
Characterization of Aspartate Kinase from Corynebacterium pekinense and the Critical Site of Arg169


Abstract
:
1. Introduction

2. Results and Discussion
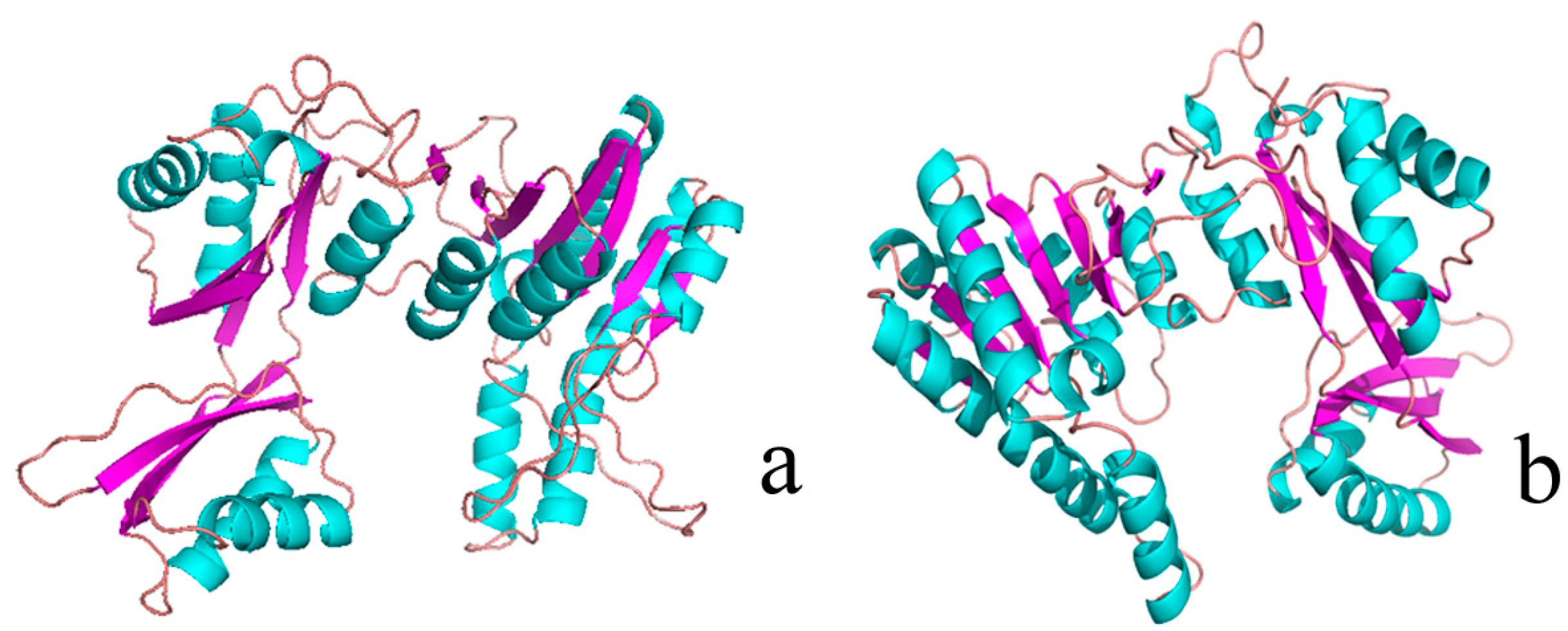
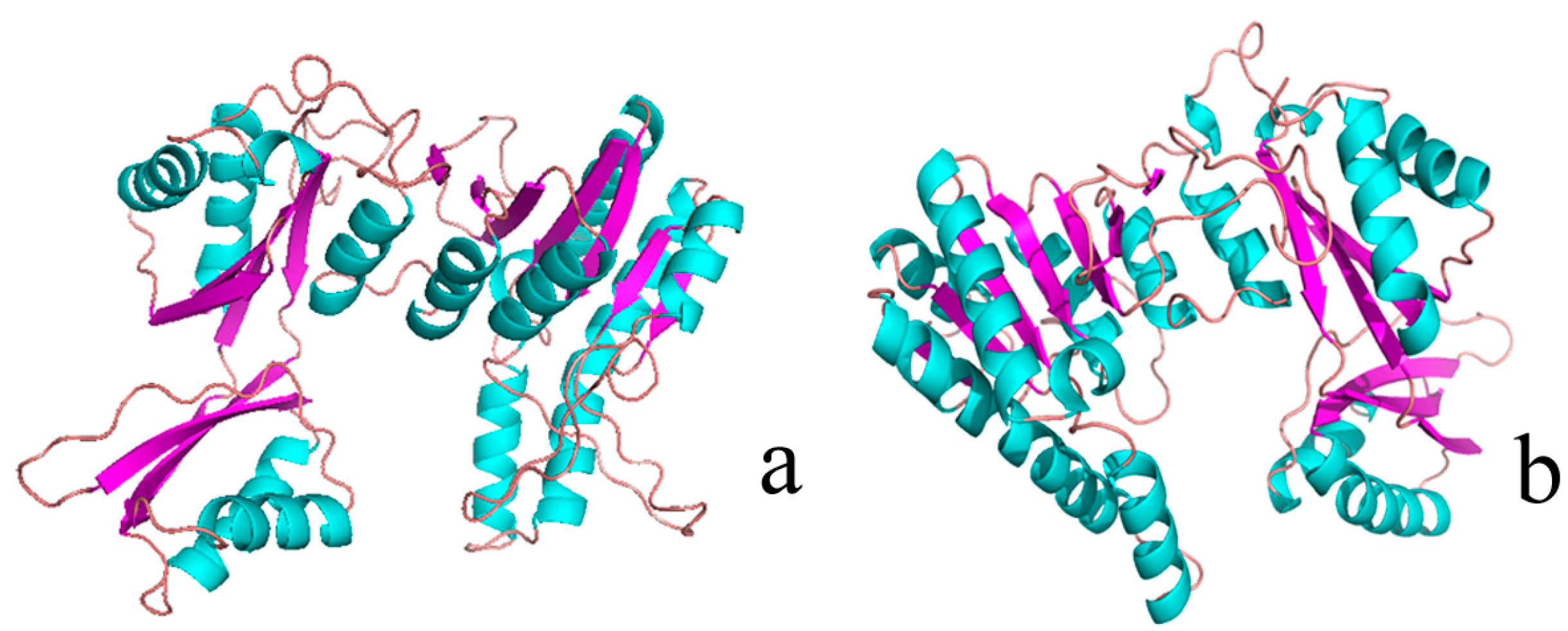
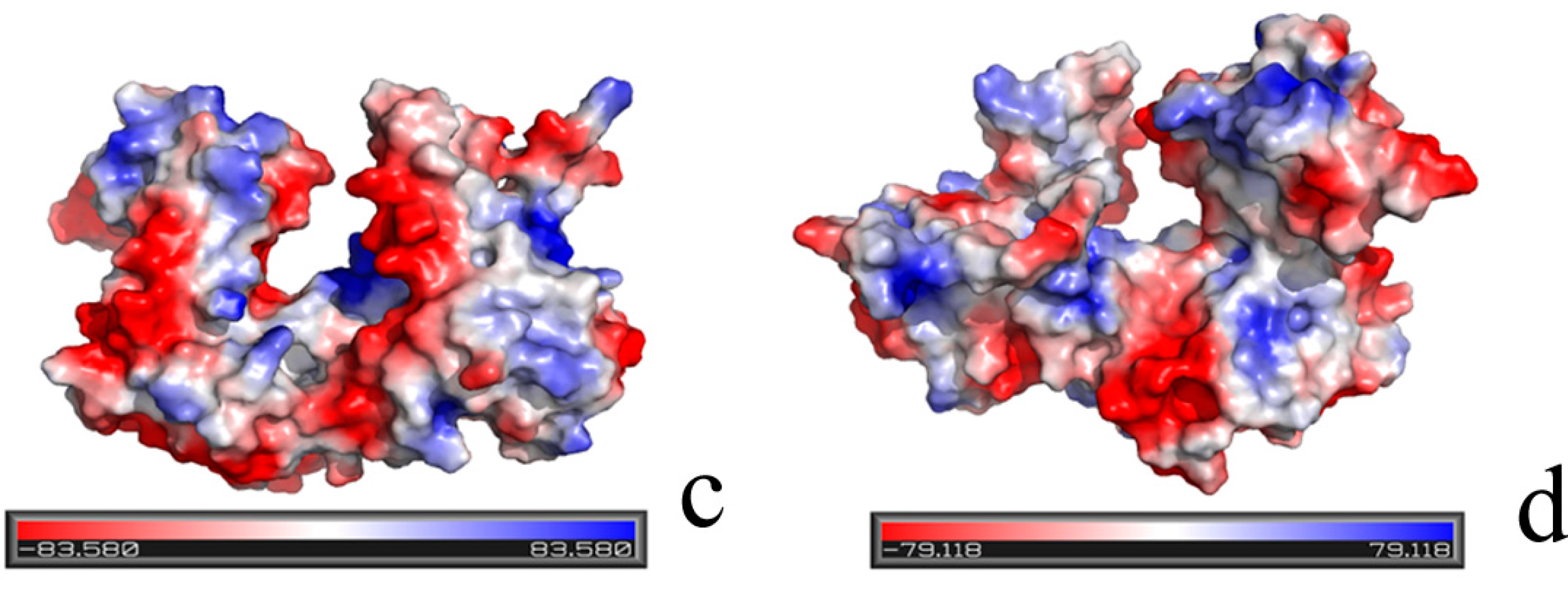
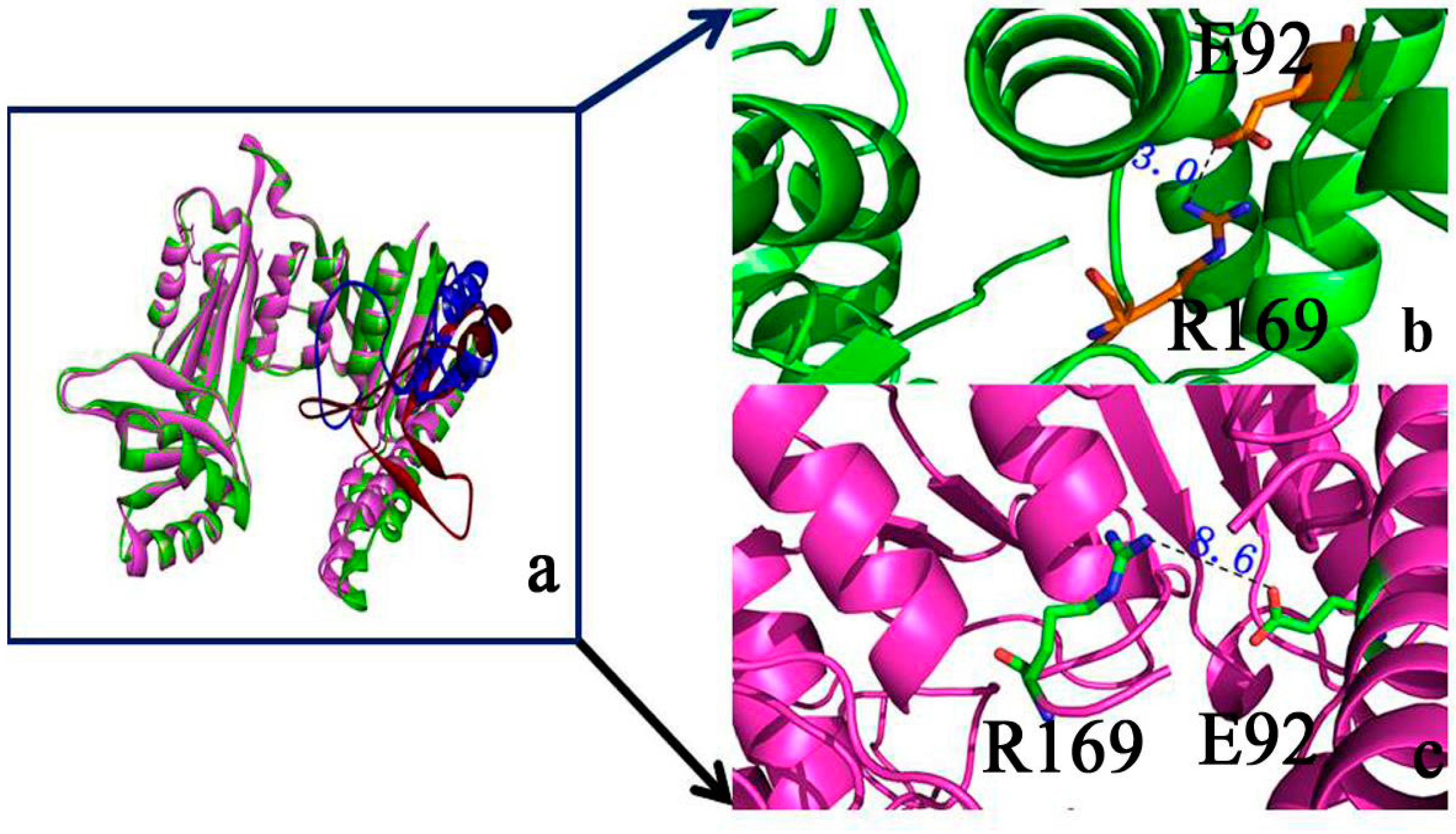
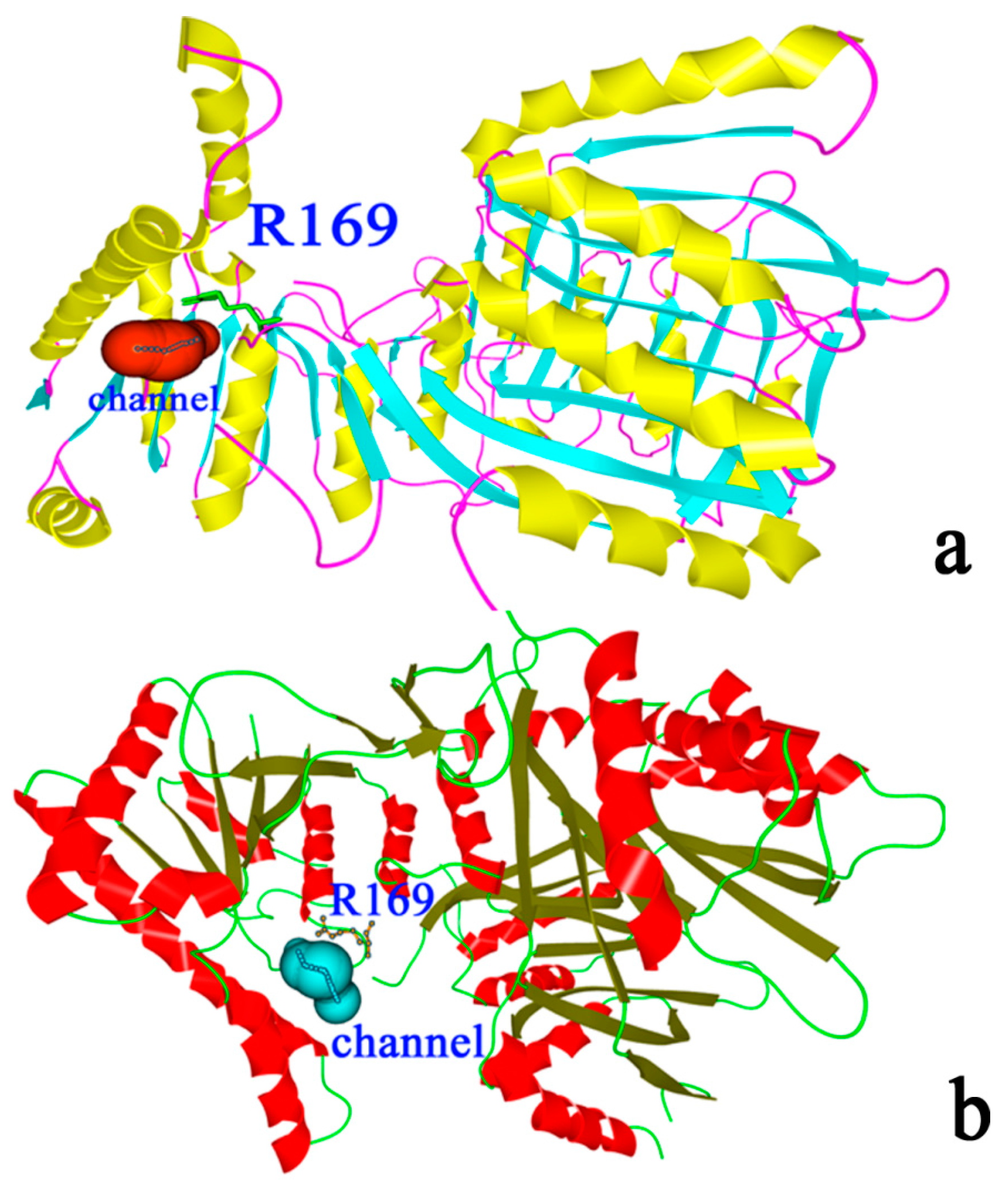
2.1. Structural Differences between the Active and Inactive Forms of Aspartate Kinase


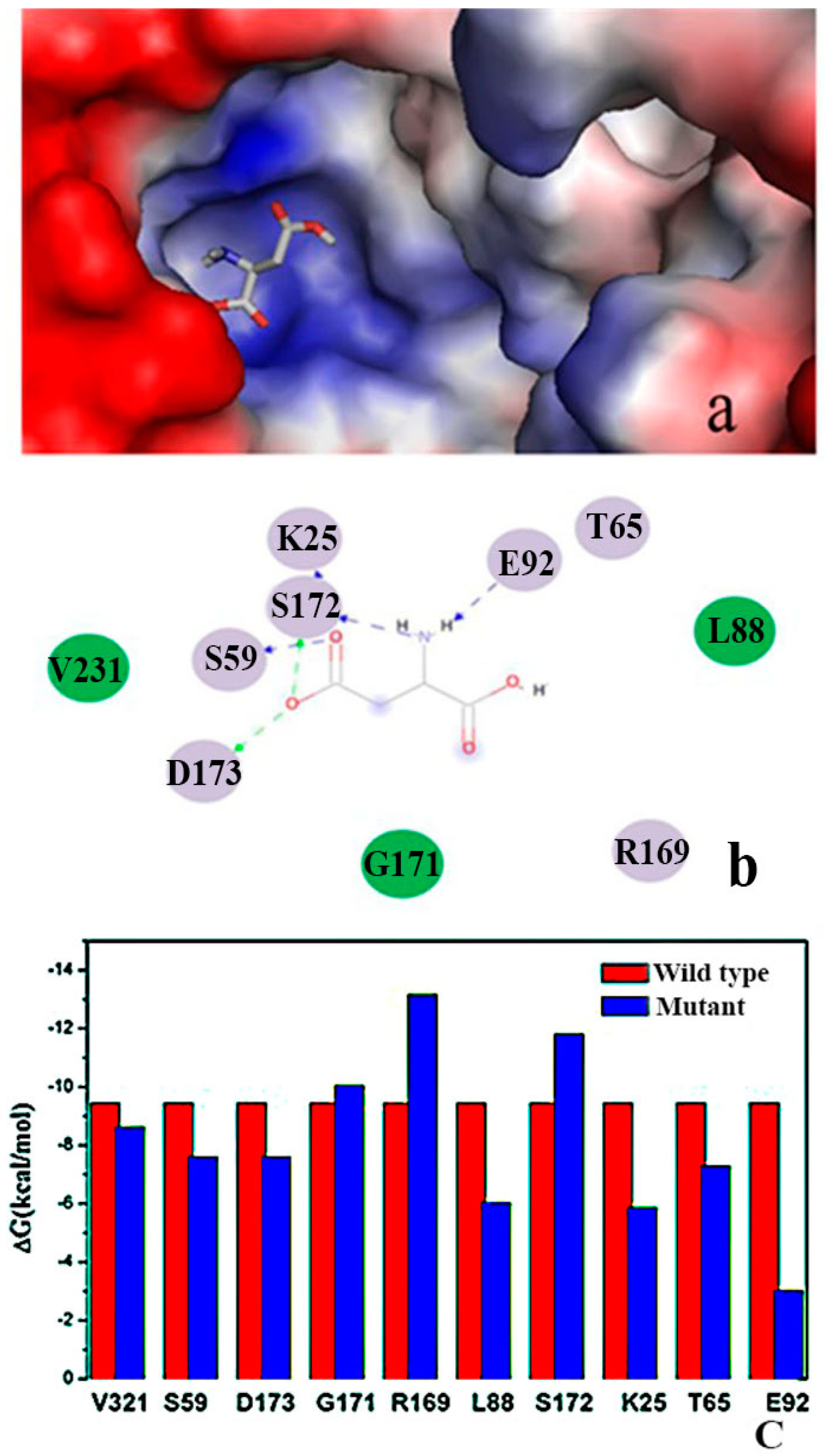
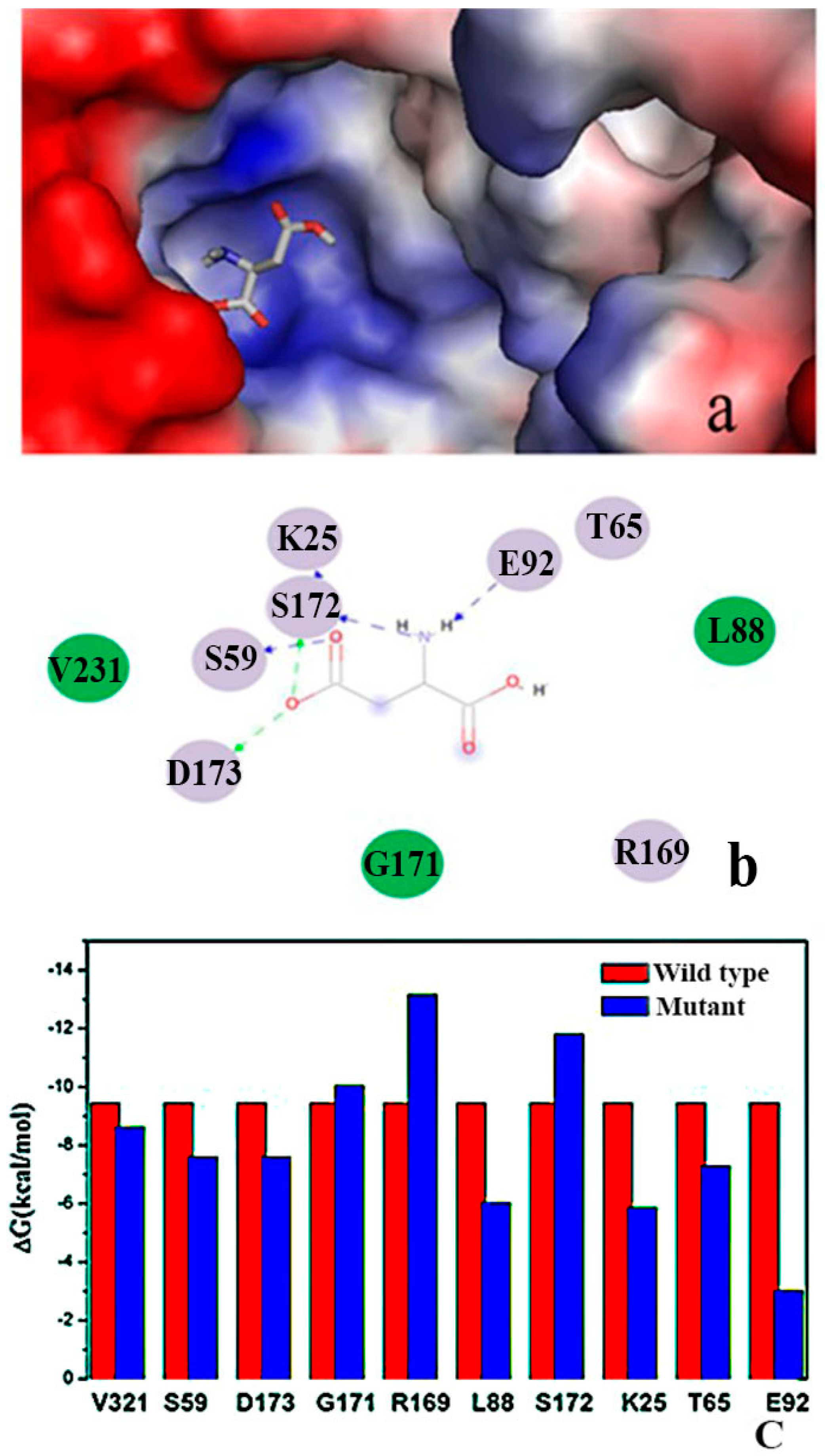
2.2. Binding Position of AK
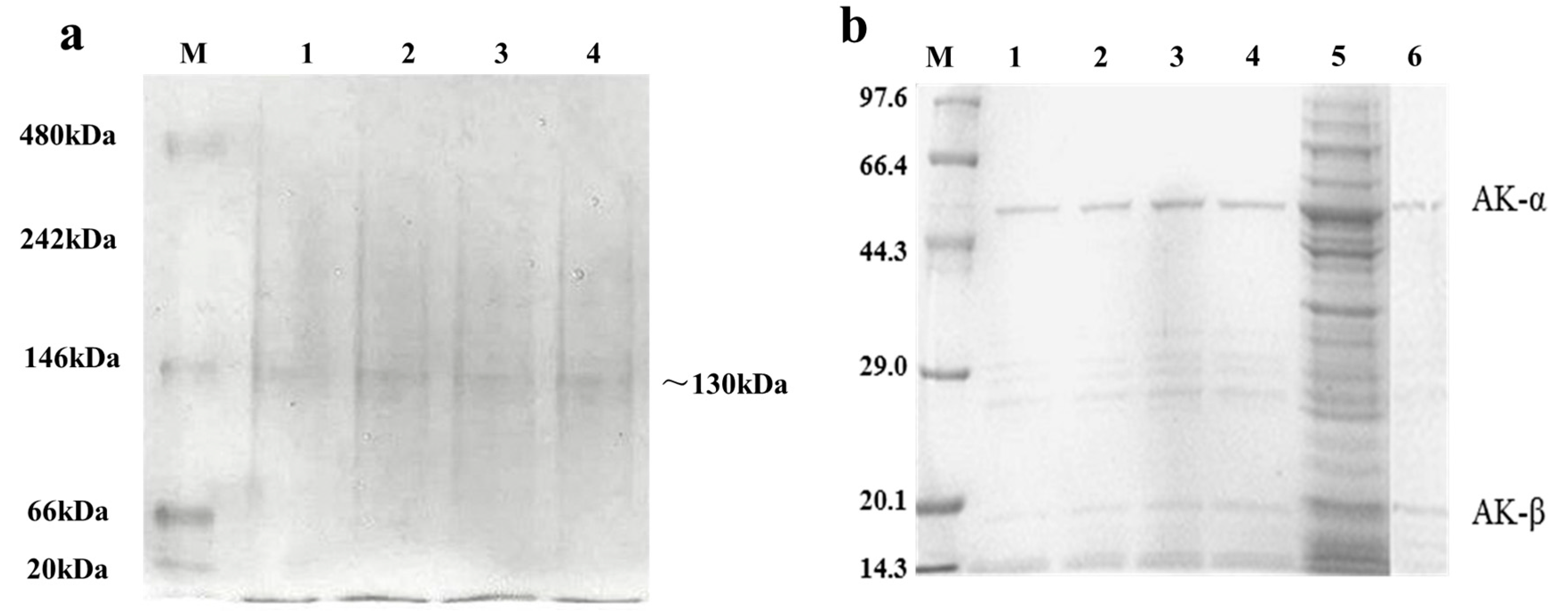
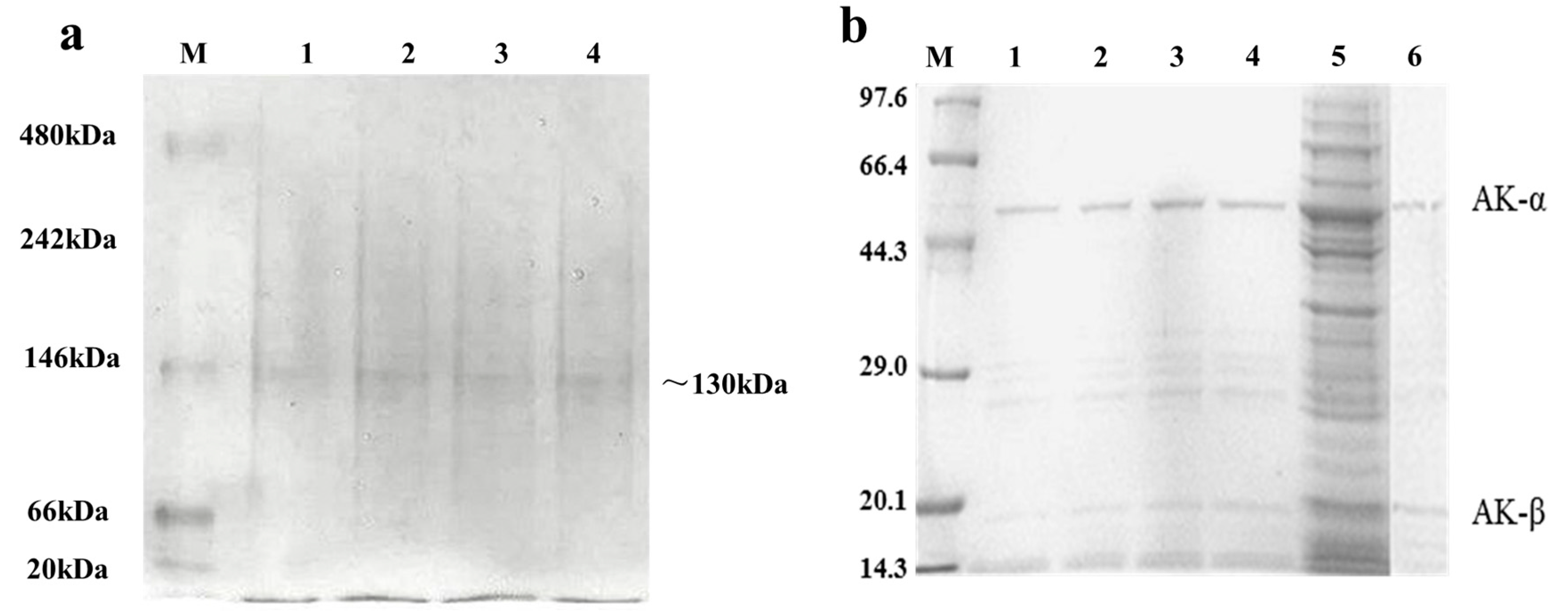
2.3. Expression and Purification of Wild Type and Mutant AK
2.4. Kinetic Assay of the Wild Type (WT) and AK Mutants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | WT | R169Y | R169P | R169D | R169H |
|---|---|---|---|---|---|
| Vmax (U/mg) | 96.07 | 452.78 | 216.55 | 208.77 | 204.25 |
| Km (mol·L−1) | 4.56 | 4.98 | 4.95 | 6.29 | 8.79 |
| nH | 2.82 | 1.13 | 1.95 | 0.91 | 1.09 |
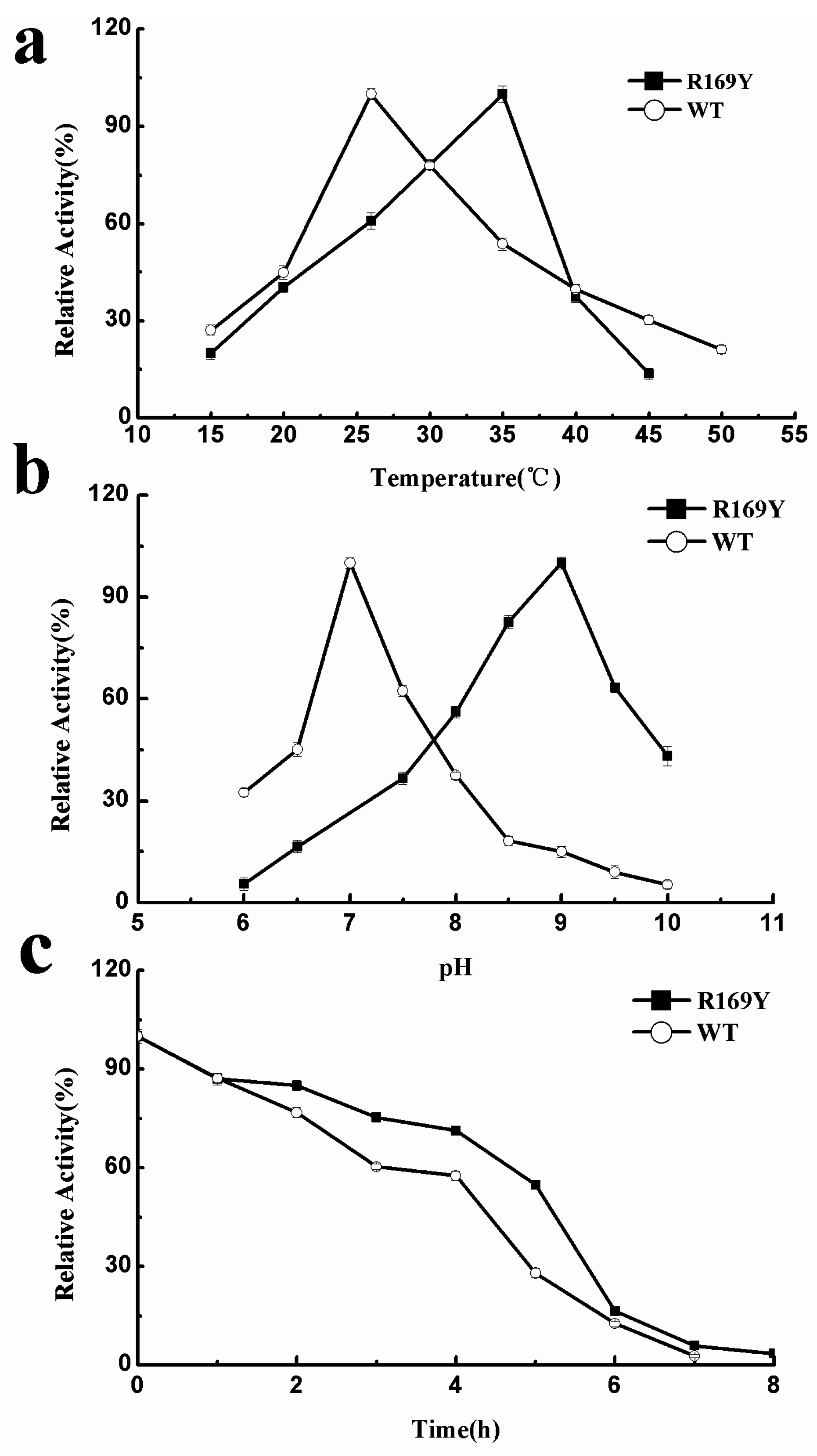
2.5. Optimum Temperature, pH, and Thermostability of Wild Type (WT) and R169Y
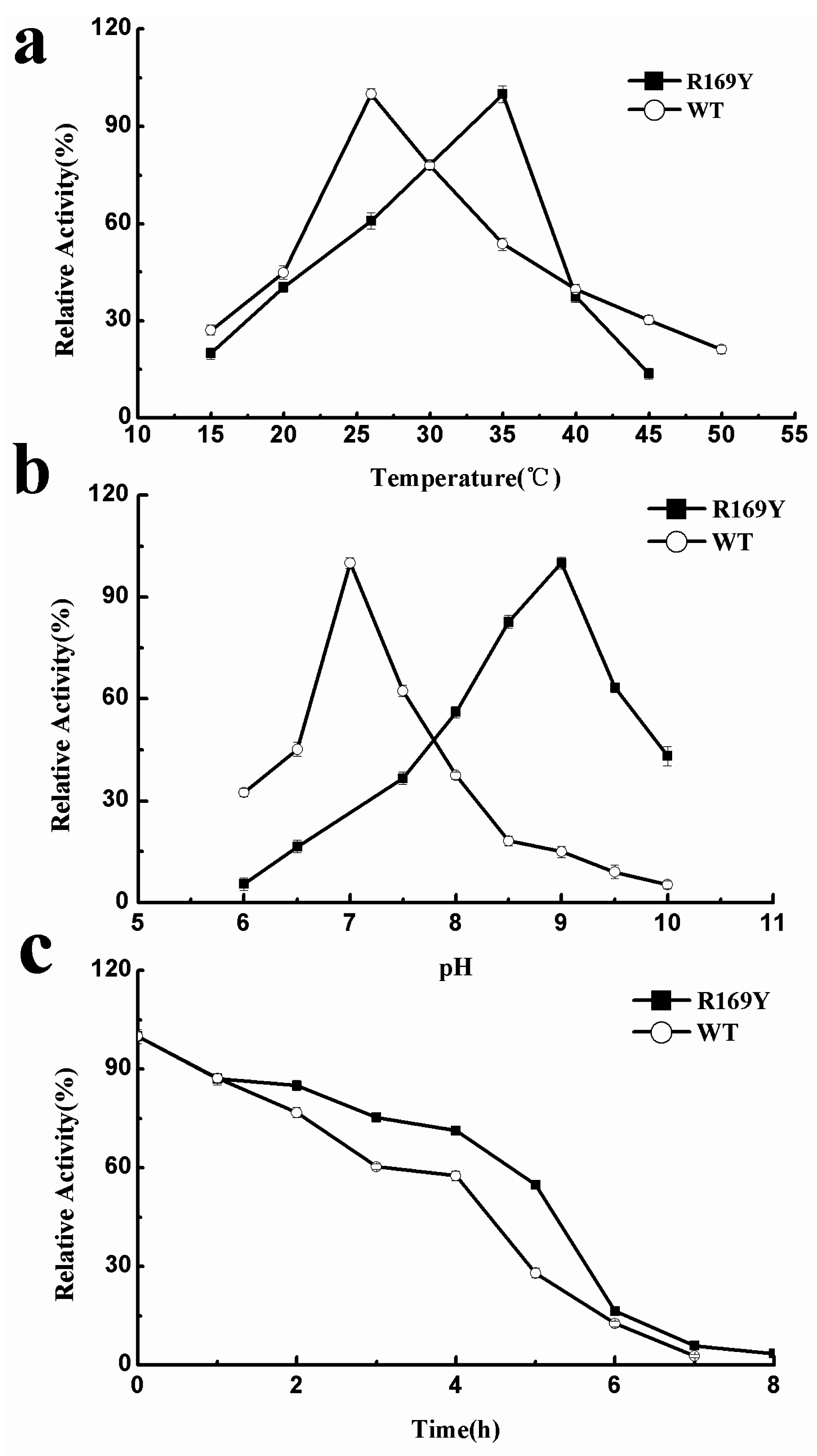
2.6. Effect of Metal Ions and Organic Solvents on WT and R169Y
| Enzyme | WT | R169Y | ||||||
|---|---|---|---|---|---|---|---|---|
| Relative Activity (%) | Relative Activity (%) | |||||||
| Metal ions (mM) | 0.2 | 1 | 5 | 10 | 0.2 | 1 | 5 | 10 |
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Na+ | ND | 75.44 ± 0.47 | 11.40 ± 1.44 | 5.26 ± 0.33 | 46.11 ± 0.88 | 101.48 ± 2.23 | 89.20 ± 0.43 | 39.75 ± 0.74 |
| K+ | 137.72 ± 1.54 | 62.28 ± 1.03 | 55.26 ± 1.31 | 45.61 ± 1.04 | 54.51 ± 1.09 | 118.10 ± 1.94 | 105.61 ± 0.98 | 84.03 ± 1.43 |
| Ca2+ | 57.02 ± 1.09 | 38.60 ± 0.89 | 14.91 ± 0.33 | 10.53 ± 0.43 | 33.40 ± 0.95 | 41.60 ± 0.74 | 92.33 ± 1.45 | 66.24 ± 1.47 |
| Mg2+ | 54.39 ± 0.87 | 55.26 ± 1.53 | 57.90 ± 1.77 | 43.42 ± 0.74 | 35.34 ± 1.54 | 40.74 ± 2.03 | 76.12 ± 1.78 | 62.84 ± 1.45 |
