
Protein Recognition in Drug-Induced DNA Alkylation: When the Moonlight Protein GAPDH Meets S23906-1/DNA Minor Groove Adducts


Abstract
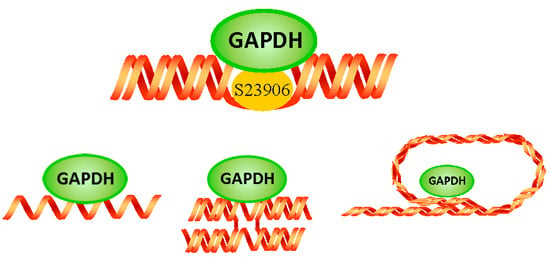
:
1. Introduction
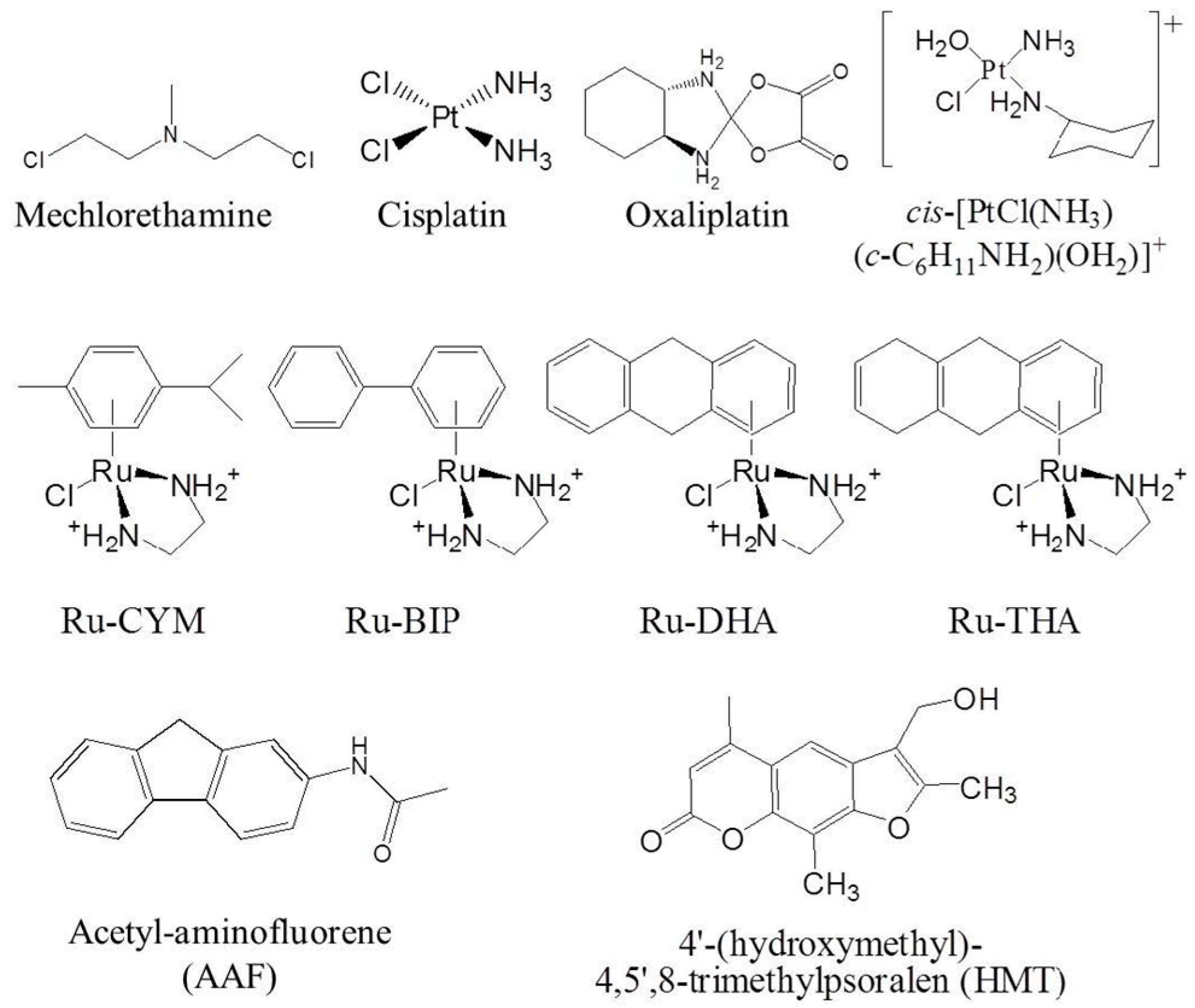
1.1. DNA Alkylation and Destabilization by DNA Alkylating Drugs
1.1.1. DNA Major Groove Alkylating Agents
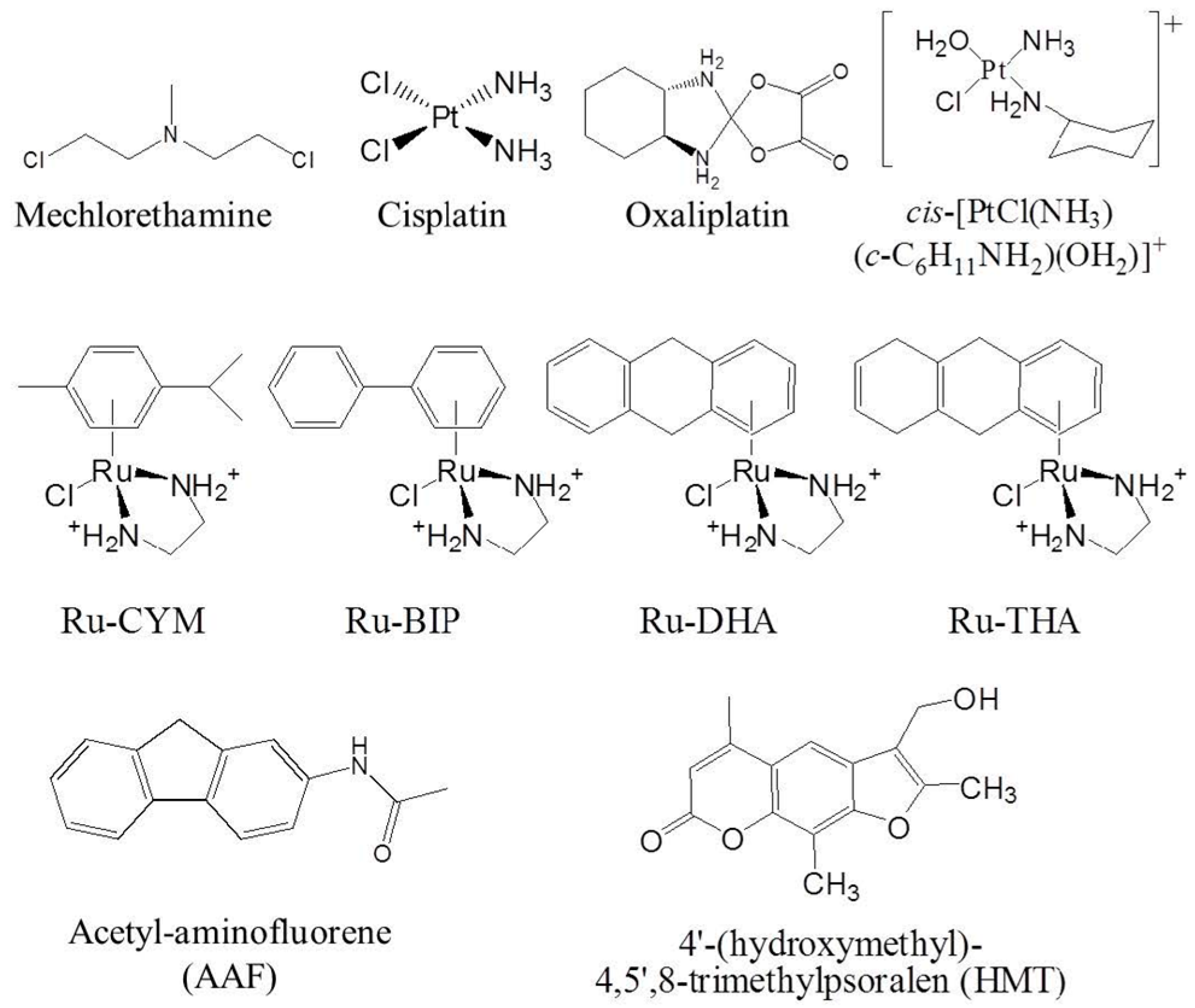
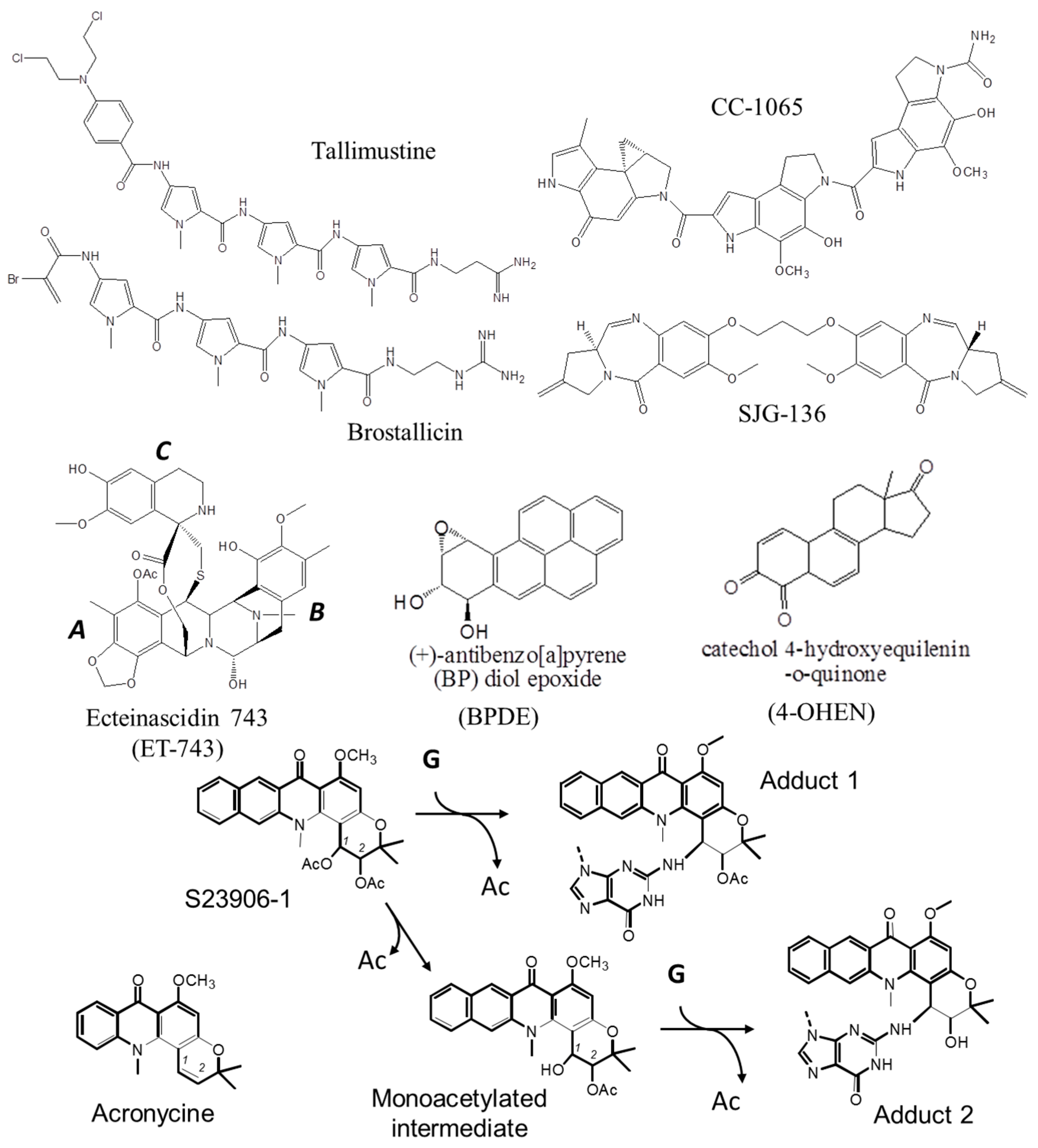
1.1.2. DNA Minor Groove Alkylating Agents
