
Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Nicotinic Acetylcholine Receptors (nAChRs) with a Focus on the α7-Subunit
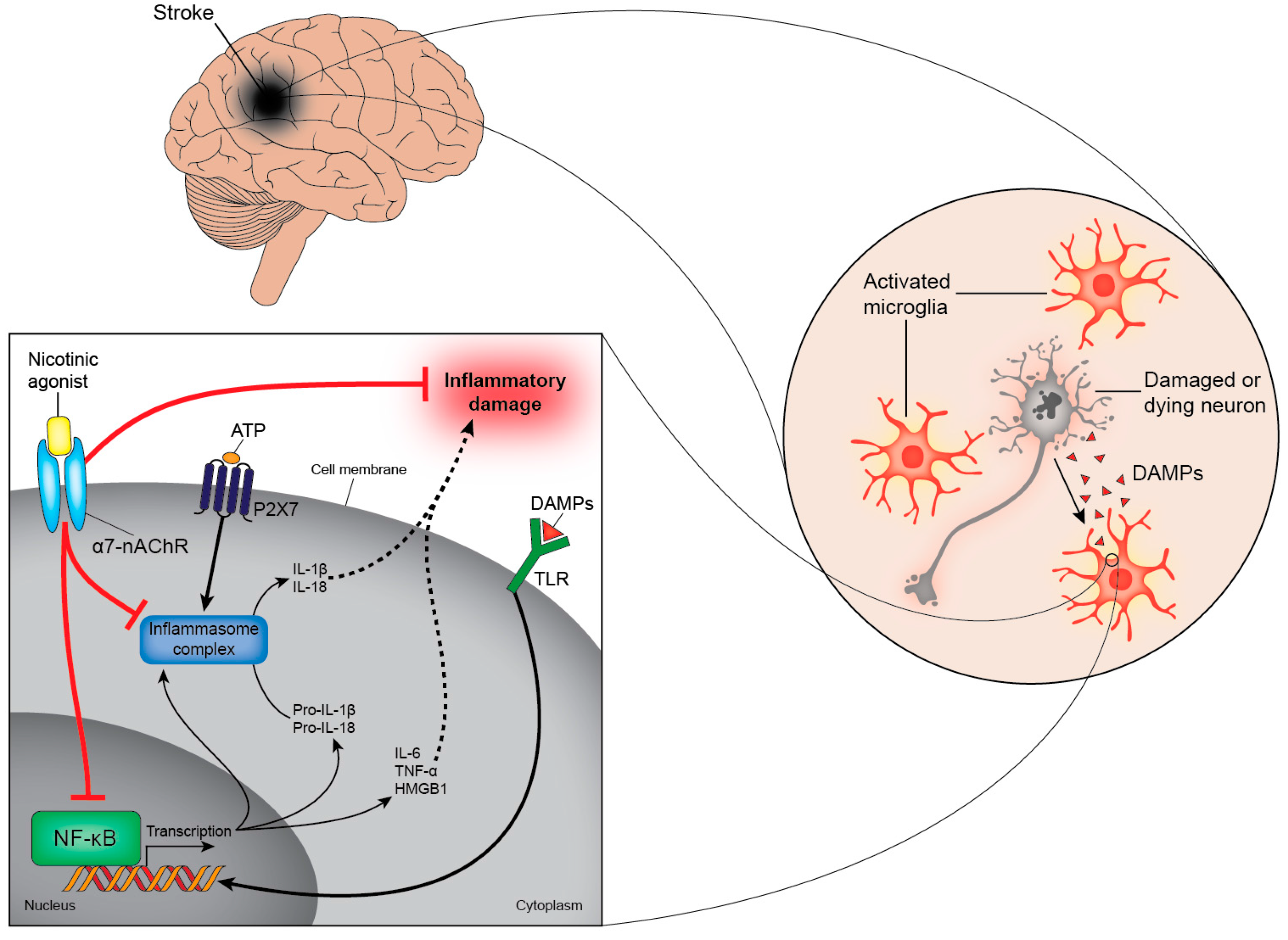
3. An Inflammatory Reflex Shapes Immune Responses Post-Stroke via Activation of α7-nAChRs
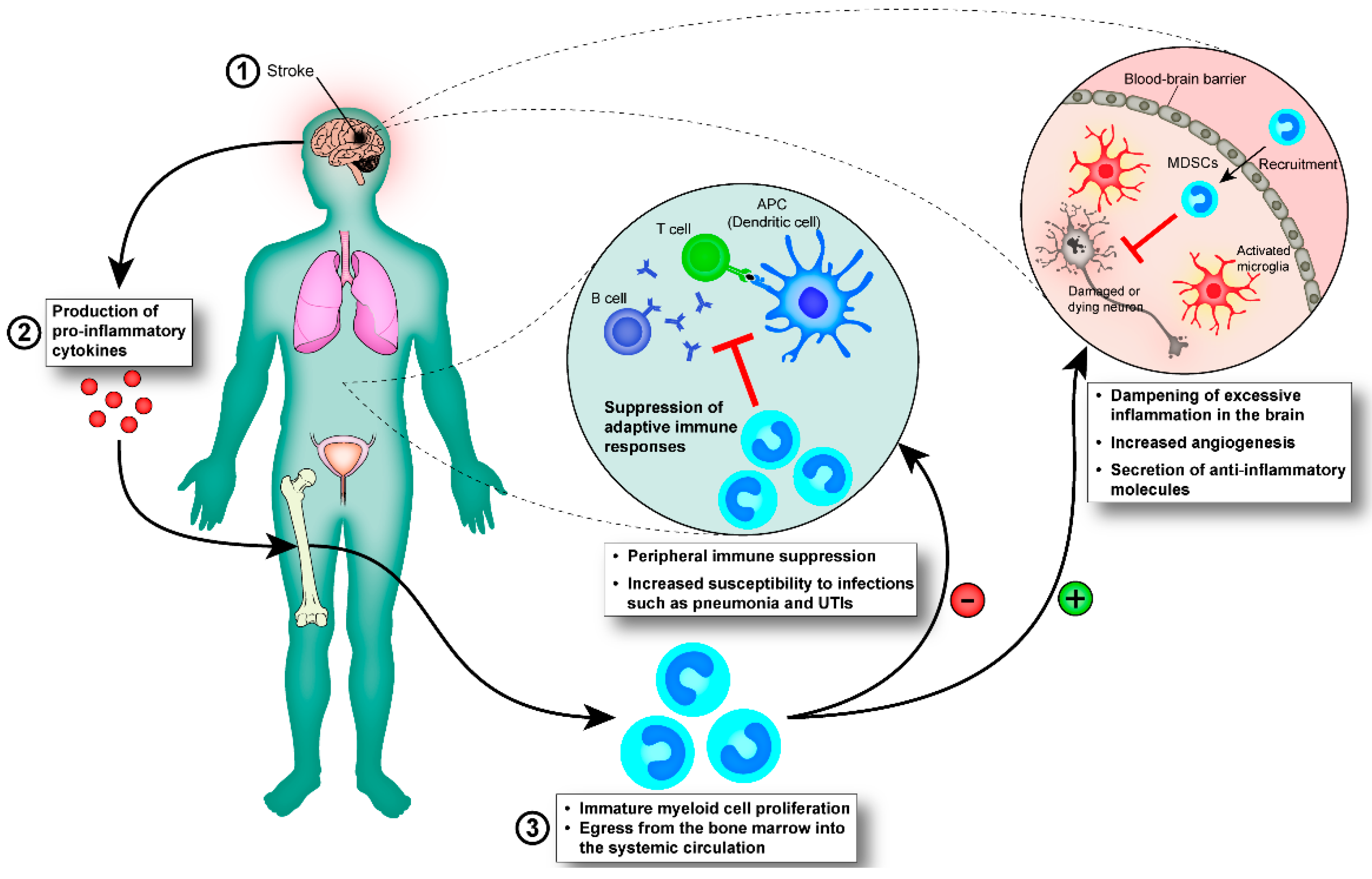
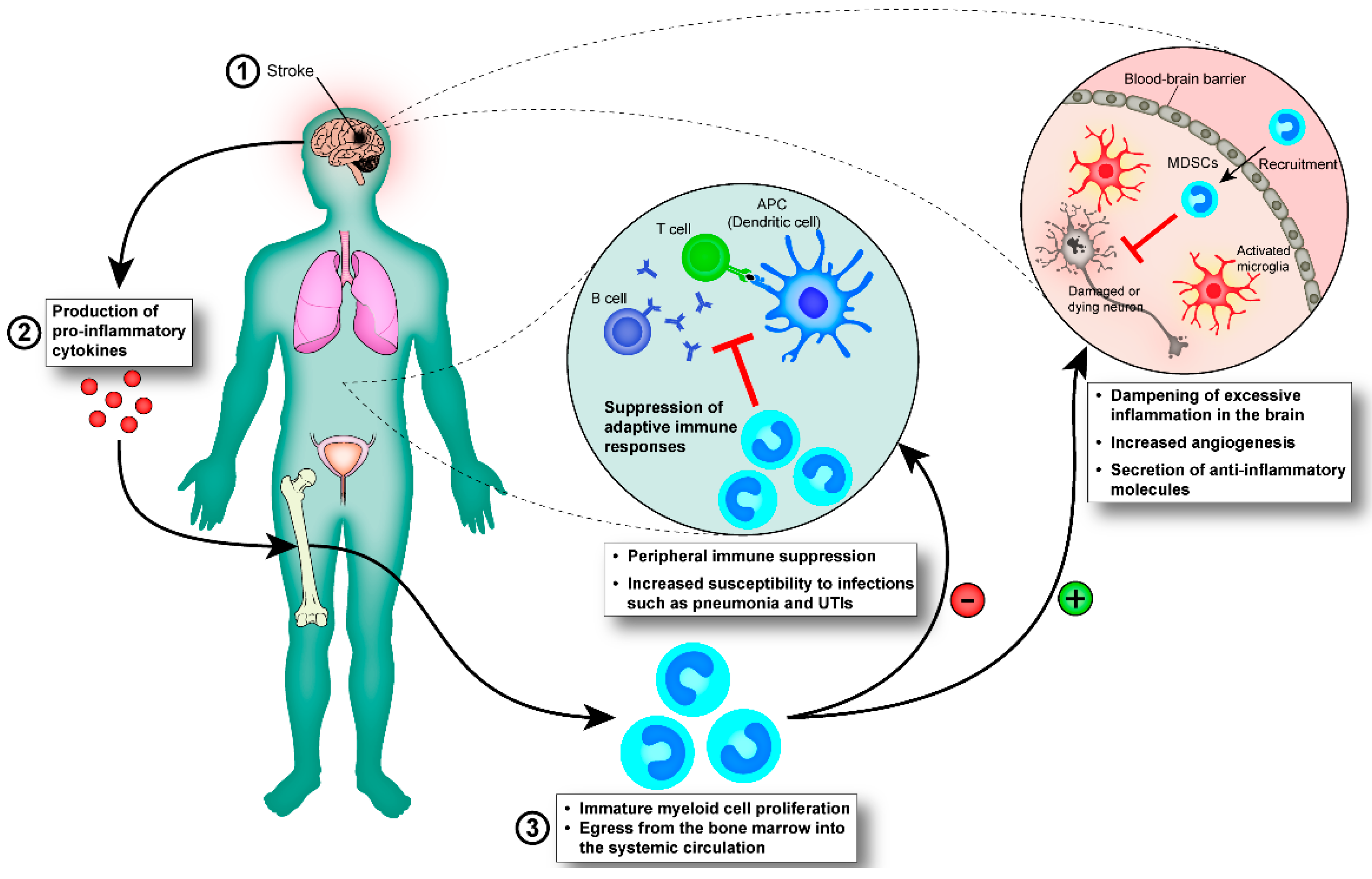
4. Inflammatory Response of the Immune System Post-Stroke
4.1. Early Innate Immune Responses Post-Stroke
4.1.1. Myeloid Cells
4.1.2. Immature Myeloid Cells
4.2. Infection Post-Stroke
4.3. Pattern Recognition Receptors in Stroke
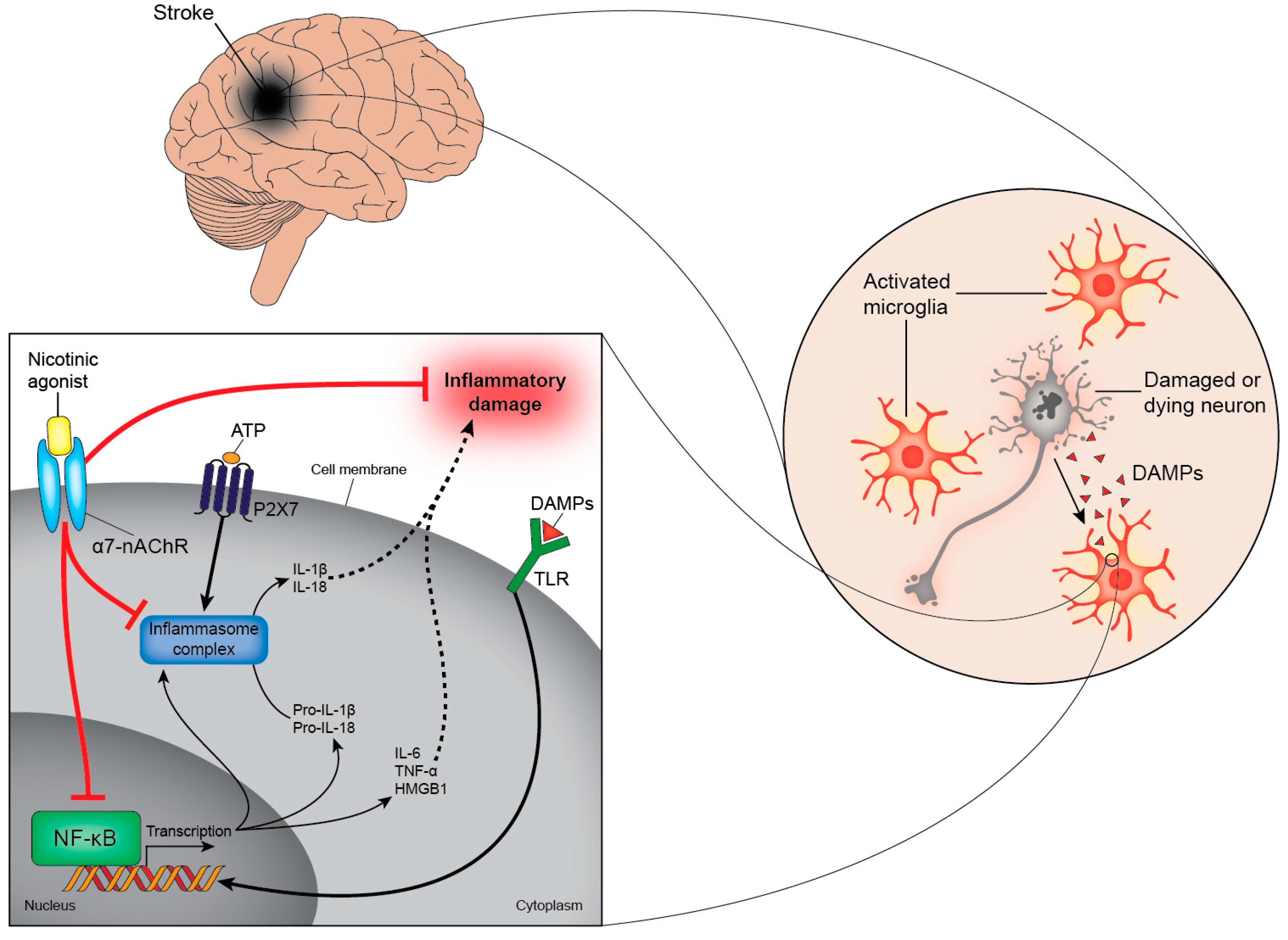
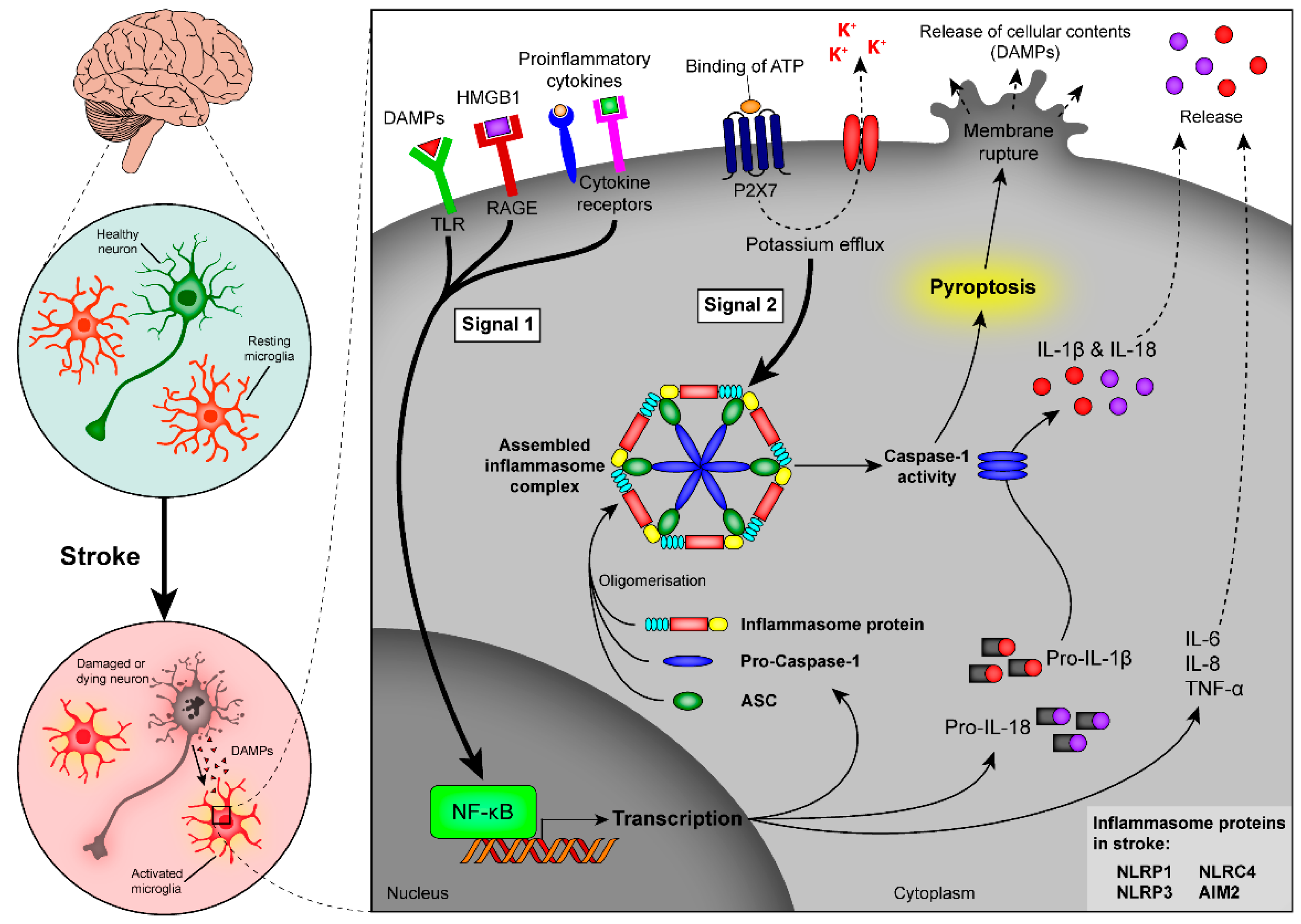
4.4. Purinergic Signalling in Stroke
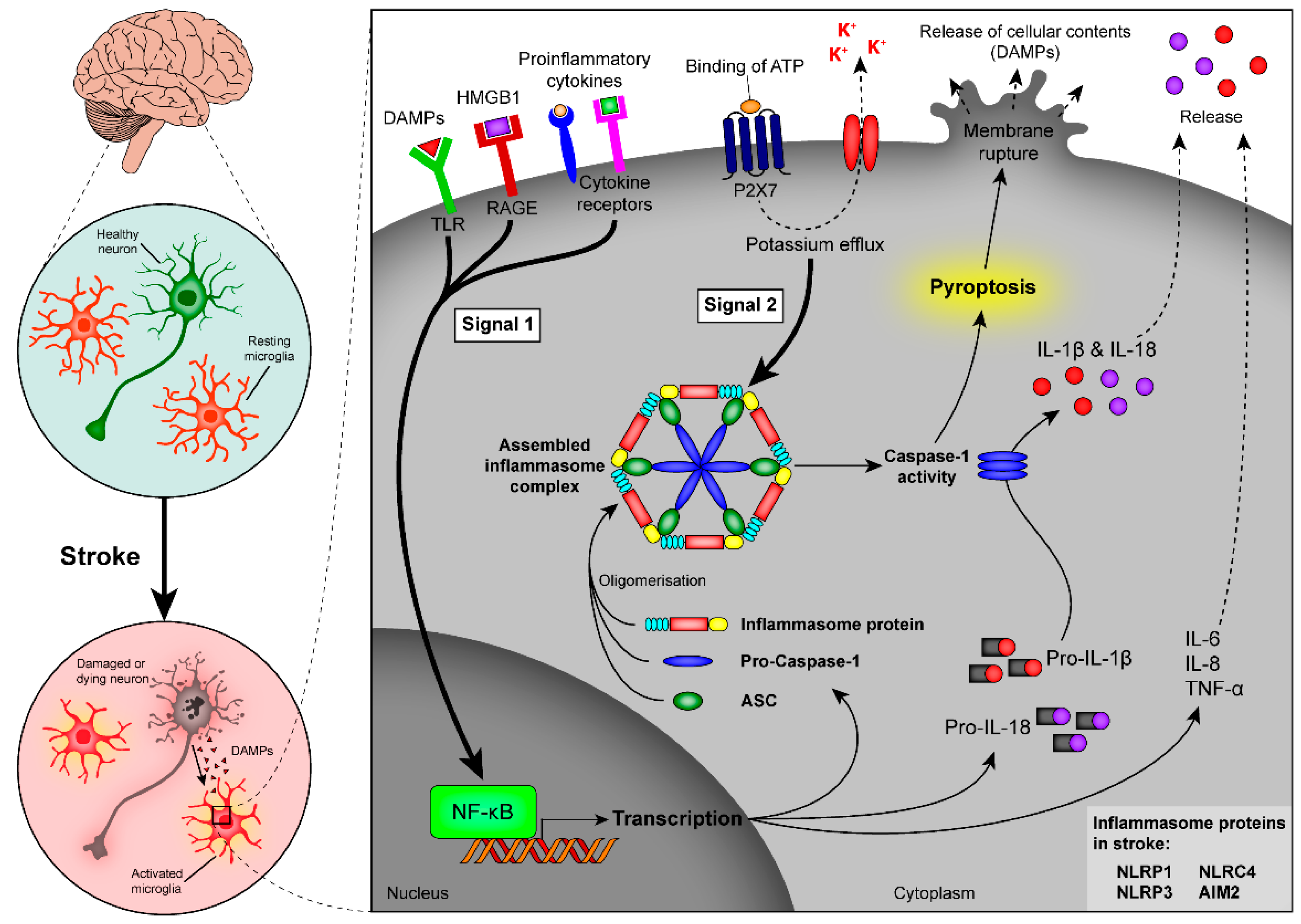
4.5. Role of Inflammasomes in Stroke
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Martinon, F. Detection of immune danger signals by nalp3. J. Leukoc. Biol. 2008, 83, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.J.; Walton, M.; Harper, J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheumatol. 2009, 60, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Chamorro, A.; Urra, X.; Planas, A.M. Infection after acute ischemic stroke: A manifestation of brain-induced immunodepression. Stroke 2007, 38, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Dalpke, A.; Mracsko, E.; Antoine, D.J.; Roth, S.; Zhou, W.; Yang, H.; Na, S.Y.; Akhisaroglu, M.; Fleming, T.; et al. Damp signaling is a key pathway inducing immune modulation after brain injury. J. Neurosci. 2015, 35, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [PubMed]
- Cai, P.Y.; Bodhit, A.; Derequito, R.; Ansari, S.; Abukhalil, F.; Thenkabail, S.; Ganji, S.; Saravanapavan, P.; Shekar, C.C.; Bidari, S.; et al. Vagus nerve stimulation in ischemic stroke: Old wine in a new bottle. Front. Neurol. 2014, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed]
- Zoli, M.; Pistillo, F.; Gotti, C. Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 2015, 96, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Nashmi, R.; Lester, H.A. Cns localization of neuronal nicotinic receptors. J. Mol. Neurosci. 2006, 30, 181–184. [Google Scholar] [CrossRef]
- Gotti, C.; Riganti, L.; Vailati, S.; Clementi, F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr. Pharm. Des. 2006, 12, 407–428. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Egleton, R.D.; Davis, T.P. Modulation of cerebral microvascular permeability by endothelial nicotinic acetylcholine receptors. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H212–H219. [Google Scholar] [CrossRef] [PubMed]