| Zn2+ | 90.35 ± 1.89 | 82.46 ± 1.34 | 78.07 ± 1.64 | 71.93 ± 0.45 | 49.39 ± 0.43 | 75.54 ± 1.54 | 164.02 ± 1.95 | 95.87 ± 0.74 |
| Mn2+ | 78.07 ± 1.54 | 70.18 ± 2.04 | 40.35 ± 0.54 | 10.53 ± 0.32 | 64.27 ± 1.54 | 36.71 ± 0.87 | 18.14 ± 0.32 | ND |
| Cu2+ | 57.02 ± 0.43 | 42.11 ± 1.54 | 40.64 ± 0.68 | 32.46 ± 0.39 | 24.74 ± 1.43 | 93.12 ± 1.87 | 106.84 ± 0.45 | 73.51 ± 1.54 |
| Fe3+ | 78.07 ± 0.58 | 44.74 ± 1.54 | 33.33 ± 1.81 | 16.67 ± 0.85 | 86.10 ± 1.73 | 68.01 ± 0.39 | 55.73 ± 1.82 | ND |
| Ni2+ | 159.94 ± 1.54 | 143.57 ± 1.05 | 129.30 ± 1.74 | 122.22 ± 2.57 | 21.34 ± 0.38 | 75.66 ± 1.47 | 164.90 ± 0.78 | 62.08 ± 1.94 |
| Enzyme | WT | R169Y | ||||||
|---|---|---|---|---|---|---|---|---|
| Relative Activity (%) | Relative Activity (%) | |||||||
| Concentration (%) | Concentration (%) | |||||||
| Organic solvents | 1 | 5 | 10 | 20 | 1 | 5 | 10 | 20 |
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| methanol | 45.56 ± 1.74 | 64.44 ± 0.64 | 113.33 ± 0.54 | 46.67 ± 0.92 | 43.04 ± 0.29 | 5.0 ± 0.32 | ND | ND |
| ethanol | 143.33 ± 0.43 | 182.22 ± 1.54 | 154.44 ± 2.01 | 22.22 ± 0.41 | 30.91 ± 1.94 | 26.67 ± 1.02 | 18.84 ± 0.32 | 7.71 ± 0.55 |
| isopropyl alcohol | 30.0 ± 1.56 | 46.67 ± 2.04 | 74.45 ± 1.09 | 67.78 ± 1.87 | 39.41 ± 0.54 | 14.07 ± 0.25 | ND | ND |
| n-butanol | 23.33 ± 0.31 | 20.0 ± 0.51 | ND | ND | 30.95 ± 0.93 | ND | ND | ND |
| acetonitrile | 41.11 ± 0.43 | 11.11 ± 0.23 | 13.33 ± 0.96 | ND | 42.39 ± 2.06 | 36.33 ± 1.03 | 29.48 ± 0.62 | 16.96 ± 1.09 |
| dimethyl sulfoxide | 62.22 ± 1.73 | 31.11 ± 1.47 | 13.33 ± 0.95 | ND | 26.24 ± 1.53 | 18.46 ± 0.34 | 16.15 ± 0.49 | 11.44 ± 1.55 |
| glycerol | 25.56 ± 1.07 | ND | ND | ND | 66.61 ± 1.33 | 53.83 ± 0.52 | 37.01 ± 1.54 | 3.94 ± 0.21 |
2.7. Effect of Inhibitors on WT and R169Y
2.8. Enzymology Properties of Arg169
| Enzyme | WT | R169Y | ||||||
|---|---|---|---|---|---|---|---|---|
| Relative Activity (%) | Relative Activity (%) | |||||||