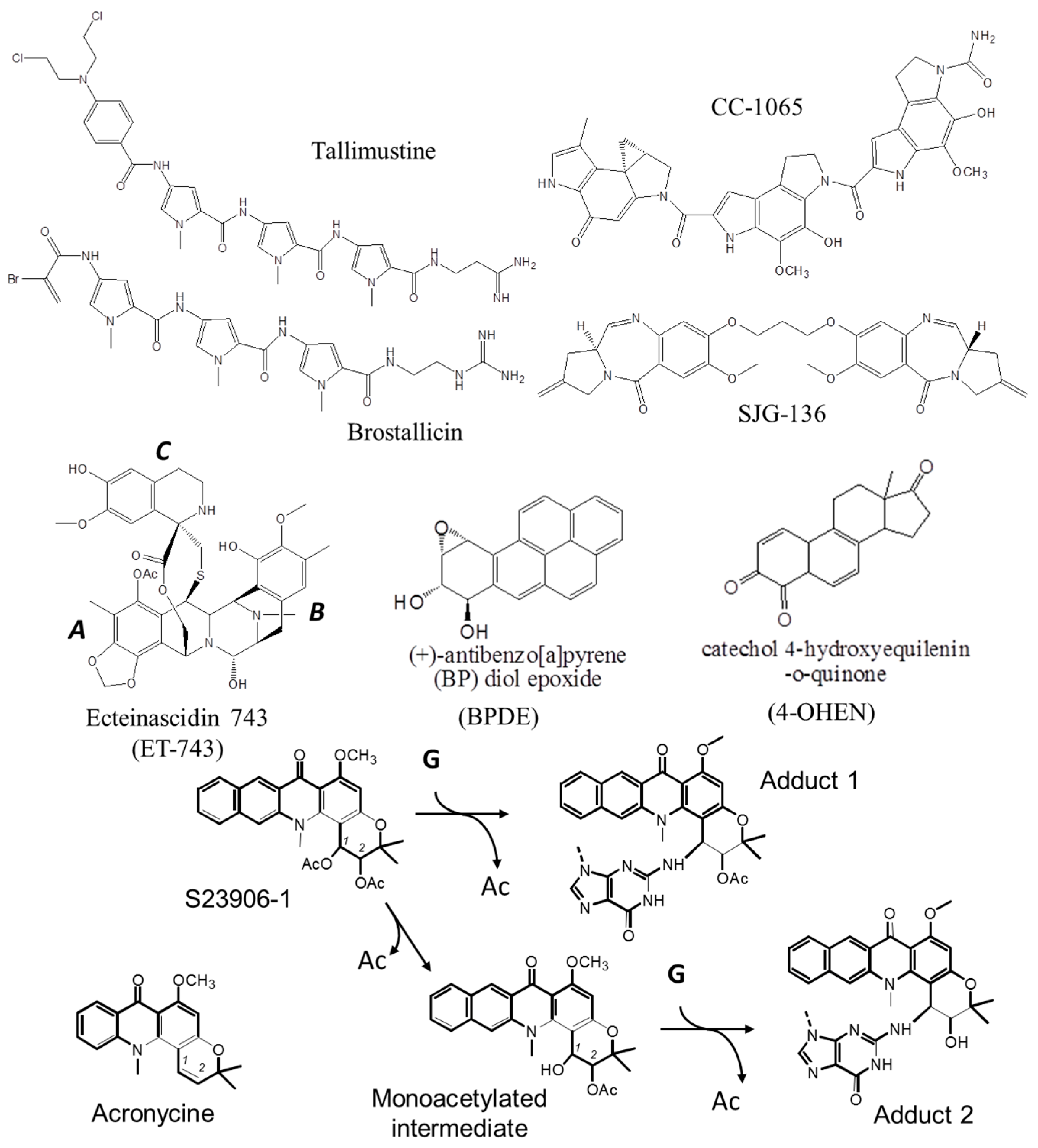
1.2. Protein Recognition of DNA Adducts
1.2.1. Protein Recognition of Major Groove DNA Adducts
1.2.2. Protein Recognition of Major Groove DNA Adducts
2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) Binding to Damaged DNA: The Example of S23906-1/DNA Adduct Recognition
2.1. S23906-1
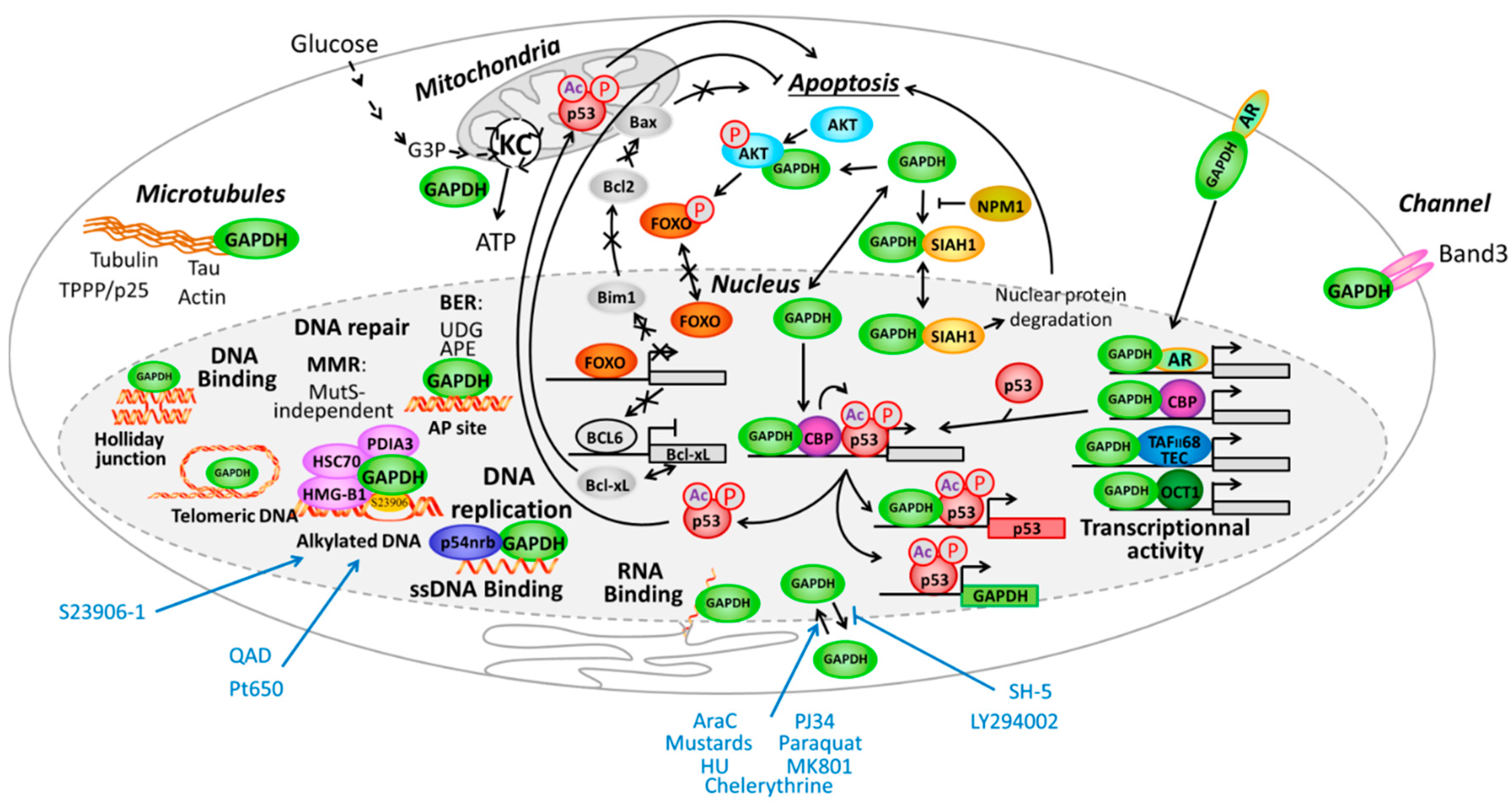
2.2. GAPDH
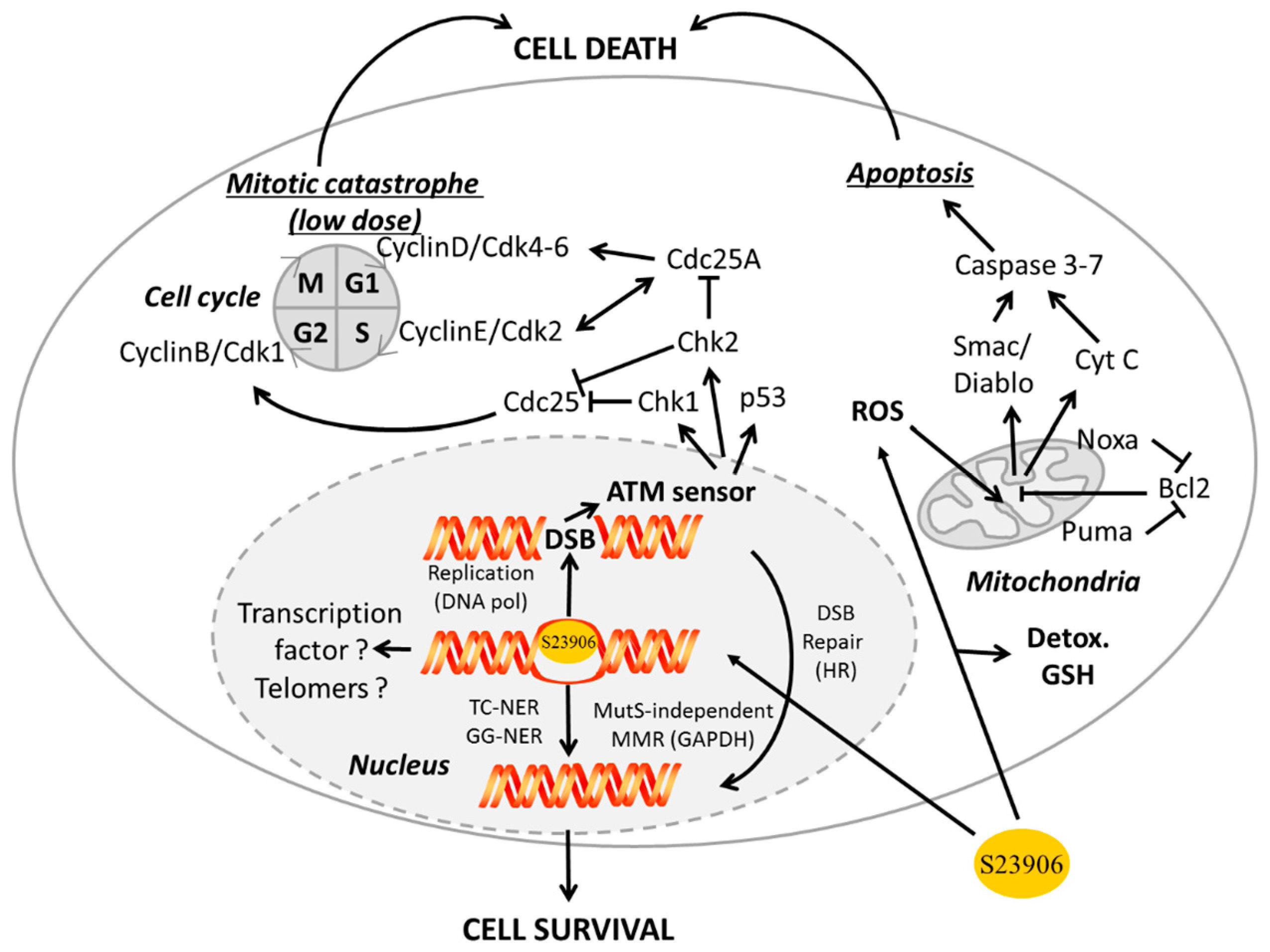
- In the cytoplasm: GAPDH was implicated in intracellular membrane