- Akaike, A.; Takada-Takatori, Y.; Kume, T.; Izumi, Y. Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: Role of alpha4 and alpha7 receptors in neuroprotection. J. Mol. Neurosci. 2010, 40, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; Leon, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nachrs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Yakel, J.L. Nicotinic ach receptors in the hippocampal circuit; functional expression and role in synaptic plasticity. J. Physiol. 2014, 592, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nat. Rev. Drug Discov. 2005, 4, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Hamano, R.; Takahashi, H.K.; Iwagaki, H.; Yoshino, T.; Nishibori, M.; Tanaka, N. Stimulation of alpha7 nicotinic acetylcholine receptor inhibits cd14 and the toll-like receptor 4 expression in human monocytes. Shock 2006, 26, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Del Bufalo, A.; Frustaci, A.; Fini, M.; Cesario, A. Beyond acetylcholinesterase inhibitors for treating alzheimer's disease: Alpha7-nachr agonists in human clinical trials. Curr. Pharm. Des. 2014, 20, 6014–6021. [Google Scholar] [CrossRef] [PubMed]
- Keefe, R.S.; Meltzer, H.A.; Dgetluck, N.; Gawryl, M.; Koenig, G.; Moebius, H.J.; Lombardo, I.; Hilt, D.C. Randomized, double-blind, placebo-controlled study of encenicline, an alpha7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacology 2015, 40, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kang, K.M.; Kim, J.H.; Bahn, J.J.; Jeon, D.; Kim, M.; Lee, S.K.; Roh, J.K. Cholinergic anti-inflammatory pathway in intracerebral hemorrhage. Brain Res. 2010, 1309, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Martelli, D.; McKinley, M.J.; McAllen, R.M. The cholinergic anti-inflammatory pathway: A critical review. Auton. Neurosci. 2014, 182, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Verhagen, J.; Blaser, K.; Akdis, M.; Akdis, C.A. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: The role of t regulatory cells. Immunology 2006, 117, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, G.; Zhang, P.; Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2015, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, D. Antigen presenting cells. Immunol. Res. 1989, 8, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain cns microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Schilling, M.; Besselmann, M.; Muller, M.; Strecker, J.K.; Ringelstein, E.B.; Kiefer, R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2005, 196, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; Reis e Sousa, C. Adaptive immunity after cell death. Trends Immunol. 2013, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Offner, H.; Subramanian, S.; Parker, S.M.; Afentoulis, M.E.; Vandenbark, A.A.; Hurn, P.D. Experimental stroke induces massive, rapid activation of the peripheral immune system. J. Cereb. Blood Flow Metab. 2006, 26, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Germain, R.N.; Margulies, D.H. The biochemistry and cell biology of antigen processing and presentation. Annu. Rev. Immunol. 1993, 11, 403–450. [Google Scholar] [CrossRef] [PubMed]