| Concentration (mM) | Concentration (mM) | |||||||
| Inhibitors | 0.2 | 1 | 5 | 10 | 0.2 | 1 | 5 | 10 |
| Control | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Thr | 73.97 ± 0.46 | 55.86 ± 1.63 | 27.07 ± 0.54 | 18.19 ± 0.59 | 94.25 ± 1.45 | 79.20 ± 0.54 | 50.34 ± 1.04 | 33.82 ± 0.29 |
| Lys | 38.47 ± 0.92 | 20.36 ± 1.03 | 19.18 ± 0.41 | ND | 83.09 ± 1.93 | 186.05 ± 1.22 | 68.46 ± 1.29 | 50.63 ± 0.34 |
| Met | 53.36 ± 0.56 | 50.89 ± 0.43 | 49.58 ± 0.94 | 43.62 ± 1.65 | 99.88 ± 1.98 | 167.43 ± 1.93 | 116.36 ± 1.19 | 68.62 ± 1.01 |
| Thr + Lys | 88.30 ± 1.14 | 33.95 ± 1.45 | 28.63 ± 0.19 | 30.18 ± 0.82 | 84.23 ± 0.17 | 41.13 ± 0.26 | 37.76 ± 1.38 | 27.93 ± 0.42 |
| Thr + Met | 73.32 ± 1.04 | 46.52 ± 1.82 | 29.38 ± 1.05 | 15.05 ± 0.04 | 73.04 ± 0.32 | 48.50 ± 0.91 | 32.88 ± 0.96 | 27.46 ± 1.04 |
| Met + Lys | 30.86 ± 1.74 | 22.63 ± 0.43 | ND | ND | 102.38 ± 2.03 | 138.47 ± 1.92 | 146.76 ± 1.03 | 183.12 ± 1.13 |
| Thr + Lys + Met | 93.96 ± 1.54 | 64.35 ± 1.83 | 49.04 ± 1.62 | 35.92 ± 0.81 | 192.5 ± 2.04 | 141.99 ± 0.63 | 30.68 ± 0.14 | 16.87 ± 0.26 |
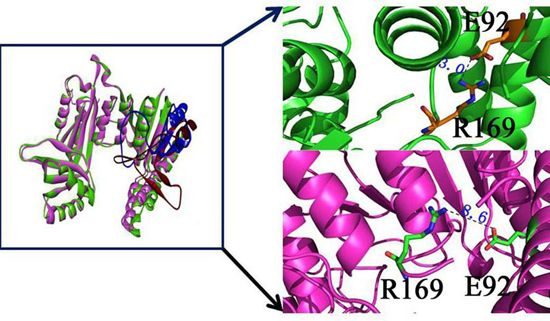
2.9. “Triple Function” of Arg169

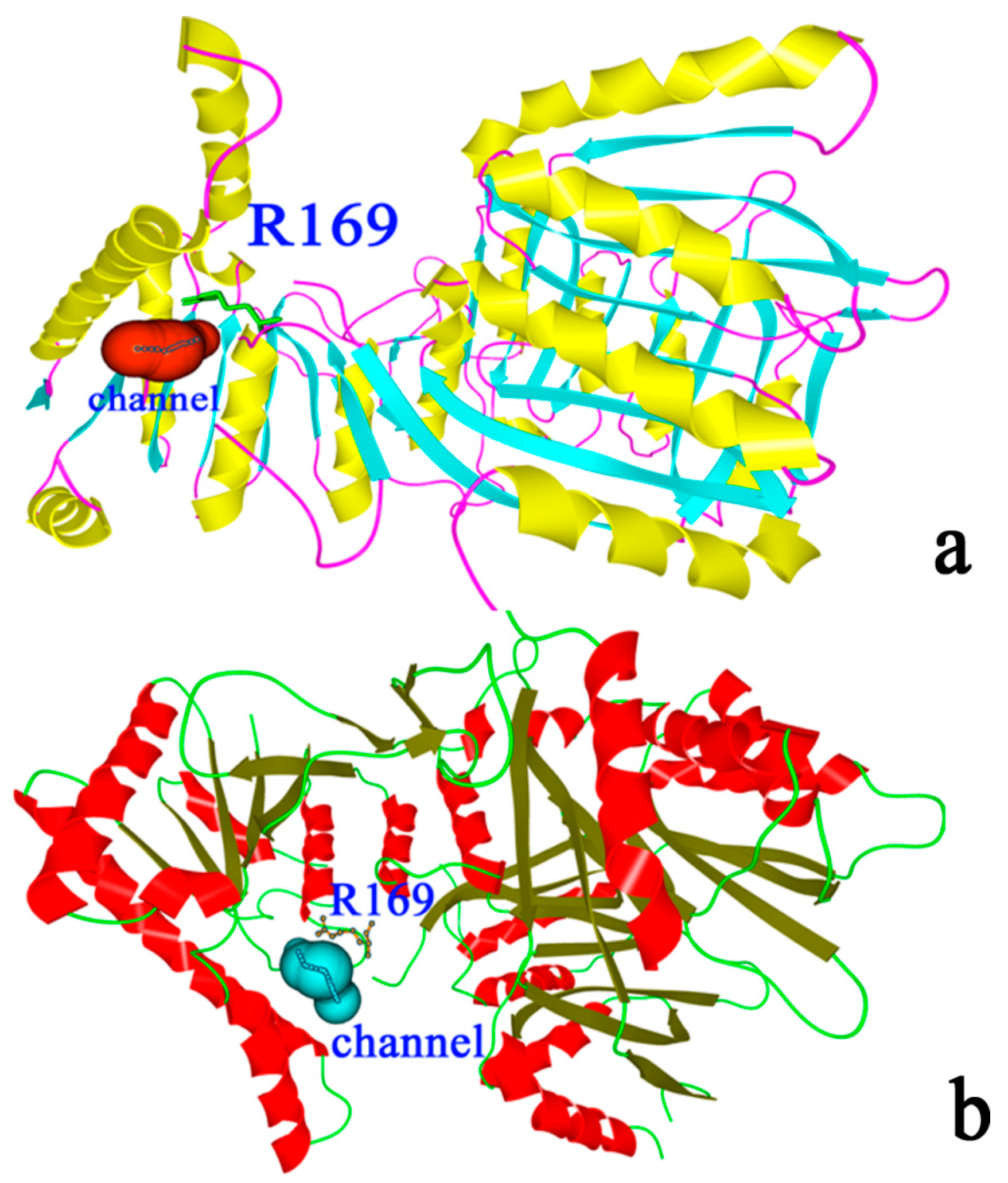
3. Experimental Section
3.1. Reagents, Strains, and Plasmids
3.2. Construction of Mutant Strains
3.3. Expression and Purification of the Recombinant AK and Mutants
3.4. Analysis of Native PAGE, SDS-PAGE, and Western Blot
3.5. Steady-State Kinetics Assay of WT and Mutant AKs
3.6. Characterization of AK from WT and Mutant R169Y
3.7. Homology Modeling and Structural Analysis
3.8. Molecular Docking
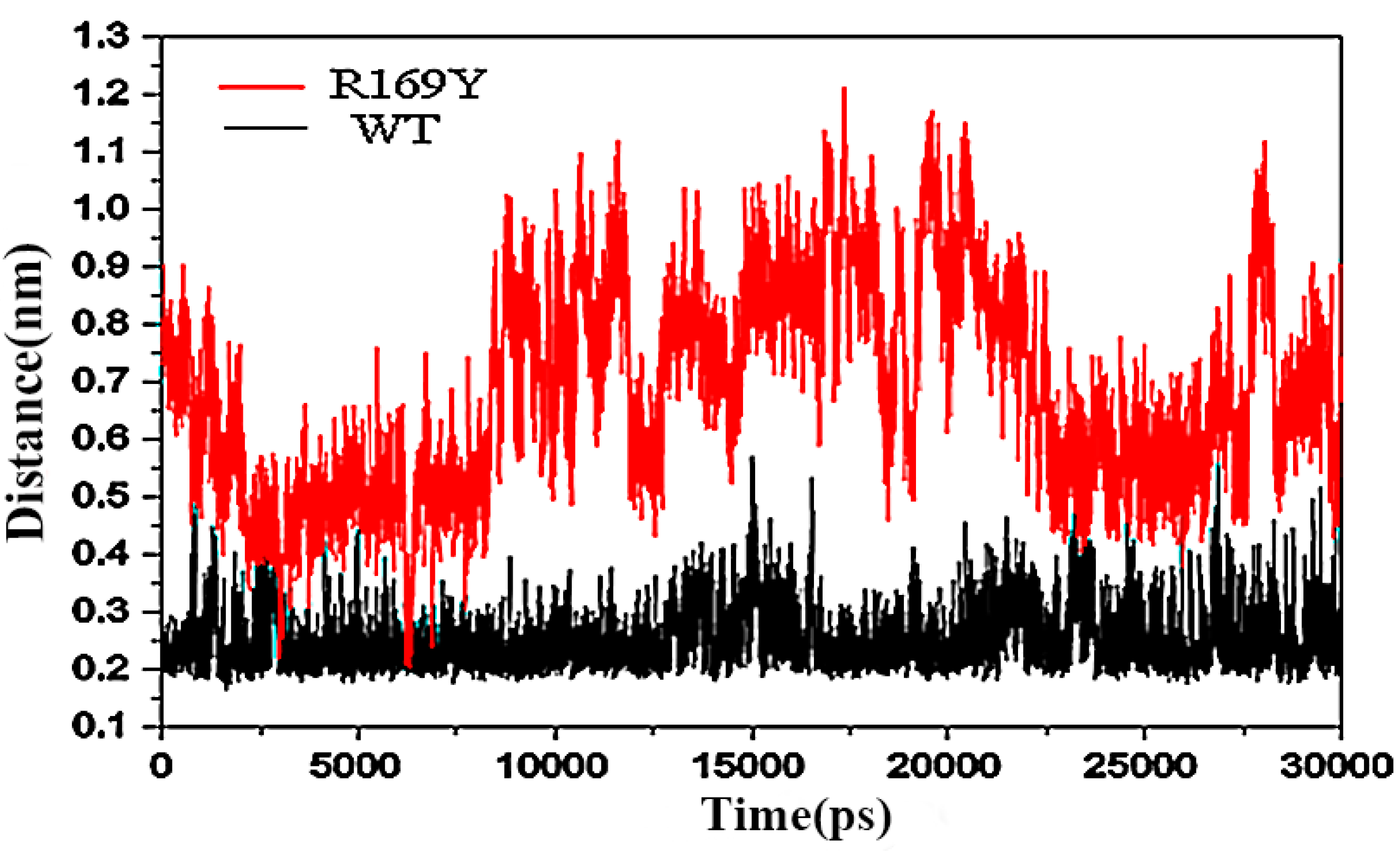
3.9. Molecular Dynamics (MD) Simulation and Molecular Mechanics-Poisson-Boltzmann Surface Area (MM-PBSA) Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Z.; Sugima, R.; Sun, J.B.; Zeng, A.P. Integrating molecular dynamics and co-evolutionary analysis for reliable target prediction and deregulation of the allosteric inhibition of aspartokinase for amino acid production. J. Biotechnol. 