarcoma: Validation and clinical experience with 75 cases. Mod. Pathol. 2014, 27, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Kobos, R.; Nagai, M.; Tsuda, M.; Merl, M.Y.; Saito, T.; Laé, M.; Mo, Q.; Olshen, A.; Lianoglou, S.; Leslie, C.; et al. Combining integrated genomics and functional genomics to dissect the biology of a cancer-associated, aberrant transcription factor, the ASPSCR1-TFE3 fusion oncoprotein. J. Pathol. 2013, 229, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Ross, H.; Argani, P. Xp11 translocation renal cell carcinoma. Pathology 2010, 42, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Gulley, M.L.; Kaiser-Rogers, K.A. A rational approach to genetic testing for sarcoma. Diagn. Mol. Pathol. 2009, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.; Traves, V.; Cruz, J.; Llombart, B.; Navarro, S.; Llombart-Bosch, A. Superficial small round-cell tumors with special reference to the ewing’s sarcoma family of tumors and the spectrum of differential diagnosis. Semin. Diagn. Pathol. 2013, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.D.; Burchill, S.A.; Abrams, K.R.; Heney, D.; Sutton, A.J.; Jones, D.R.; Lambert, P.C.; Young, B.; Wailoo, A.J.; Lewis, I.J. A systematic review of molecular and biological markers in tumours of the ewing’s sarcoma family. Eur. J. Cancer 2003, 39, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat. Genet. 2004, 36, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Tamayo, P.; Rifkin, R.; Mukherjee, S.; Yeang, C.H.; Angelo, M.; Ladd, C.; Reich, M.; Latulippe, E.; Mesirov, J.P.; et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 2001, 98, 15149–15154. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-S.; Lin, C.-T.; Tseng, G.C.; Chung, I.-F.; Pal, N.R. Discovery of dominant and dormant genes from expression data using a novel generalization of snr for multi-class problems. BMC Bioinform. 2008, 9, 425. [Google Scholar] [CrossRef]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Perez-Diez, A.; Morgun, A.; Shulzhenko, N. Microarrays for cancer diagnosis and classification. Adv. Exp. Med. Biol. 2007, 593, 74–85. [Google Scholar] [PubMed]
- Statnikov, A.; Aliferis, C.F. Are random forests better than support vector machines for microarray-based cancer classification? AMIA Annu. Symp. Proc. 2007, 686–690. [Google Scholar]
- Brownlee, N.A.; Perkins, L.A.; Stewart, W.; Jackle, B.; Pettenati, M.J.; Koty, P.P.; Iskandar, S.S.; Garvin, A.J. Recurring translocation (10;17) and deletion (14q) in clear cell sarcoma of the kidney. Arch. Pathol. Lab. Med. 2007, 131, 446–451. [Google Scholar] [PubMed]
- Rakheja, D.; Weinberg, A.G.; Tomlinson, G.E.; Partridge, K.; Schneider, N.R. Translocation (10;17)(q22;p13): A recurring translocation in clear cell sarcoma of kidney. Cancer Genet. Cytogenet. 2004, 154, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Meis-Kindblom, J.M. Clear cell sarcoma of tendons and aponeuroses: A historical perspective and tribute to the man behind the entity. Adv. Anat. Pathol. 2006, 13, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Mandahl, N. Absence of mutations of the braf gene in malignant melanoma of soft parts (clear cell sarcoma of tendons and aponeuroses). Cancer Genet. Cytogenet. 2005, 156, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Covinsky, M.; Gong, S.; Rajaram, V.; Perry, A.; Pfeifer, J. EWS-ATF1 fusion transcripts in gastrointestinal tumors previously diagnosed as malignant melanoma. Hum. Pathol. 2005, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Cano, S.J. Pathological Bases for a Robust Application of Cancer Molecular Classification. Int. J. Mol. Sci. 2015, 16, 8655-8675. https://doi.org/10.3390/ijms16048655
Diaz-Cano SJ. Pathological Bases for a Robust Application of Cancer Molecular Classification. International Journal of Molecular Sciences. 2015; 16(4):8655-8675. https://doi.org/10.3390/ijms16048655
Chicago/Turabian StyleDiaz-Cano, Salvador J. 2015. "Pathological Bases for a Robust Application of Cancer Molecular Classification" International Journal of Molecular Sciences 16, no. 4: 8655-8675. https://doi.org/10.3390/ijms16048655
APA StyleDiaz-Cano, S. J. (2015). Pathological Bases for a Robust Application of Cancer Molecular Classification. International Journal of Molecular Sciences, 16(4), 8655-8675. https://doi.org/10.3390/ijms16048655