- Yuseff, M.-I.; Pierobon, P.; Reversat, A.; Lennon-Dumenil, A.-M. How b cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schwab, C.; Parent, A.; Doudet, D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Ann. Neurol. 2003, 54, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Huh, Y.; Jung, J.W.; Park, C.; Ryu, J.R.; Shin, C.Y.; Kim, W.K.; Ryu, J.H. Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neurosci. Lett. 2003, 349, 63–67. [Google Scholar] [CrossRef]
- Liguz-Lecznar, M.; Kossut, M. Influence of inflammation on poststroke plasticity. Neural Plast. 2013, 2013, 258582. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.; Chopp, M. Vegf enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, M.; Leypoldt, F.; Steinbach, K.; Behrens, D.; Choe, C.U.; Siler, D.A.; Arumugam, T.V.; Orthey, E.; Gerloff, C.; Tolosa, E.; et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009, 40, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Greifzu, F.; Schmidt, S.; Schmidt, K.F.; Kreikemeier, K.; Witte, O.W.; Lowel, S. Global impairment and therapeutic restoration of visual plasticity mechanisms after a localized cortical stroke. Proc. Natl. Acad. Sci. USA 2011, 108, 15450–15455. [Google Scholar] [CrossRef] [PubMed]
- Saino, O.; Taguchi, A.; Nakagomi, T.; Nakano-Doi, A.; Kashiwamura, S.; Doe, N.; Nakagomi, N.; Soma, T.; Yoshikawa, H.; Stern, D.M.; et al. Immunodeficiency reduces neural stem/progenitor cell apoptosis and enhances neurogenesis in the cerebral cortex after stroke. J. Neurosci. Res. 2010, 88, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.T. Themes and strategies for studying the biology of stroke recovery in the poststroke epoch. Stroke 2008, 39, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Overman, J.J.; Carmichael, S.T. Plasticity in the injured brain: More than molecules matter. Neuroscientist 2014, 20, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Narla, S.; Klejbor, I.; Birkaya, B.; Lee, Y.W.; Morys, J.; Stachowiak, E.K.; Terranova, C.; Bencherif, M.; Stachowiak, M.K. Alpha7 nicotinic receptor agonist reactivates neurogenesis in adult brain. Biochem. Pharmacol. 2013, 86, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D.; Micieli, G.; Tassorelli, C.; Cuartero, M.I.; Ballesteros, I.; Certo, M.; Moro, M.A.; Lizasoain, I.; Bagetta, G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Brew, B.J. Microglia, macrophages, perivascular macrophages, and pericytes: A review of function and identification. J. Leukoc. Biol. 2004, 75, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Pang, S.; Yu, Y.; Wu, X.; Guo, J.; Zhang, S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 2013, 136, 3578–3588. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Liu, Y.-J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Hofer, T.P. Towards a refined definition of monocyte subsets. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, Y. The yin and yang of innate immunity in stroke. BioMed Res. Int. 2014, 2014, 807978. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Zinselmeyer, B.H.; Corps, K.N.; McGavern, D.B. In vivo dynamics of innate immune sentinels in the cns. Intravital 2012, 1, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Delano, M.J.; Kelly-Scumpia, K.M.; Moreno, C.; Scumpia, P.O.; Laface, D.M.; Heyworth, P.G.; Efron, P.A.; Moldawer, L.L. A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol. Med. 2011, 17, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor gr+cd11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (mdsc), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Dolcetti, L.; Peranzoni, E.; Ugel, S.; Marigo, I.; Fernandez Gomez, A.; Mesa, C.; Geilich, M.; Winkels, G.; Traggiai, E.; Casati, A.; et al. Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by gm-csf. Eur. J. Immunol. 2010, 40, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Damuzzo, V.; Pinton, L.; Desantis, G.; Solito, S.; Marigo, I.; Bronte, V.; Mandruzzato, S. Complexity and challenges in defining myeloid-derived suppressor cells. Cytom. B Clin. Cytom. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Ionita, C.C.; Siddiqui, A.H.; Levy, E.I.; Hopkins, L.N.; Snyder, K.V.; Gibbons, K.J. Acute ischemic stroke and infections. J. Stroke Cerebrovasc. Dis. 2011, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gesuete, R.; Storini, C.; Fantin, A.; Stravalaci, M.; Zanier, E.R.; Orsini, F.; Vietsch, H.; Mannesse, M.L.; Ziere, B.; Gobbi, M.; et al. Recombinant c1 inhibitor in brain ischemic injury. Ann. Neurol. 2009, 66, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, M.; Lippiello, P.M.; Lucas, R.; Marrero, M.B. Alpha7 nicotinic receptors as novel therapeutic targets for inflammation-based diseases. Cell. Mol. Life Sci. 2011, 68, 931–949. [Google Scholar] [CrossRef] [PubMed]
- Kourtis, I.C.; Hirosue, S.; de Titta, A.; Kontos, S.; Stegmann, T.; Hubbell, J.A.; Swartz, M.A. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS ONE 2013, 8, e61646. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Weimar, C.; Roth, M.P.; Zillessen, G.; Glahn, J.; Wimmer, M.L.; Busse, O.; Haberl, R.L.; Diener, H.C. Complications following acute ischemic stroke. Eur. Neurol. 