2011, 154, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tomita, T.; Kurihara, T.; Fushinobu, S.; Kuzuyama, T.; Nishiyama, M. Structural insight into concerted inhibition of α2β2-type aspartate kinase from Corynebacterium glutamicum. J. Mol. Biol. 2007, 368, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Chaitanya, M.; Babajan, B.; Anuradha, C.; Naveen, M.; Rajasekhar, C.; Madhusudana, P.; Kumar, C.S. Exploring the molecular basis for selective binding of Mycobacterium tuberculosis Asp kinase toward its natural substrates and feedback inhibitors: A docking and molecular dynamics study. J. Mol. Model. 2010, 16, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Ogawa-Miyata, Y.; Kojima, H.; Sano, K. Mutation analysis of the feedback inhibition site of aspartokinase III of Escherichia coli K-12 and its use in l-threonine production. Biosci. Biotechnol. Biochem. 2001, 65, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tomita, T.; Kono, H.; Fushinobu, S.; Kuzuyama, T.; Nishiyama, M. Crystal structures of the regulatory subunit of Thr-sensitive aspartate kinase from Thermus thermophilus. FEBS J. 2009, 276, 3124–3136. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Huang, J.; Crowley, J.; Ruschke, L.; Goldman, B.S.; Wen, L. Metabolically engineered soybean seed with enhanced threonine levels: Biochemical characterization and seed-specific expression of lysine-insensitive variants of aspartate kinases from the enteric bacterium Xenorhabdus bovienii. Plant Biotechnol. J. 2011, 9, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Kotaka, M.; Ren, J.; Lockyer, M.; Hawkins, A.R.; Stammers, D.K. Structures of R- and T-state Escherichia coli aspartokinase III mechanisms of the allosteric transition and inhibition by lysine. J. Biol. Chem. 2006, 281, 31544–31552. [Google Scholar] [CrossRef] [PubMed]
