
An Investigation of Molecular Docking and Molecular Dynamic Simulation on Imidazopyridines as B-Raf Kinase Inhibitors

Abstract
:
1. Introduction
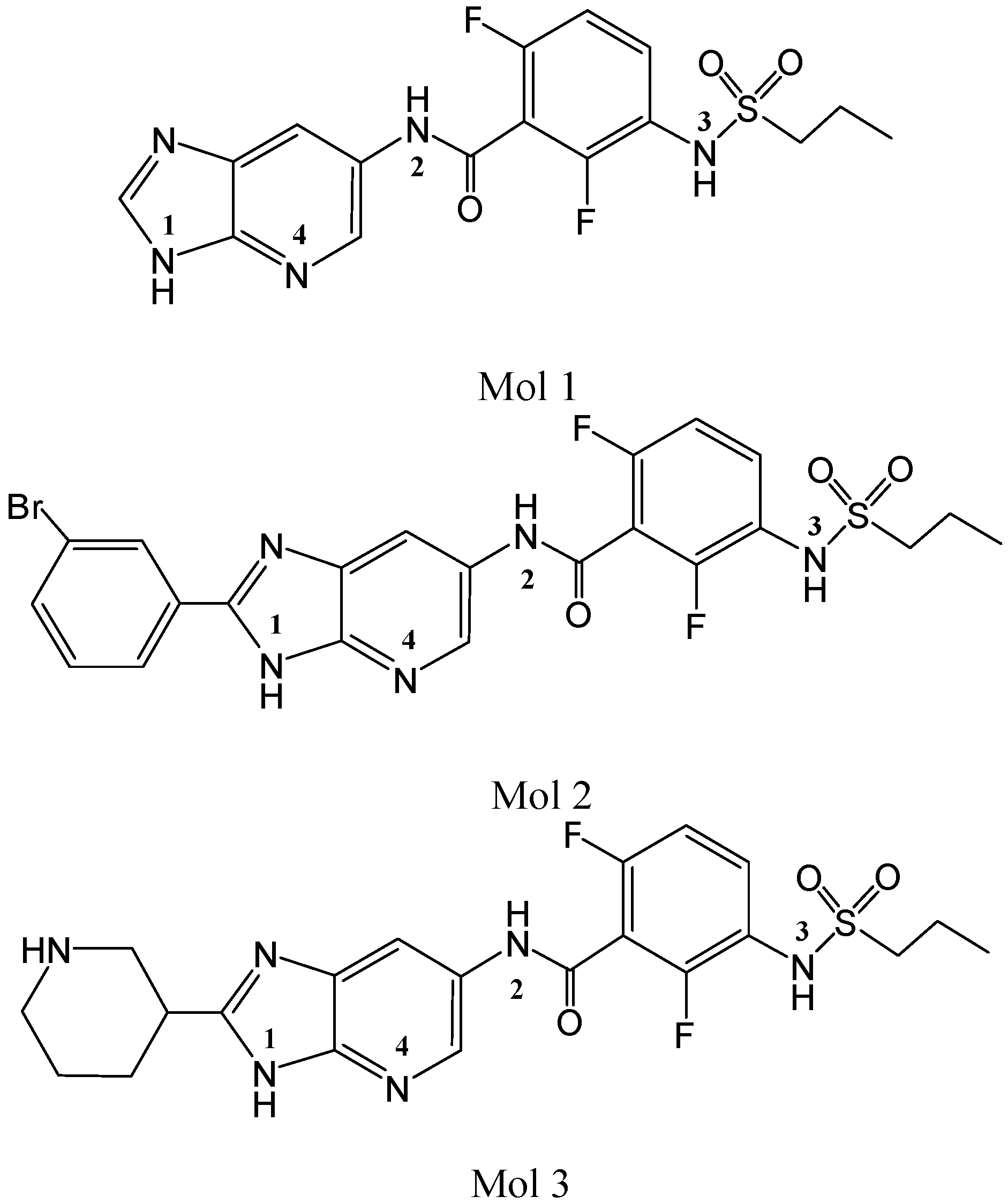
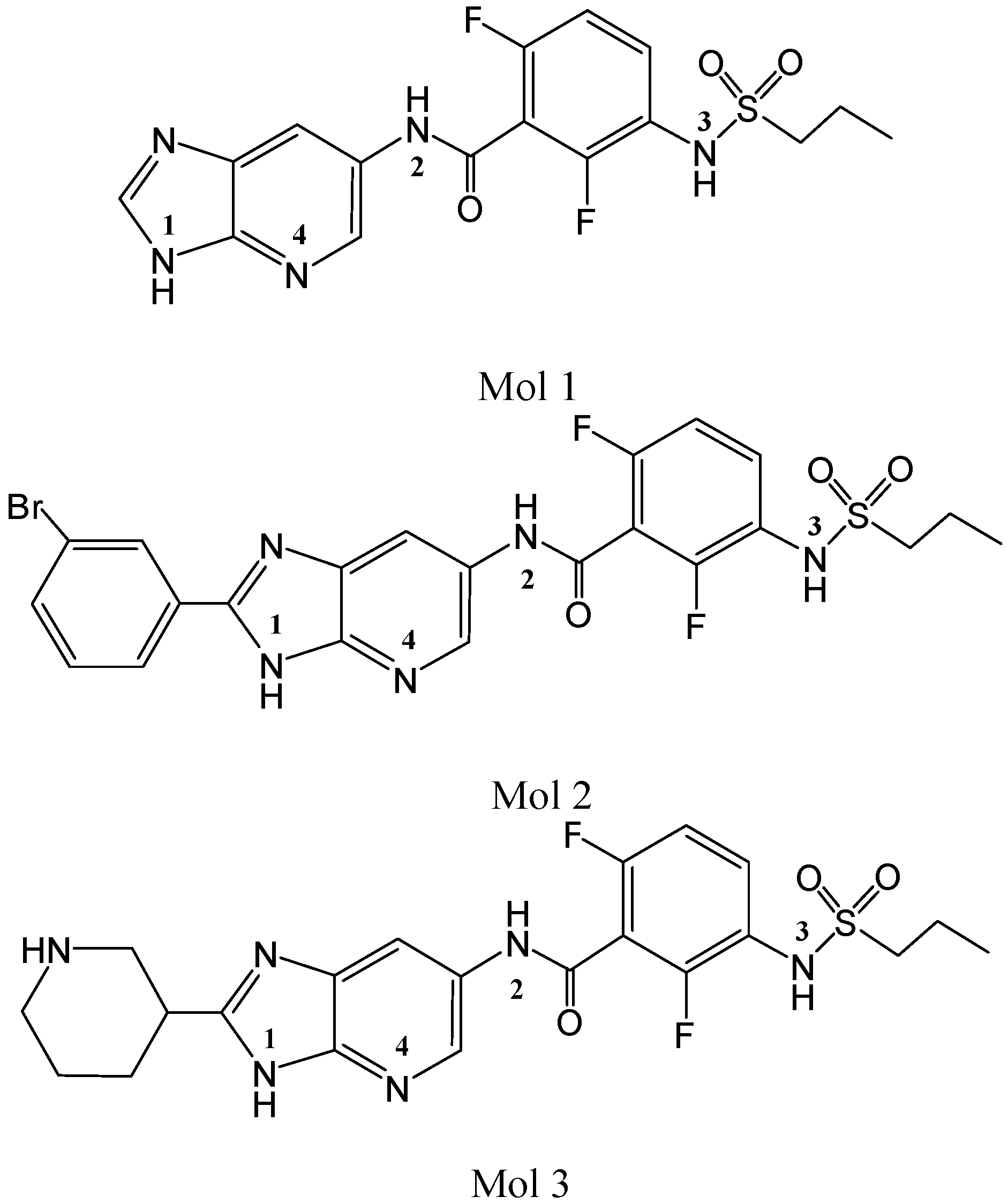
2. Results and Discussion
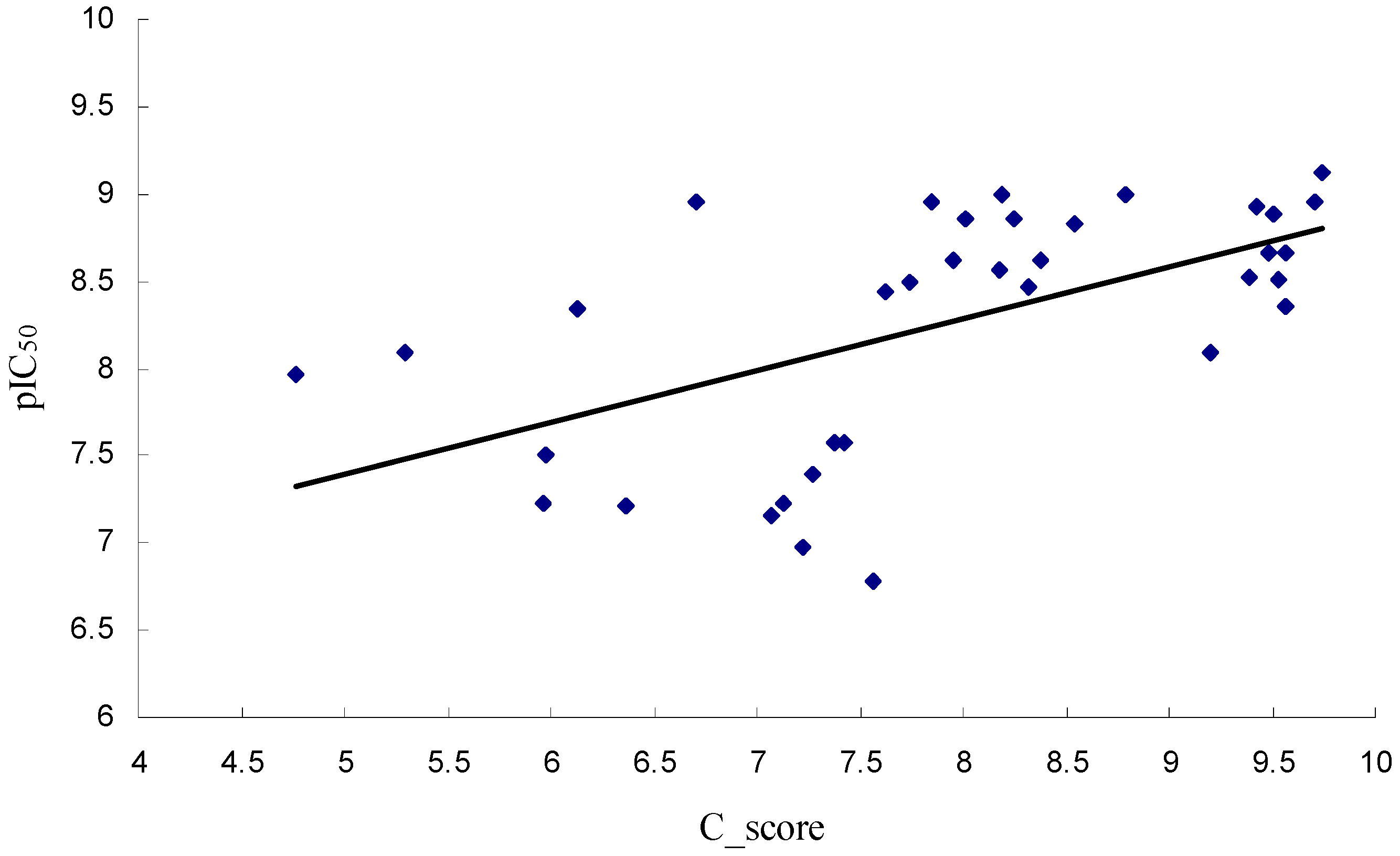
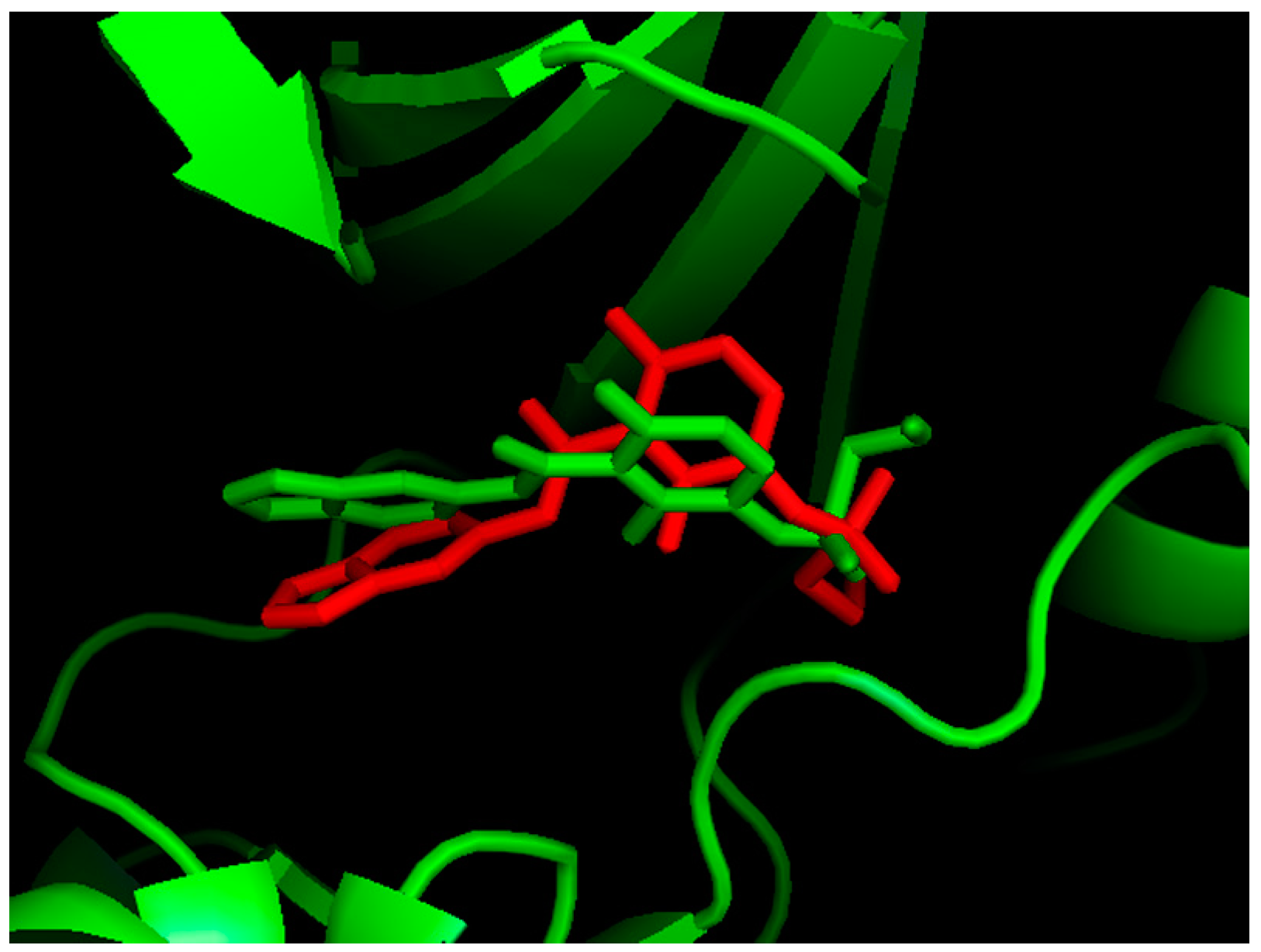
2.1. Molecular Docking
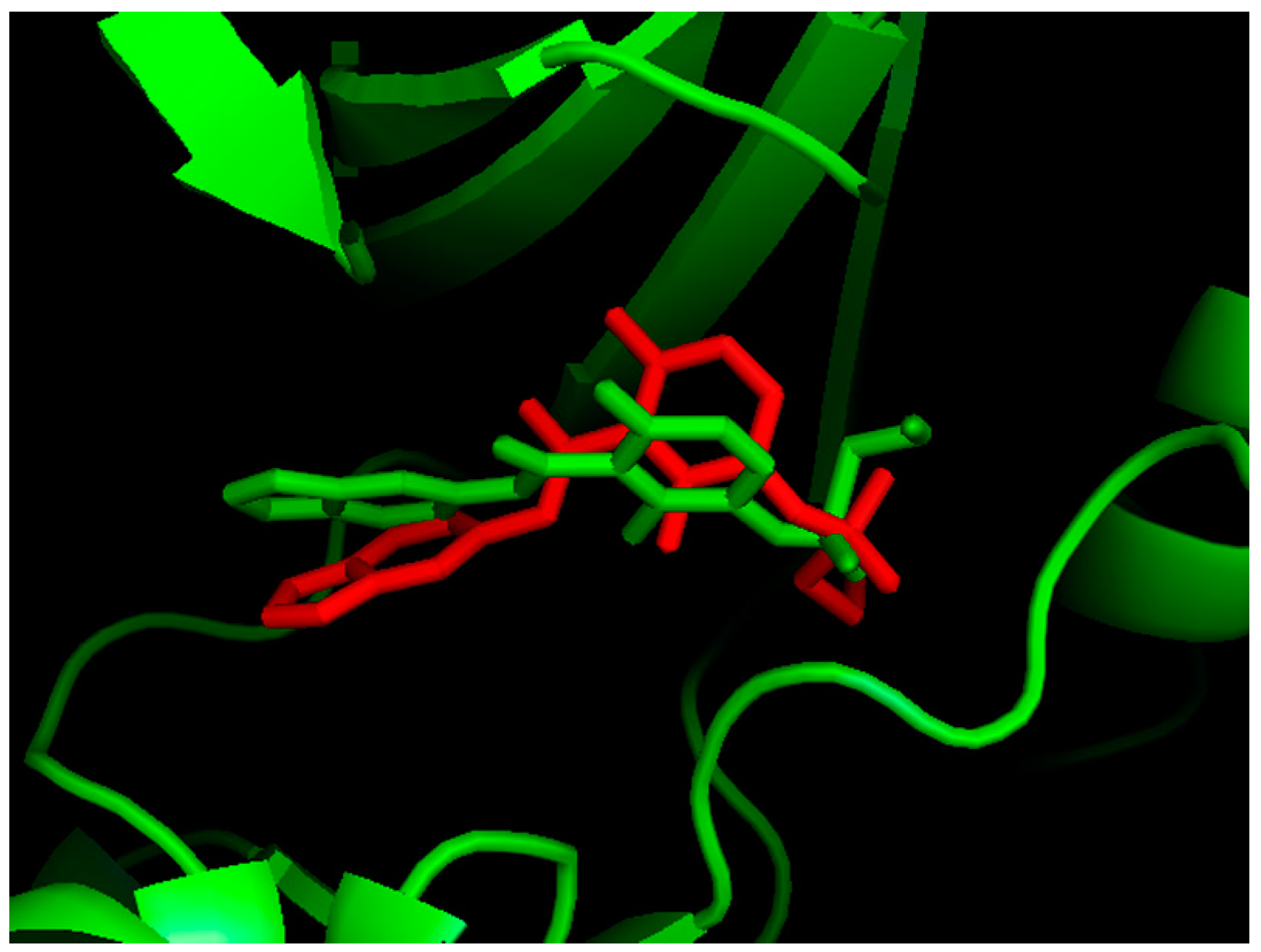
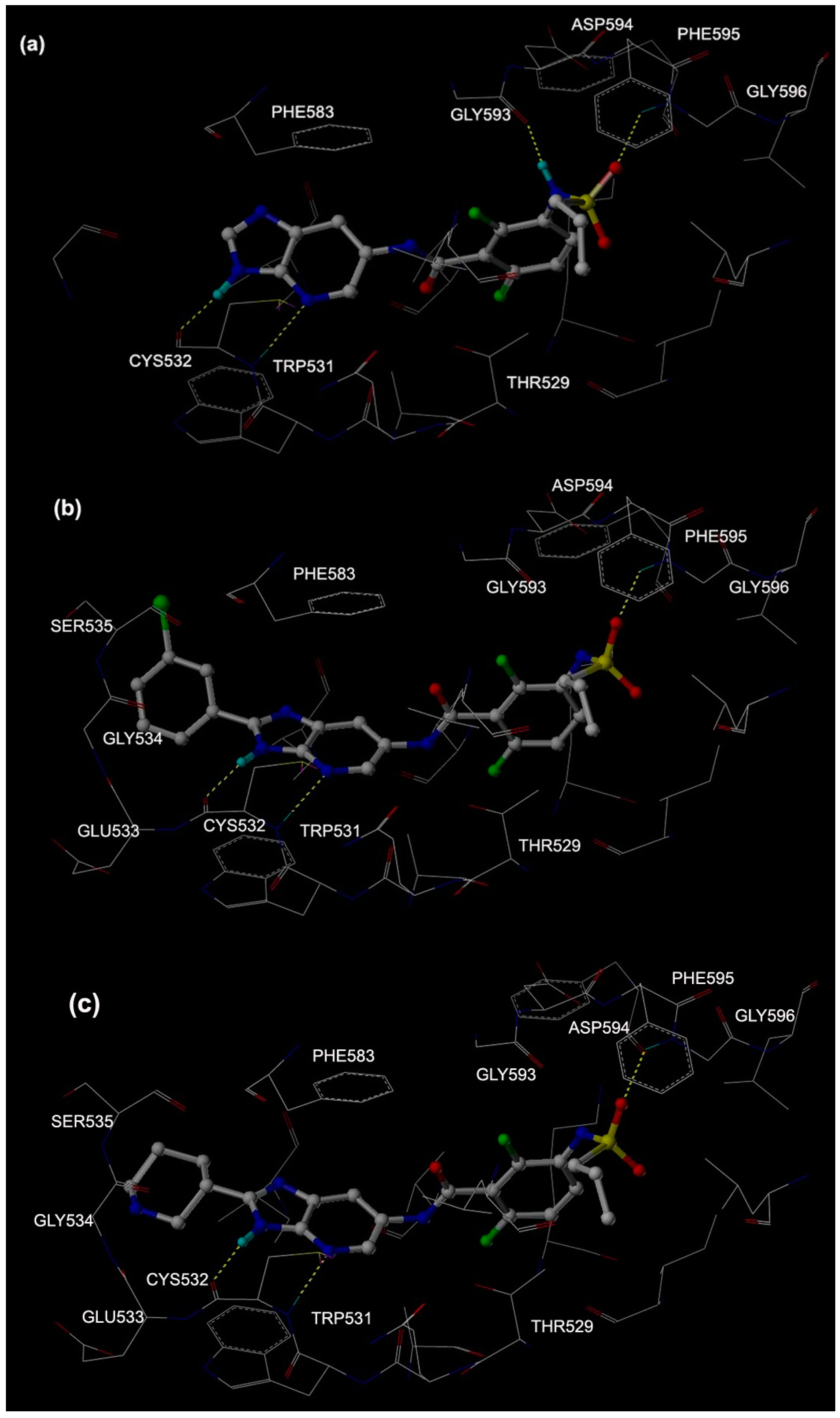
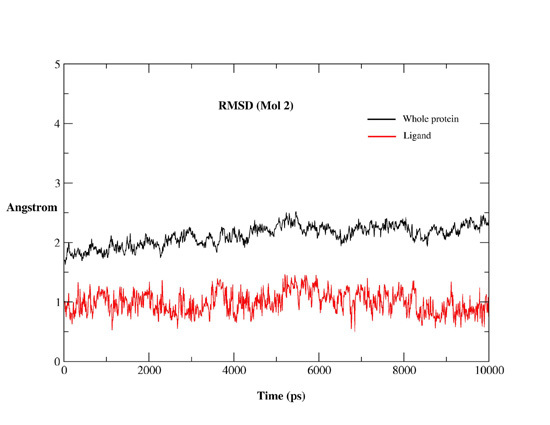
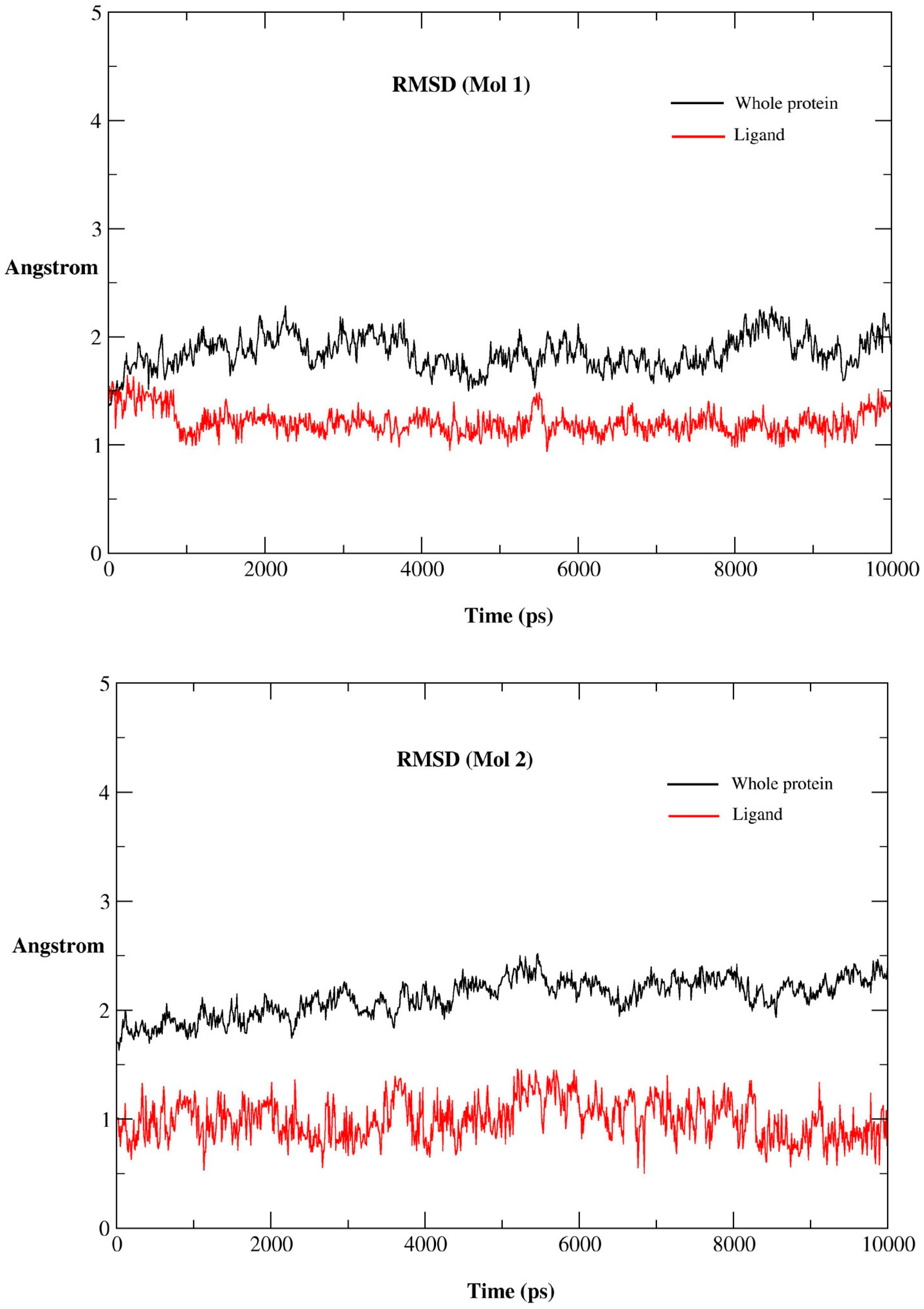
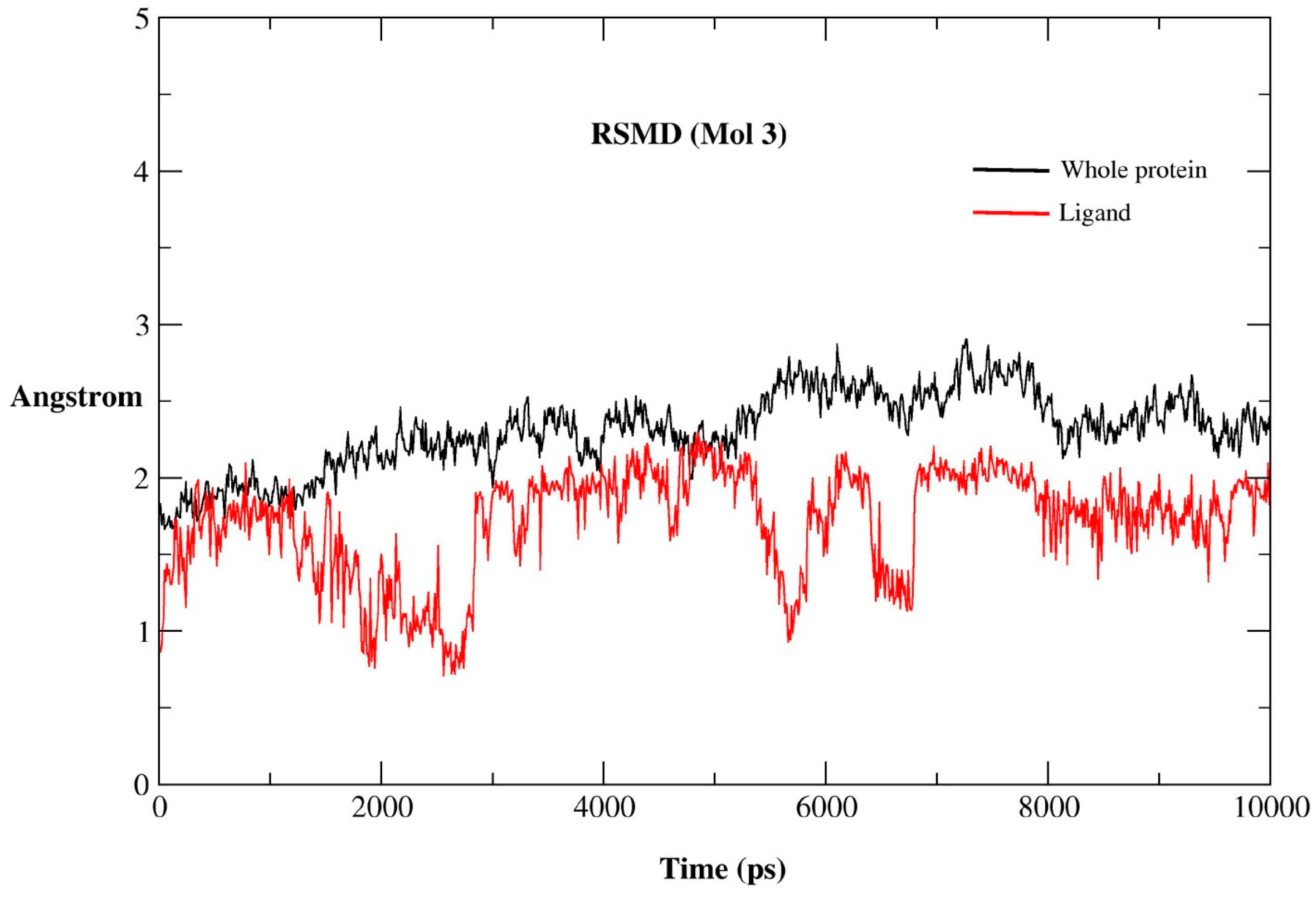
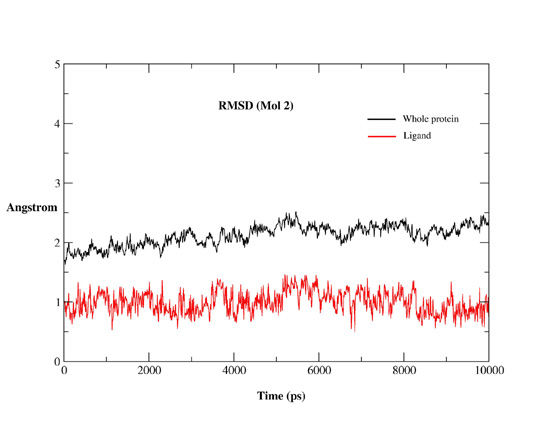
2.2. MD Simulations
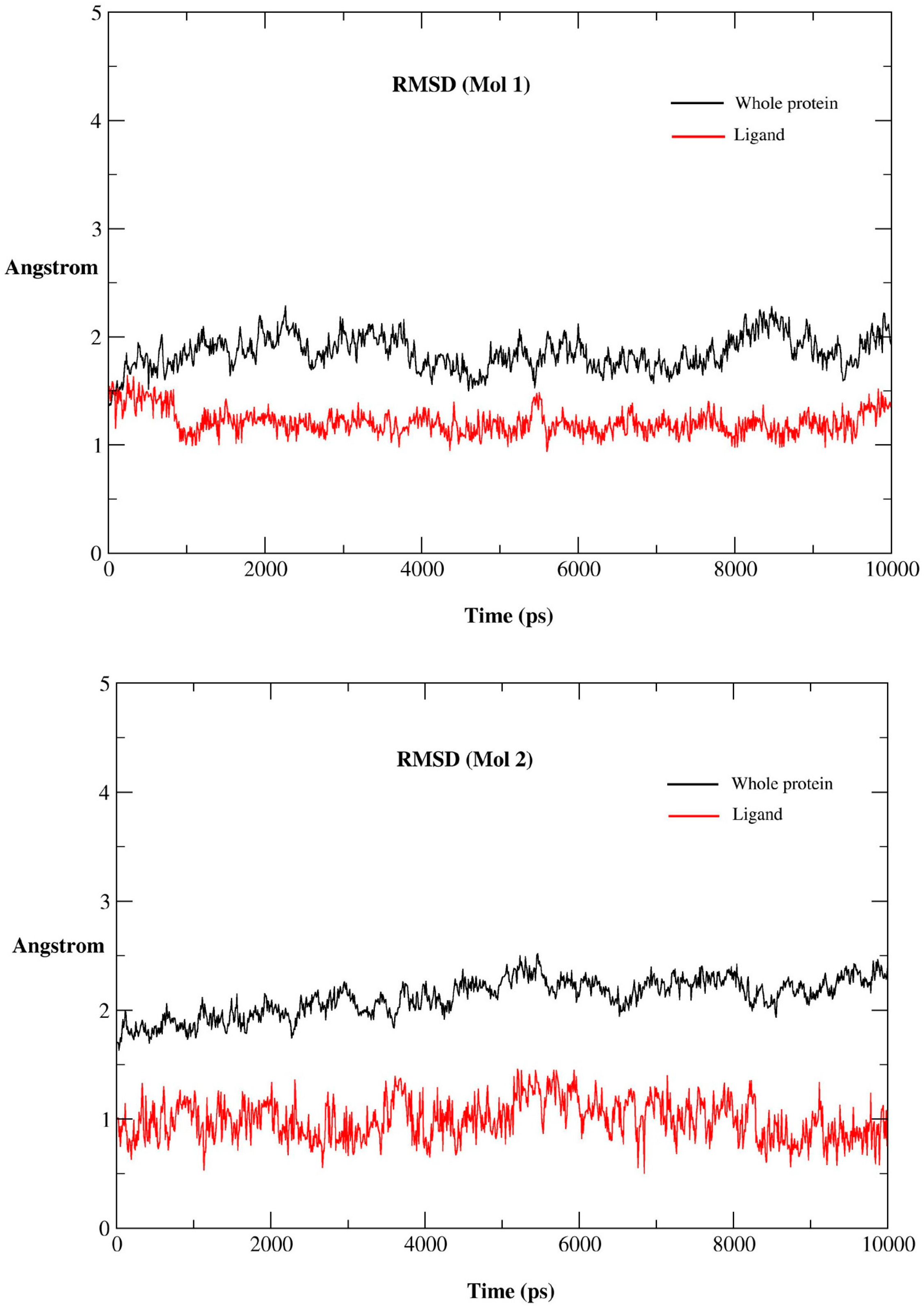
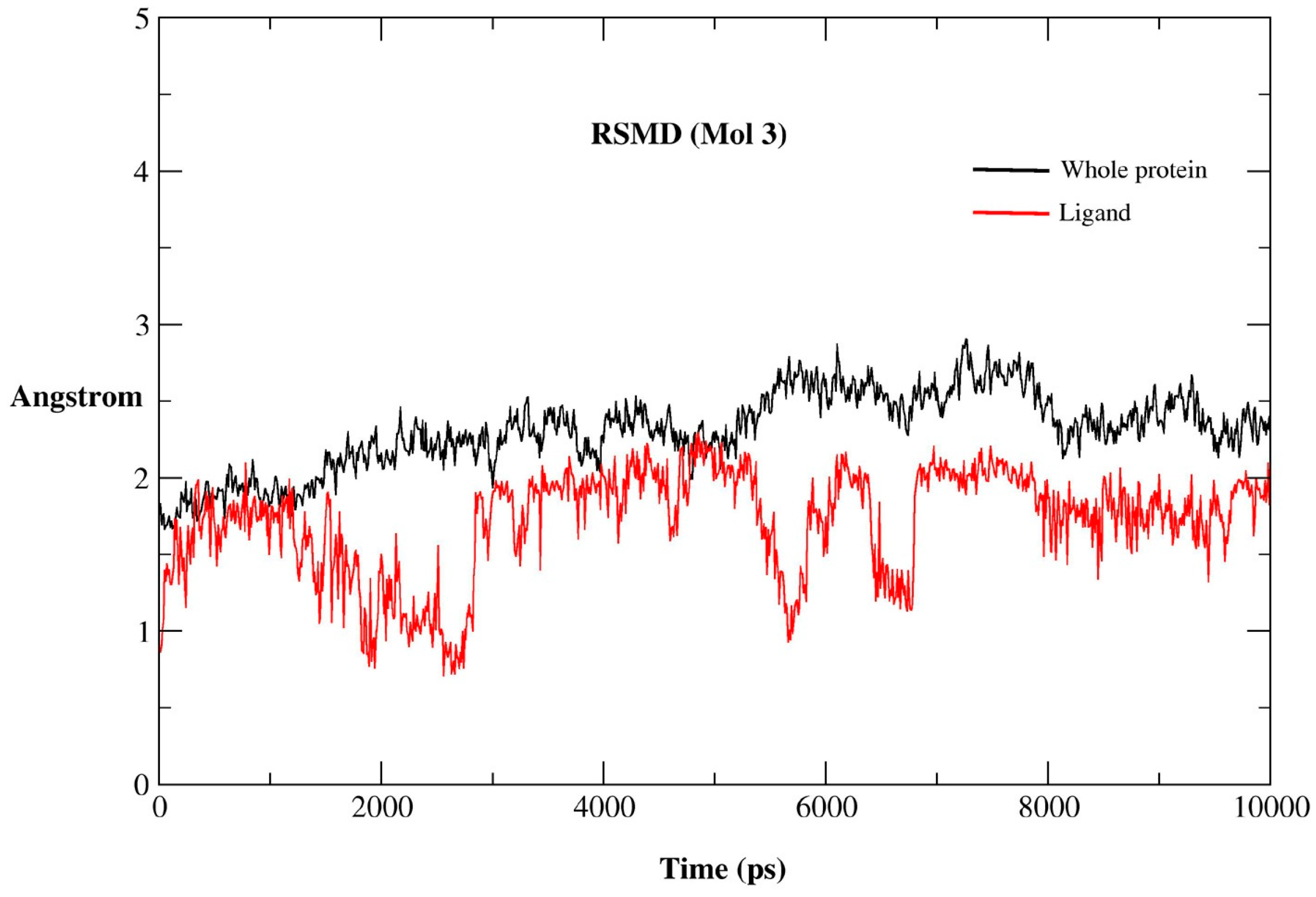
2.2.1. MD Simulations Features
2.2.2. RMSF for Residues of the Binding Pocket

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residues | Mol 1 | Mol 2 | Mol 3 |
|---|---|---|---|