2002, 48, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, H.; Du, X.; Lit, L.; Walker, W.; Lu, A.; Ran, R.; Gregg, J.P.; Reilly, M.; Pancioli, A.; et al. Gene expression in blood changes rapidly in neutrophils and monocytes after ischemic stroke in humans: A microarray study. J. Cereb. Blood Flow Metab. 2006, 26, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Ve, T.; Gay, N.J.; Mansell, A.; Kobe, B.; Kellie, S. Adaptors in toll-like receptor signaling and their potential as therapeutic targets. Curr. Drug Targets 2012, 13, 1360–1374. [Google Scholar] [CrossRef] [PubMed]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Hattori, K.; Hamanaka, J.; Murase, T.; Egashira, Y.; Mishiro, K.; Ishiguro, M.; Tsuruma, K.; Hirose, Y.; Tanaka, H.; et al. Pharmacological inhibition of tlr4-nox4 signal protects against neuronal death in transient focal ischemia. Sci. Rep. 2012, 2, 896. [Google Scholar] [CrossRef] [PubMed]
- Moraga, A.; Pradillo, J.M.; Cuartero, M.I.; Hernandez-Jimenez, M.; Oses, M.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 modulates cell migration and cortical neurogenesis after focal cerebral ischemia. FASEB J. 2014, 28, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Rajbhandari, L.; Tegenge, M.A.; Shrestha, S.; Ganesh Kumar, N.; Malik, A.; Mithal, A.; Hosmane, S.; Venkatesan, A. Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia 2014, 62, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rahman, D.; Lehner, T. A comparative study of stress-mediated immunological functions with the adjuvanticity of alum. J. Biol. Chem. 2012, 287, 17152–17160. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Fossati, S.; Bianchi, M.E.; Patrone, M.; Pedrazzi, M.; Sparatore, B.; Moroni, F.; Chiarugi, A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007, 103, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Neill, L.A.; Bowie, A.G. The family of five: Tir-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Seifert, H.A.; Collier, L.A.; Chapman, C.B.; Benkovic, S.A.; Willing, A.E.; Pennypacker, K.R. Pro-inflammatory interferon gamma signaling is directly associated with stroke induced neurodegeneration. J. Neuroimmune Pharmacol. 2014, 9, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic agonists inhibit hmgb1 release and improve survival in experimental sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, L.; Wang, L.; Degos, V.; Maze, M.; Su, H. Alpha-7 nicotinic acetylcholine receptor agonist treatment reduces neuroinflammation, oxidative stress, and brain injury in mice with ischemic stroke and bone fracture. J. Neurochem. 2014, 131, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Duris, K.; Manaenko, A.; Suzuki, H.; Rolland, W.B.; Krafft, P.R.; Zhang, J.H. Alpha7 nicotinic acetylcholine receptor agonist pnu-282987 attenuates early brain injury in a perforation model of subarachnoid hemorrhage in rats. Stroke 2011, 42, 3530–3536. [Google Scholar] [CrossRef] [PubMed]