- Mas-Droux, C.; Curien, G.; Robert-Genthon, M.; Laurencin, M.; Ferrer, J.-L.; Dumas, R. A novel organization of ACT domains in allosteric enzymes revealed by the crystal structure of Arabidopsis aspartate kinase. Plant Cell Online 2006, 18, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Nishida, H.; Narumi, I. Phylogenetic and disruption analyses of aspartate kinase of Deinococcus radiodurans. Biosci. Biotechnol. Biochem. 2007, 71, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Tomita, T.; Kuzuyama, T.; Nishiyama, M. Mechanism of concerted inhibition of α2β2-type hetero-oligomeric aspartate kinase from Corynebacterium glutamicum. J. Biol. Chem. 2010, 285, 27477–27486. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.H.; Adluru, N.; Chung, M.K.; Charchut, S.; GadElkarim, J.J.; Altshuler, L.; Moody, T.; Kumar, A.; Singh, V.; Leow, A.D. Multi-resolutional brain network filtering and analysis via wavelets on non-Euclidean space. Med. Image Comput. Comput. Assist. Interv. 2013, 16 Pt 3, 643–651. [Google Scholar] [PubMed]
- Futera, Z.; Burda, J.V. Reaction mechanism of Ru(II) piano-stool complexes: Umbrella sampling QM/MM MD study. J. Comput. Chem. 2014, 35, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Robin, A.Y.; Cobessi, D.; Curien, G.; Robert-Genthon, M.; Ferrer, J.L.; Dumas, R. A new mode of dimerization of allosteric enzymes with ACT domains revealed by the crystal structure of the aspartate kinase from Cyanobacteria. J. Mol. Biol. 2010, 399, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, M.; Kukimoto, M.; Beppu, T.; Horinouchi, S. An operon encoding aspartokinase and purine phosphoribosyltransferase in Thermus flavus. Microbiology 1995, 141, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Kobashi, N.; Nishiyama, M.; Tanokura, M. Aspartate kinase-independent lysine synthesis in an extremely thermophilic bacterium, Thermus thermophilus: Lysine is synthesized via α-aminoadipic acid not via diaminopimelic acid. J. Bacteriol. 1999, 181, 1713–1718. [Google Scholar] [PubMed]