trafficking, endoplasmic reticulum to Golgi transport and maintenance/regulation of protein polymerization. It interacts as a tetramer with tubulin to facilitate polymerization [119,120] and forms a complex with microtubule-associated protein 1B [121] or Rab2 [122]. Similarly GAPDH interacts with TPPP/p25 protein in Lewy bodies [123] and is found in neurofibrillary Tau proteins in brains from Alzheimer patients [124] and finally interacts with actin [125].
- In mitochondria: GAPDH is a glycolytic enzyme directly implicated in the 6th step of glycolysis to catalyze, in an NAD+-dependent manner, the conversion of glyceraldehyde-3-phosphate to d-glycerate-1,3-bisphosphate I to then be further converted in several steps to pyruvate that finally entered the Krebs cycle to produce energy.
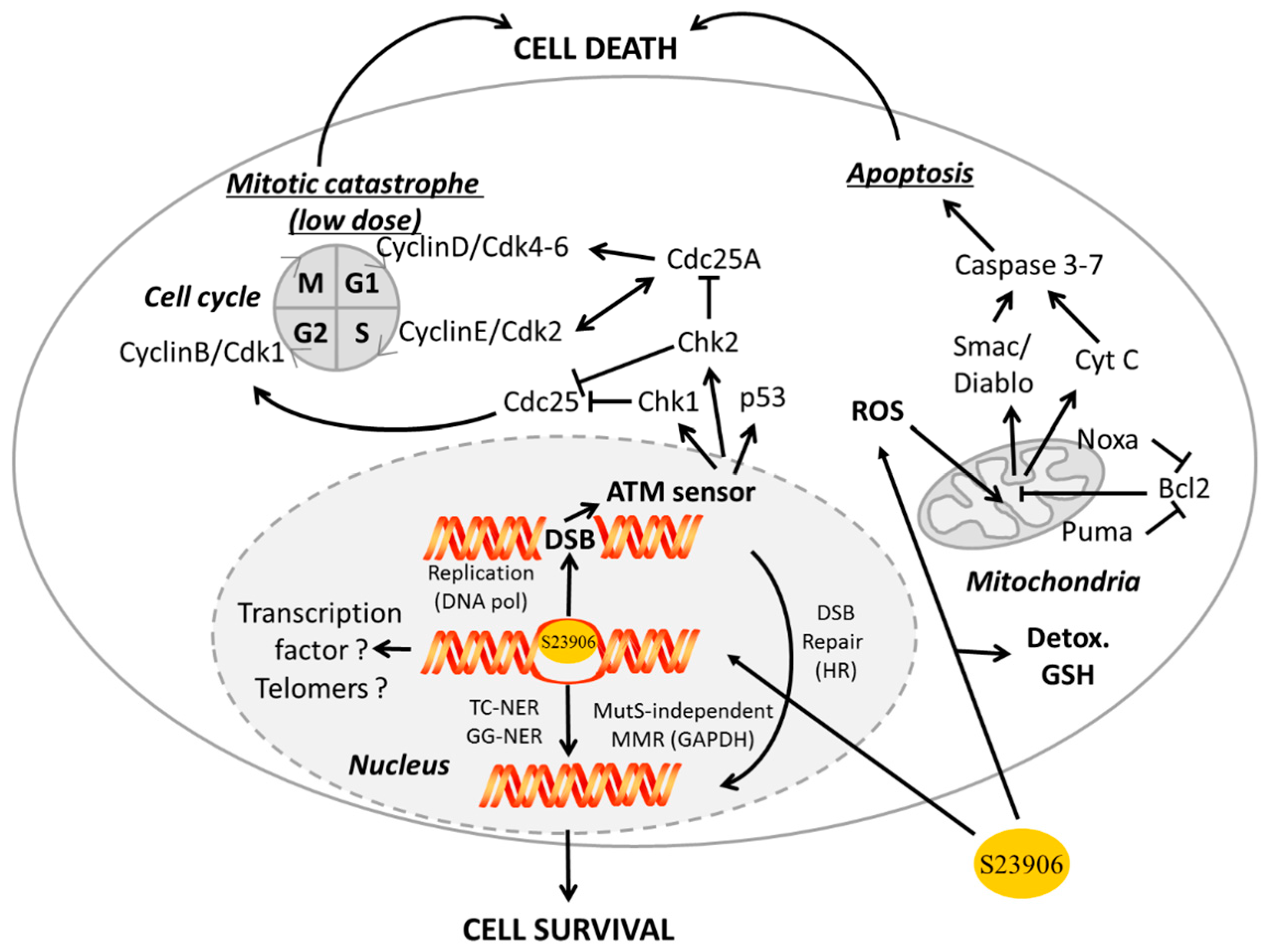
- Associated with apoptosis through different ways:
- GAPDH interacts with phospho-AKT (P-AKT) to block its dephosphorylation, thus preventing Forkhead box class O protein (FOXO) nuclear translocation and further expression of the transcription inhibitor BCL6 that is usually responsible for inhibiting Bcl-xL expression. In that way the expression of anti-apoptotic protein Bcl-xL is reduce and apoptosis is enhanced upon control by GAPDH [126]. Since FOXO also activates Bim1, which controls apoptosis through Bcl2 and Bax, another way for GAPDH to potentially inhibit apoptosis is additionally presented in Figure 4.