- Krafft, P.R.; Altay, O.; Rolland, W.B.; Duris, K.; Lekic, T.; Tang, J.; Zhang, J.H. Alpha7 nicotinic acetylcholine receptor agonism confers neuroprotection through gsk-3beta inhibition in a mouse model of intracerebral hemorrhage. Stroke 2012, 43, 844–850. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, W.J.; van der Zanden, E.P.; The, F.O.; Bijlsma, M.F.; van Westerloo, D.J.; Bennink, R.J.; Berthoud, H.R.; Uematsu, S.; Akira, S.; van den Wijngaard, R.M.; et al. Stimulation of the vagus nerve attenuates macrophage activation by activating the jak2-stat3 signaling pathway. Nat. Immunol. 2005, 6, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Sig Choi, J.; Yu, Y.M.; Nam, K.; Piao, C.S.; Kim, S.W.; Lee, M.H.; Han, P.L.; Park, J.S.; Lee, J.K. Hmgb1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Biscetti, F.; Straface, G.; de Cristofaro, R.; Lancellotti, S.; Rizzo, P.; Arena, V.; Stigliano, E.; Pecorini, G.; Egashira, K.; de Angelis, G.; et al. High-mobility group box-1 protein promotes angiogenesis after peripheral ischemia in diabetic mice through a vegf-dependent mechanism. Diabetes 2010, 59, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Bianchi, M.E. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem. Pharmacol. 2004, 68, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Nakano, T.; Irie, K.; Higuchi, S.; Fujioka, M.; Orito, K.; Iwasaki, K.; Jin, G.; Lo, E.H.; Mishima, K.; et al. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2010, 30, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Lykhmus, O.; Voytenko, L.; Koval, L.; Mykhalskiy, S.; Kholin, V.; Peschana, K.; Zouridakis, M.; Tzartos, S.; Komisarenko, S.; Skok, M. A7 nicotinic acetylcholine receptor-specific antibody induces inflammation and amyloid β42 accumulation in the mouse brain to impair memory. PLoS ONE 2015, 10, e0122706. [Google Scholar] [CrossRef] [PubMed]
- Pendlebury, S.T.; Rothwell, P.M. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: A systematic review and meta-analysis. Lancet Neurol. 2009, 8, 1006–1018. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Godoy, J.A.; Vargas, J.Y.; Arrazola, M.S.; Rios, J.A.; Carvajal, F.J.; Serrano, F.G.; Farias, G.G. Nicotine prevents synaptic impairment induced by amyloid-beta oligomers through alpha7-nicotinic acetylcholine receptor activation. Neuromol. Med. 2013, 15, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Govind, A.P.; Walsh, H.; Green, W.N. Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J. Neurosci. 2012, 32, 2227–2238. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, Y.; Liu, Y. The role of nucleotides and purinergic signaling in apoptotic cell clearance-implications for chronic inflammatory diseases. Front. Immunol. 2014, 5, 656. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, M.F.; Khakh, B.S. Atp-gated p2x cation-channels. Neuropharmacology 2009, 56, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. Atp mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to atp and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [PubMed]
- Kufer, T.A.; Sansonetti, P.J. Nlr functions beyond pathogen recognition. Nat. Immunol. 2011, 12, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tschopp, J.; Schroder, K. Nlrp3 inflammasome activation: The convergence of multiple signalling pathways on ros production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Hecker, A.; Kullmar, M.; Wilker, S.; Richter, K.; Zakrzewicz, A.; Atanasova, S.; Mathes, V.; Timm, T.; Lerner, S.; Klein, J.; et al. Phosphocholine-modified macromolecules and canonical nicotinic agonists inhibit atp-induced il-1beta release. J. Immunol. 2015, 195, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Kwan, K.; Levine, Y.A.; Olofsson, P.S.; Yang, H.; Li, J.; Joshi, S.; Wang, H.; Andersson, U.; Chavan, S.S.; et al. Alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 2014, 20, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Boutin, H.; LeFeuvre, R.A.; Horai, R.; Asano, M.; Iwakura, Y.; Rothwell, N.J. Role of il-1alpha and il-1beta in ischemic brain damage. J. Neurosci. 2001, 21, 5528–5534. [Google Scholar] [PubMed]
- Tsuzaki, M.; Guyton, G.; Garrett, W.; Archambault, J.M.; Herzog, W.; Almekinders, L.; Bynum, D.; Yang, X.; Banes, A.J. Il-1 beta induces cox2, mmp-1, -3 and -13, adamts-4, il-1 beta and il-6 in human tendon cells. J. Orthop. Res. 2003, 21, 256–264. [Google Scholar] [CrossRef]
- Lima-Junior, D.S.; Costa, D.L.; Carregaro, V.; Cunha, L.D.; Silva, A.L.N.; Mineo, T.W.P.; Gutierrez, F.R.S.; Bellio, M.; Bortoluci, K.R.; Flavell, R.A.; et al. Inflammasome-derived il-1[beta] production induces nitric oxide-mediated resistance to leishmania. Nat. Med. 2013, 19, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Deroide, N.; Li, X.; Lerouet, D.; Van Vre, E.; Baker, L.; Harrison, J.; Poittevin, M.; Masters, L.; Nih, L.; Margaill, I.; et al. Mfge8 inhibits inflammasome-induced il-1beta production and limits postischemic cerebral injury. J. Clin. Investig. 2013, 123, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase ii randomised controlled trial. J. Neuroinflamm. 2014, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Helmy, A.; Guilfoyle, M.R.; Carpenter, K.L.; Pickard, J.D.; Menon, D.K.; Hutchinson, P.J. Recombinant human interleukin-1 receptor antagonist in severe traumatic brain injury: A phase ii randomized control trial. J. Cereb. Blood Flow Metab. 