| ILE 463 | 0.075 | 0.109 | 0.127 |
| PHE 468 | 0.081 | 0.148 | 0.207 |
| VAL 471 | 0.275 | 0.194 | 0.265 |
| ALA 481 | 0.614 | 0.701 | 0.704 |
| LYS 483 | 0.452 | 0.512 | 0.563 |
| LEU 505 | 0.540 | 0.201 | 0.805 |
| LEU 514 | 0.621 | 0.573 | 0.597 |
| ILE 527 | 0.678 | 0.712 | 0.597 |
| THR 529 | 0.420 | 0.343 | 0.413 |
| GLN 530 | 0.166 | 0.148 | 0.452 |
| TRP 531 | 0.663 | 0.215 | 0.266 |
| CYS 532 | 0.245 | 0.328 | 0.442 |
| GLU 533 | 0.232 | 0.163 | 0.141 |
| GLY 534 | 0.712 | 0.330 | 0.203 |
| SER 535 | 0.158 | 0.244 | 0.217 |
| PHE 583 | 0.653 | 0.746 | 0.958 |
| GLY 593 | 0.177 | 0.248 | 0.512 |
| ASP 594 | 0.197 | 0.241 | 0.218 |
| PHE 595 | 0.086 | 0.098 | 0.064 |
| GLY 596 | 0.835 | 0.566 | 0.748 |
| LEU 597 | 0.405 | 0.610 | 0.604 |
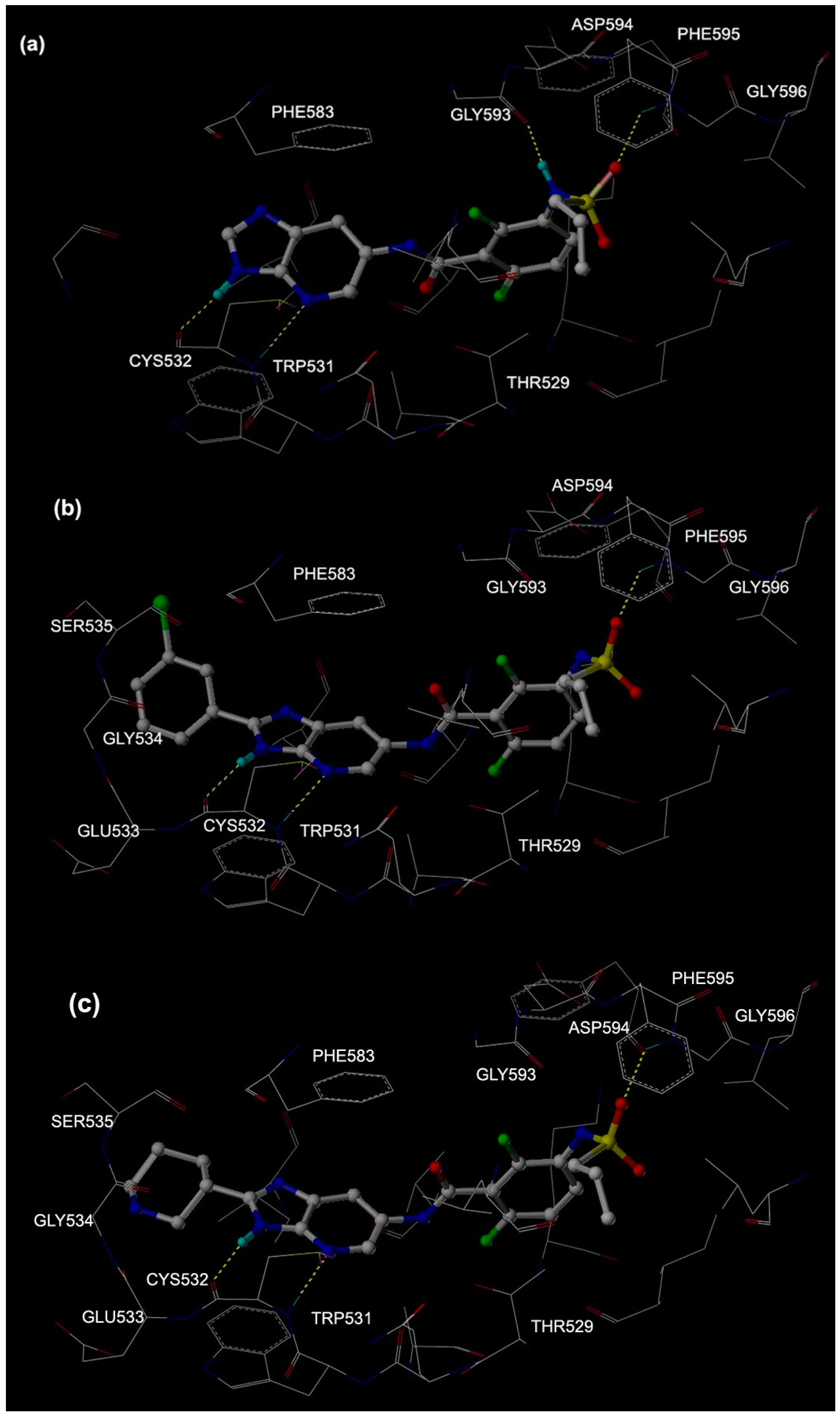
2.2.3. H-Bonds in MD Simulations
| Complex | Acceptor | Donor | Occupancy (%) | Distance (Å) | Angle (°) |