- Dong, X.; Quinn, P.J.; Wang, X. Metabolic engineering of Escherichia coli and Corynebacterium glutamicum for the production of l-threonine. Biotechnol. Adv. 2011, 29, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, J.; Cremer., J.; Bachmann, B.; Eggeling, L.; Sahm, H.; Pühler, A. Genetic and biochemical analysis of the aspartokinase from Corynebacterium glutamicum. Mol. Microbiol. 1991, 5, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Kato, C.; Kurihara, T.; Kobashi, N.; Yamane, H.; Nishiyama, M. Conversion of feedback regulation in aspartate kinase by domain exchange. Biochem. Biophys. Res. Commun. 2004, 316, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Manjasetty, B.A.; Chance, M.R.; Burley, S.K.; Panjikar, S.; Almo, S.C. Crystal structure of Clostridium acetobutylicum aspartate kinase (CaAK): An important allosteric enzyme for amino acids production. Biotechnol. Rep. 2014, 3, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Dumas, R.; Cobessi, D.; Robin, A.Y.; Ferrer, J.L.; Curien, G. The many faces of aspartate kinases. Arch. Biochem. Biophys. 2012, 519, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Z.; Yu, K.; Yan, L.M.; Li, Y.Y.; Chen, C.; Li, X.M. Structural view of the regulatory subunit of aspartate kinase from Mycobacterium tuberculosis. Protein Cell 2011, 2, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Mustafi, N.; Frunzke, J.; Kennerknecht, N.; Wessel, M.; Bott, M.; Wendisch, V.F. Lrp of Corynebacterium glutamicum controls expression of the brnFE operon encoding the export system for l-methionine and branched-chain amino acids. J. Biotechnol. 2012, 158, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, N.; Grünberger, A.; Kohlheyer, D.; Bott, M.; Frunzke, J. The development and application of a single-cell biosensor for the detection of l-methionine and branched-chain amino acids. Metab. Eng. 2012, 14, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shang, X.; Deng, A.; Chai, X.; Lai, S.; Zhang, G.; Wen, T. Genetic