2014, 34, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of pro il-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- de Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic neutralization of the nlrp1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Wang, Z.; Wei, X.; Han, H.; Meng, X.; Zhang, Y.; Shi, W.; Li, F.; Xin, T.; Pang, Q.; et al. Nlrp3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. blood Flow Metab. 2014, 34, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. Aim2 and nlrc4 inflammasomes contribute with asc to acute brain injury independently of nlrp3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. Aim2 recognizes cytosolic dsdna and forms a caspase-1-activating inflammasome with asc. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Lamphier, M.S.; Sirois, C.M.; Verma, A.; Golenbock, D.T.; Latz, E. Tlr9 and the recognition of self and non-self nucleic acids. Ann. N. Y. Acad. Sci. 2006, 1082, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Schielke, G.P.; Yang, G.Y.; Shivers, B.D.; Betz, A.L. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J. Cereb. Blod Flow Metab. 1998, 18, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pastrana, J.; Ferrer, L.; Li, Y.-F.; Xiong, X.; Xi, H.; Cueto, R.; Nelson, J.Z.; Sha, X.; Li, X.; Cannella, A.L.; et al. Inhibition of caspase-1 activation in endothelial cells improves angiogenesis -a novel therapeutic potential for ischemia. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ohab, J.J.; Fleming, S.; Blesch, A.; Carmichael, S.T. A neurovascular niche for neurogenesis after stroke. J. Neurosci. 2006, 26, 13007–13016. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, S.; Shields, N.J.; Balle, T.; Chebib, M.; Clarkson, A.N. Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective. Int. J. Mol. Sci. 2015, 16, 29029-29046. https://doi.org/10.3390/ijms161226141
Neumann S, Shields NJ, Balle T, Chebib M, Clarkson AN. Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective. International Journal of Molecular Sciences. 2015; 16(12):29029-29046. https://doi.org/10.3390/ijms161226141
Chicago/Turabian StyleNeumann, Silke, Nicholas J. Shields, Thomas Balle, Mary Chebib, and Andrew N. Clarkson. 2015. "Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective" International Journal of Molecular Sciences 16, no. 12: 29029-29046. https://doi.org/10.3390/ijms161226141
APA StyleNeumann, S., Shields, N. J., Balle, T., Chebib, M., & Clarkson, A. N. (2015). Innate Immunity and Inflammation Post-Stroke: An α7-Nicotinic Agonist Perspective. International Journal of Molecular Sciences, 16(12), 29029-29046. https://doi.org/10.3390/ijms161226141