|---|---|---|---|---|---|
| Mol 1 | CYS 532 C=O | Lig N1–H | 54.00 | 2.89 ± 0.07 | 18.51 ± 12.41 |
| THR 529 C=O | Lig N2–H | 41.00 | 2.90 ± 0.07 | 20.54 ± 10.55 | |
| ASP 594 C=O | Lig N3–H | 48.00 | 2.82 ± 0.07 | 19.96 ± 10.74 | |
| H2O | Lig N3–H | 36.00 | 2.86 ± 0.10 | 20.48 ± 9.44 | |
| Mol 2 | CYS 532 C=O | Lig N1–H | 66.00 | 2.84 ± 0.09 | 26.11 ± 13.57 |
| THR 529 C=O | Lig N2–H | 11.00 | 2.90 ± 0.05 | 23.84 ± 12.42 | |
| ASP 594 C=O | Lig N3–H | 69.00 | 2.81 ± 0.10 | 19.95 ± 10.67 | |
| Mol 3 | CYS 532 C=O | Lig N1–H | 10.00 | 2.91 ± 0.07 | 19.87 ± 11.91 |
| ASP 594 C=O | Lig N3–H | 97.00 | 2.77 ± 0.08 | 14.26 ± 8.55 | |
| Lig S=O | LYS 601 N–H1 | 42.50 | 2.82 ± 0.08 | 21.22 ± 11.35 | |
| Lig S=O | LYS 601 N–H2 | 26.00 | 2.82 ± 0.08 | 22.81 ± 11.08 |
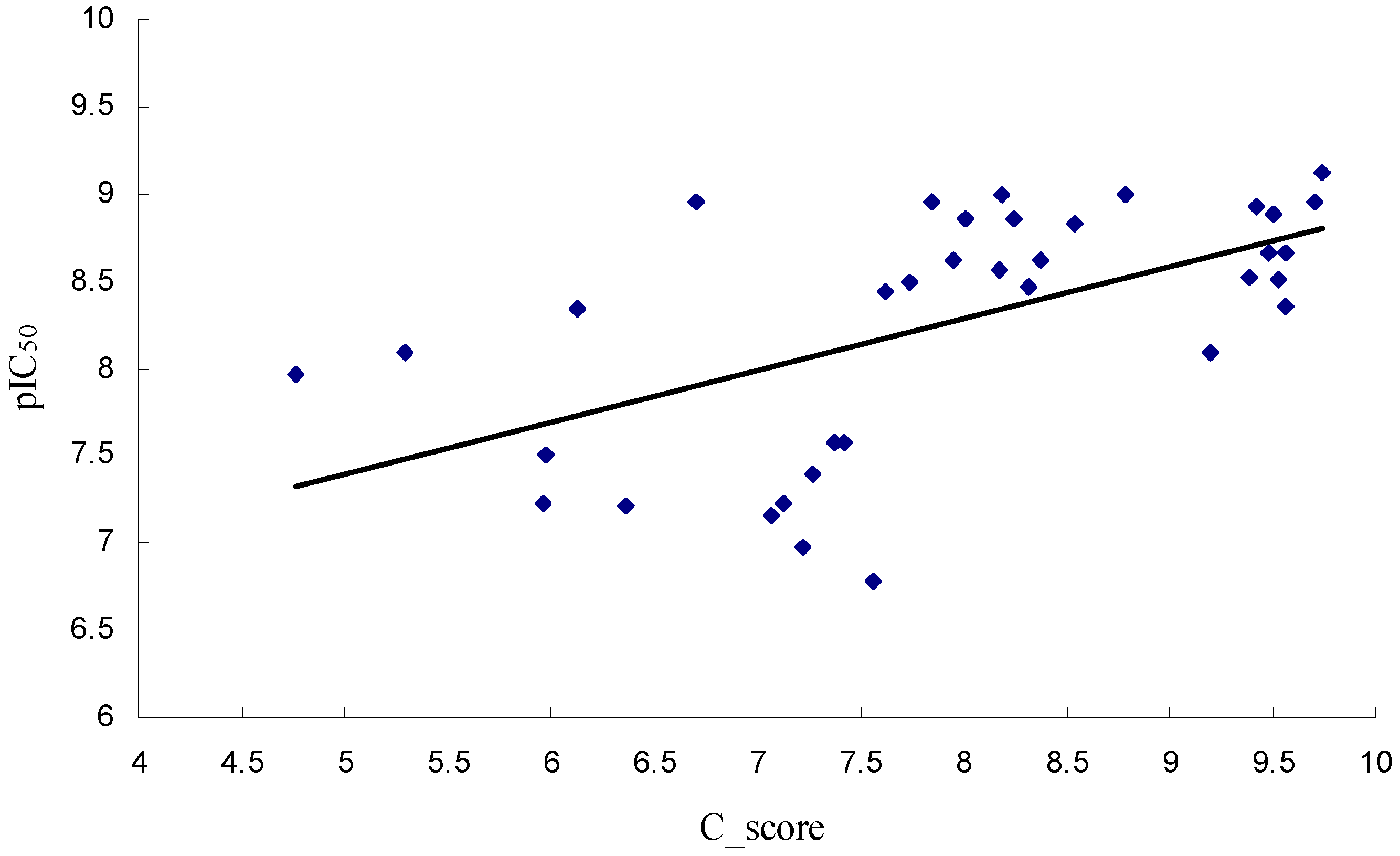
2.3. Binding Free Energies
| Energy/Activity | Mol 1 Complex | Mol 2 Complex | Mol 3 Complex |