and biochemical characterization of Corynebacterium glutamicum ATP phosphoribosyltransferase and its three mutants resistant to feedback inhibition by histidine. Biochimie 2012, 94, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Meyer, W.; Rappert, S.; Sun, J.; Zeng, A.P. Coevolutionary analysis enabled rational deregulation of allosteric enzyme inhibition in Corynebacterium glutamicum for lysine production. Appl. Environ. Microbiol. 2011, 77, 4352–4360. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, N.; Borde, A.; Wölfel, S.; Åkerman, B.; Larsson, A. Quantification of protein concentration by the Bradford method in the presence of pharmaceutical polymers. Anal. Biochem. 2011, 411, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Ochrombel, I.; Krämer, R.; Trötschel, C.; Poetsch, A.; Rückert, C.; Hueser, A.; Persicke, M.; Seiferling, D.; Kalinowski, J.; et al. Functional genomics of pH homeostasis in Corynebacterium glutamicum revealed novel links between pH response, oxidative stress, iron homeostasis and methionine synthesis. BMC Genom. 2009, 10, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Sudha. Effect of different concentrations of metal ions on α amylase production by Bacillus amyloliquefaciens. Res. Biotechnol. 2012, 3, 67–71. [Google Scholar]
- Wiggers, H.J.; Cheleski, J.; Zottis, A.; Oliva, G.; Andricopulo, A.D.; Montanari, C.A. Effects of organic solvents on the enzyme activity of Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase in calorimetric assays. Anal. Biochem. 2007, 370, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Aranvind, L.; Koonin, E.V. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999, 287, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.; Niebisch, A.; Schwaiger, A.; Viets, U.; Metzger, S.; Bramkamp, M.; Bott, M. Genetic and biochemical analysis of the serine/threonine protein kinases PknA, PknB, PknG and PknL of Corynebacterium glutamicum: Evidence for non-essentiality and for phosphorylation of OdhI and FtsZ by multiple kinases. Mol. Microbiol. 