|---|---|---|---|
| ΔEvdw | −52.94 | −59.02 | −56.25 |
| ΔEele | −45.71 | −46.95 | −48.48 |
| ΔEgas | −98.65 | −105.97 | −104.73 |
| ΔGGB | 53.66 | 56.45 | 61.94 |
| ΔGSA | −6.61 | −6.97 | −7.11 |
| ΔGsol | 47.05 | 49.48 | 54.83 |
| ΔGbind | −51.60 | −56.49 | −49.90 |
| IC50 | 61 (nM) | 0.76 (nM) | 167 (nM) |
| pIC50 | 7.215 | 9.119 | 6.777 |
3. Experimental Section
3.1. Preparation of Protein and Ligands
3.2. Molecular Docking
3.3. MD Simulations
3.4. Calculation of Binding Free Energy
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El-Azab, A.S.; Al-Omar, M.A.; Abdel-Aziz, A.M.; Abdel-Aziz, N.I.; El-Sayed, A.A.; Aleisa, A.M.; Sayed-Ahmed, M.M.; Abdel-Hamide, S.G. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. Eur. J. Med. Chem. 2010, 45, 4188–4198. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, R.; Chantani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batt, D.; Warmuth, M. B-Raf kinase inhibitors for cancer treatment. Curr. Opin. Investig. Drugs 2007, 8, 452–456. [Google Scholar] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.E.; Pritchard, C.A. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Weber, B.L.; Herlyn, M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell 2003, 4, 95–98. [Google Scholar] [CrossRef]
- Madhunapantula, S.V.; Robertson, G.P. Is B-Raf a good therapeutic target for melanoma and other malignancies? Cancer Res. 2008, 68, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Karasarides, M.; Chiloeches, A.; Hayward, R.; Niculescu-Duvaz, D.; Scanlon, I.; Friedlos, F.; Ogilvie, L.; Hedley, D.; Martin, J.; Marshall, C.J.; et al. B-RAF is a therapeutic target in melanoma. Oncogene 2004, 23, 6292–6298. [Google Scholar] [CrossRef] [PubMed]
- El-Nassan, H.B. Recent progress in the identification of BRAF inhibitors as anti-cancer agents. Eur. J. Med. Chem. 2014, 72, 170–205. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, B.J.; Wenglowsky, S.; Grina, J.; Laird, E.R.; Voegtli, W.C.; Ren, L.; Ahrendt, K.; Buckmelter, A.; Gloor, S.L.; Klopfenstein, N.; et al. Imidazo[4,5-b]pyridine inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2013, 23, 5896–5899. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A combined pharmacophore modeling, 3D QSAR and virtual screening studies on imidazopyridines as B-Raf inhibitors. Int. J. Mol. Sci. 2015, 16, 12307–12323. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sim, T. Novel small molecule Raf kinase inhibitors for targeted cancer therapeutics. Arch. Pharm. Res. 2012, 35, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.J.; Barbault, F.; Delamar, M.; Zhang, R.S. Receptor- and ligand-based 3D-QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Tripos Inc. Docking Suite Manual for Tripos 7.3; Tripos Inc.: St. Louis, MO, USA, 2006. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L., III; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Wang, J.M.; Wang, W.; Kollman, P.A. Antechamber, an accessory software package for molecular mechanical calculations. Abstr. Pap. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Hou, T.J.; Wang, J.M.; Li, Y.Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Kuhn, L.A.; Case, D.A. Change in protein flexibility upon complex formation: Analysis of Ras-Raf using molecular dynamics and a molecular framework approach. Proteins 2004, 56, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Jyrkkarinne, J.; Kublbeck, J.; Pulkkinen, J.; Honkakoski, P.; Laatikainen, R.; Poso, A.; Laitinen, T. Molecular dynamics simulations for human CAR inverse agonists. J. Chem. Inf. Model. 2012, 52, 457–464. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Li, Y.; Yu, F.; Xie, X.; Qiu, K.; Fu, J. An Investigation of Molecular Docking and Molecular Dynamic Simulation on Imidazopyridines as B-Raf Kinase Inhibitors. Int. J. Mol. Sci. 2015, 16, 27350-27361. https://doi.org/10.3390/ijms161126026
Xie H, Li Y, Yu F, Xie X, Qiu K, Fu J. An Investigation of Molecular Docking and Molecular Dynamic Simulation on Imidazopyridines as B-Raf Kinase Inhibitors. International Journal of Molecular Sciences. 2015; 16(11):27350-27361. https://doi.org/10.3390/ijms161126026
Chicago/Turabian StyleXie, Huiding, Yupeng Li, Fang Yu, Xiaoguang Xie, Kaixiong Qiu, and Jijun Fu. 2015. "An Investigation of Molecular Docking and Molecular Dynamic Simulation on Imidazopyridines as B-Raf Kinase Inhibitors" International Journal of Molecular Sciences 16, no. 11: 27350-27361. https://doi.org/10.3390/ijms161126026