2009, 74, 724–741. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.N.; Niederholtmeyer, H.; Schmitz, K.; Schoberth, S.M.; Wendisch, V.F. Polyphosphate/ATP-dependent NAD kinase of Corynebacterium glutamicum: Biochemical properties and impact of ppnK overexpression on lysine production. Appl. Microbiol. Biotechnol. 2010, 87, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Nivedha, A.K.; Makeneni, S.; Foley, B.L.; Tessier, M.B.; Woods, R.J. Importance of ligand conformational energies in carbohydrate docking: Sorting the wheat from the chaff. J. Comput. Chem. 2014, 35, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.R.C.W.; Walker, R.C.; Zhang, W.; et al. AMBER 10; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Fu, G.; Liu, H.; Doerksen, R.J. Molecular modeling to provideinsight into the substrate binding and catalytic mechanism of human biliverdin-IXα reductase. J. Phys. Chem. B 2012, 116, 9580–9594. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Kollman, P.A. Binding of a diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, W.; Li, H.; Li, H.; Liu, C.; Liu, J. Characterization of Aspartate Kinase from Corynebacterium pekinense and the Critical Site of Arg169. Int. J. Mol. Sci. 2015, 16, 28270-28284. https://doi.org/10.3390/ijms161226098
Min W, Li H, Li H, Liu C, Liu J. Characterization of Aspartate Kinase from Corynebacterium pekinense and the Critical Site of Arg169. International Journal of Molecular Sciences. 2015; 16(12):28270-28284. https://doi.org/10.3390/ijms161226098
Chicago/Turabian StyleMin, Weihong, Huiying Li, Hongmei Li, Chunlei Liu, and Jingsheng Liu. 2015. "Characterization of Aspartate Kinase from Corynebacterium pekinense and the Critical Site of Arg169" International Journal of Molecular Sciences 16, no. 12: 28270-28284. https://doi.org/10.3390/ijms161226098