- In parallel, interaction of GAPDH with P-AKT inhibits GAPDH nuclear translocation and subsequent acetylation/phosphorylation of p53 that further translocates in the mitochondria to initiate apoptosis [127].
- GAPDH forms a complex with the E3 ubiquitin ligase SIAH1. This GAPDH/SIAH1 complex translocated in the nucleus where it increases nuclear protein degradation associated with SIAH1 activity to further induce apoptosis [128,129]. This cascade could be either activated by paraquat [130], or inhibited upon S-nitrosylation of GAPDH by nucleophosmin (NPM1) [131]. Interestingly, both GAPDH and SIAH1 expression are controlled by p53 [132,133].
- Receptor mediated cell signaling, such as for the androgen-receptor that forms a complex with GAPDH to be then translocated to the nucleus [134]. This is also the case for interaction of GAPDH with the macrophage transferrin receptor, which forms a complex that is then translocated to the endosome compartment [135].
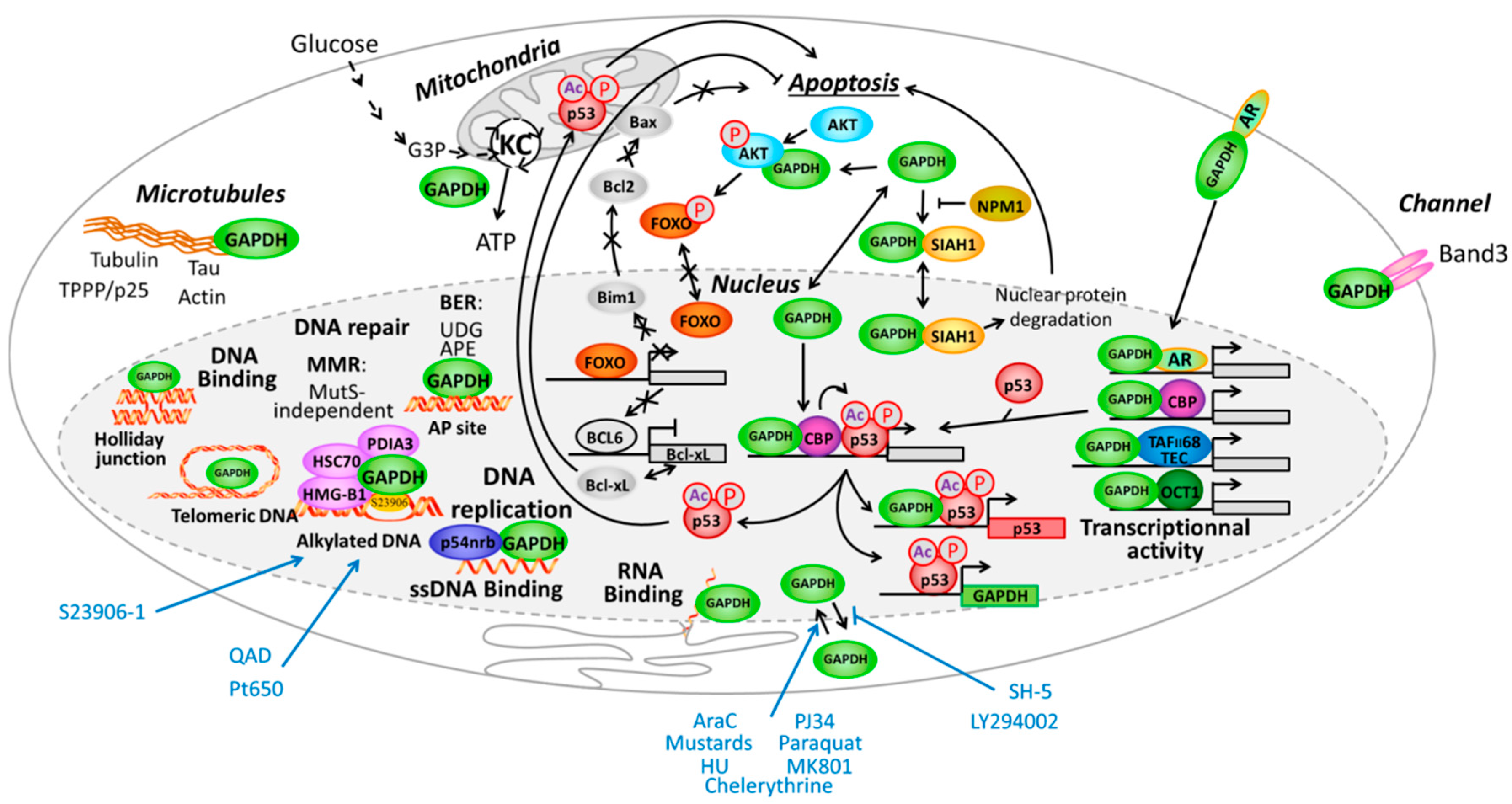
2.2.1. GAPDH at the Interface of Nucleus and Cytoplasm Compartments
2.2.2. GAPDH Modulates Transcription Factor Activity
2.2.3. GAPDH and DNA Binding
2.2.4. GAPDH and DNA Replication
2.2.5. GAPDH Implication in DNA Repair
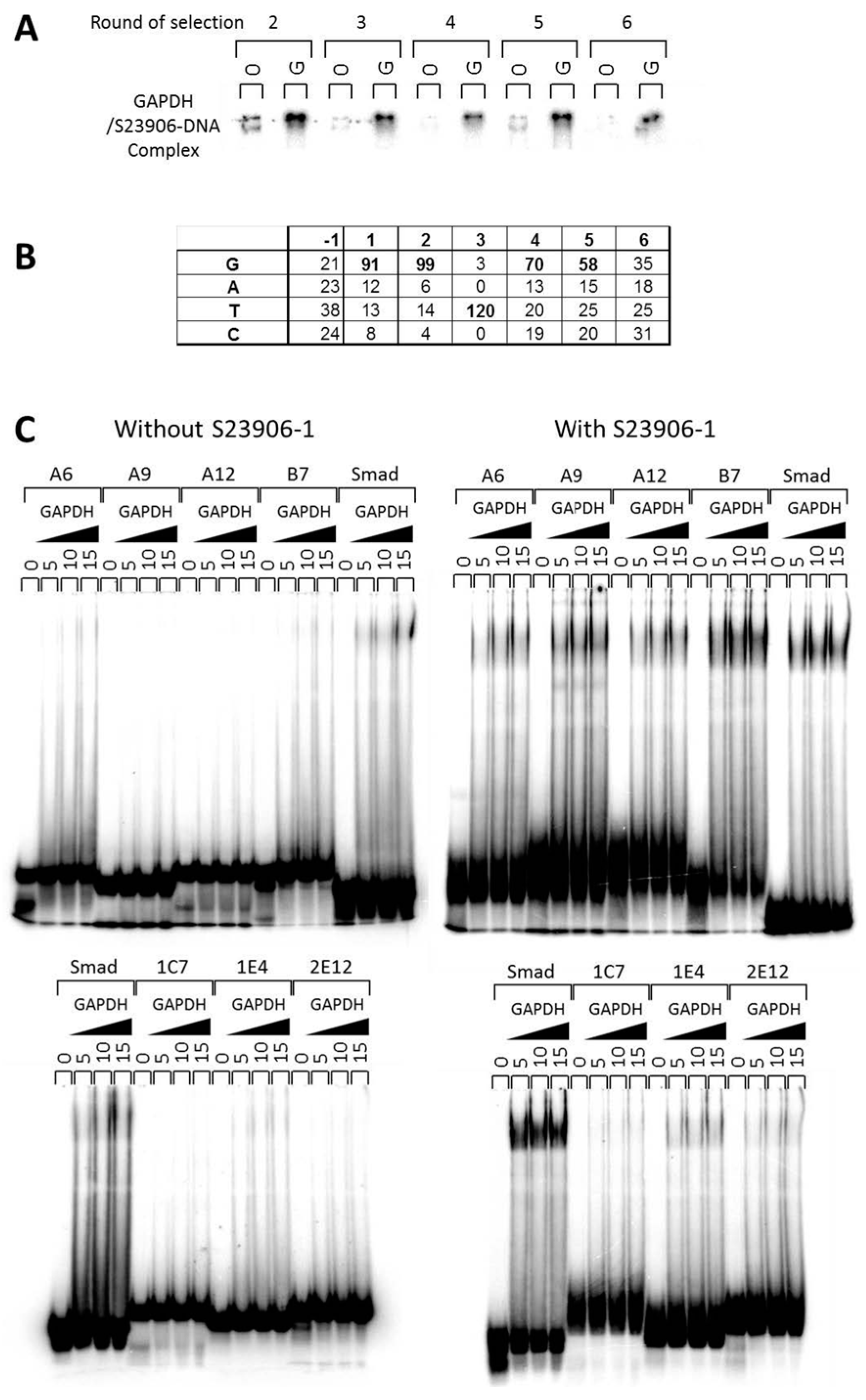
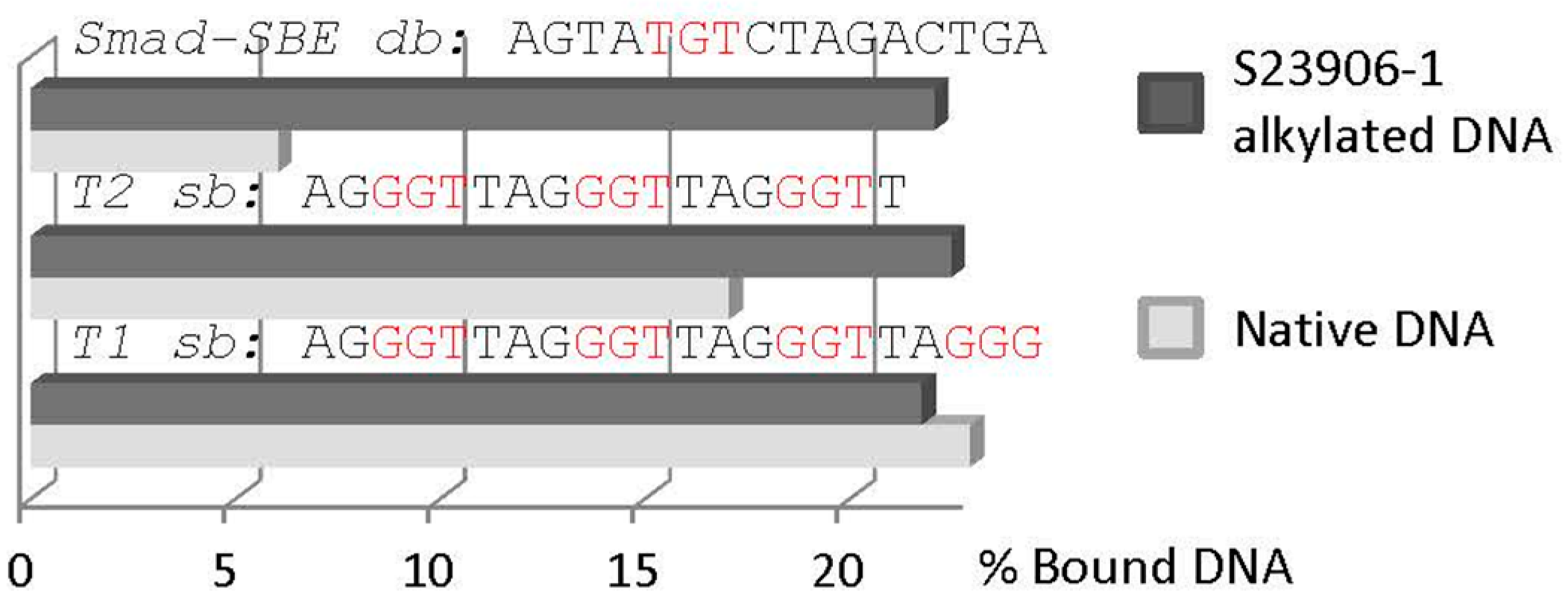
2.3 GAPDH Binds to S23906-1/DNA Adducts and Affects Its Cytotoxic Activity
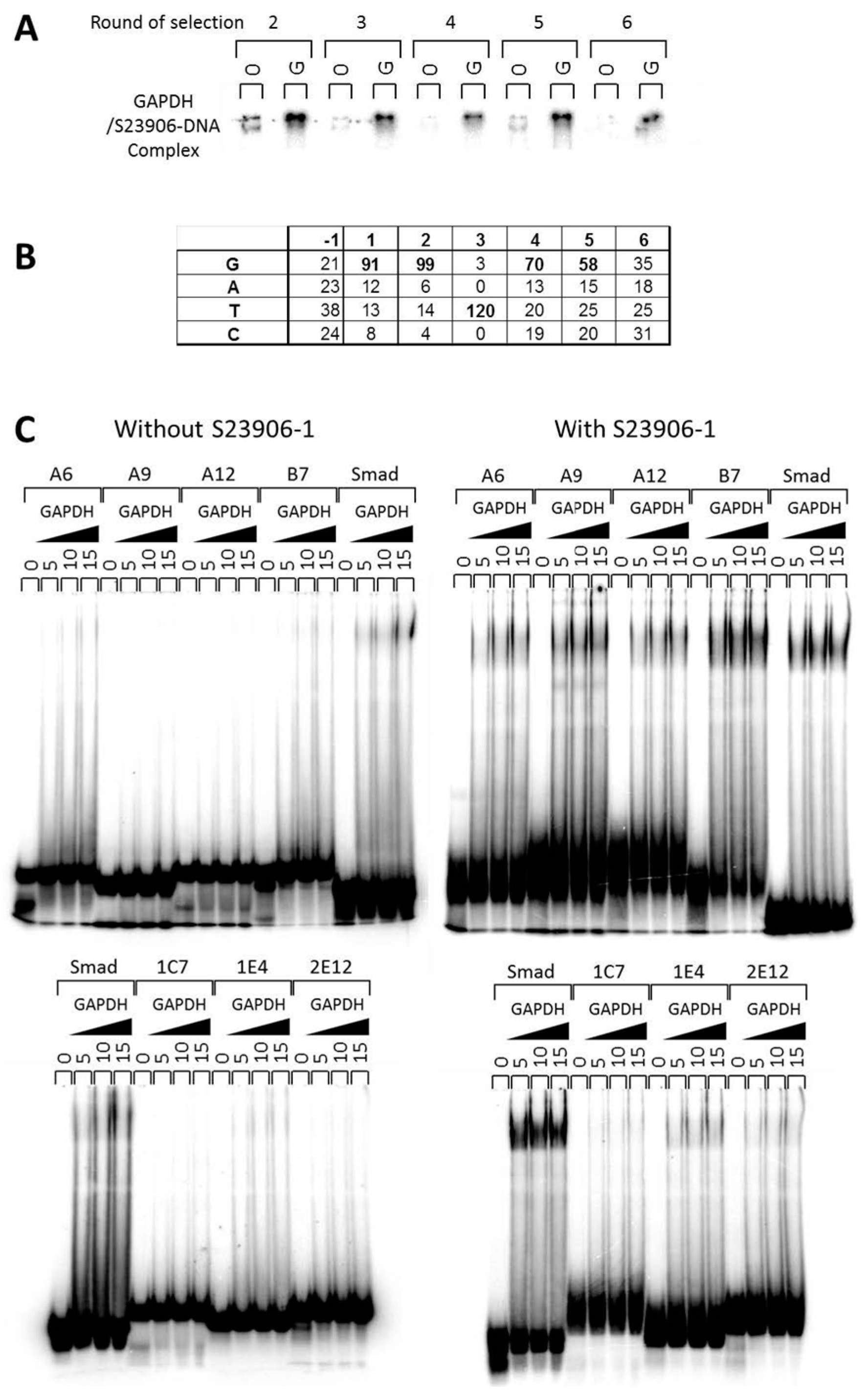
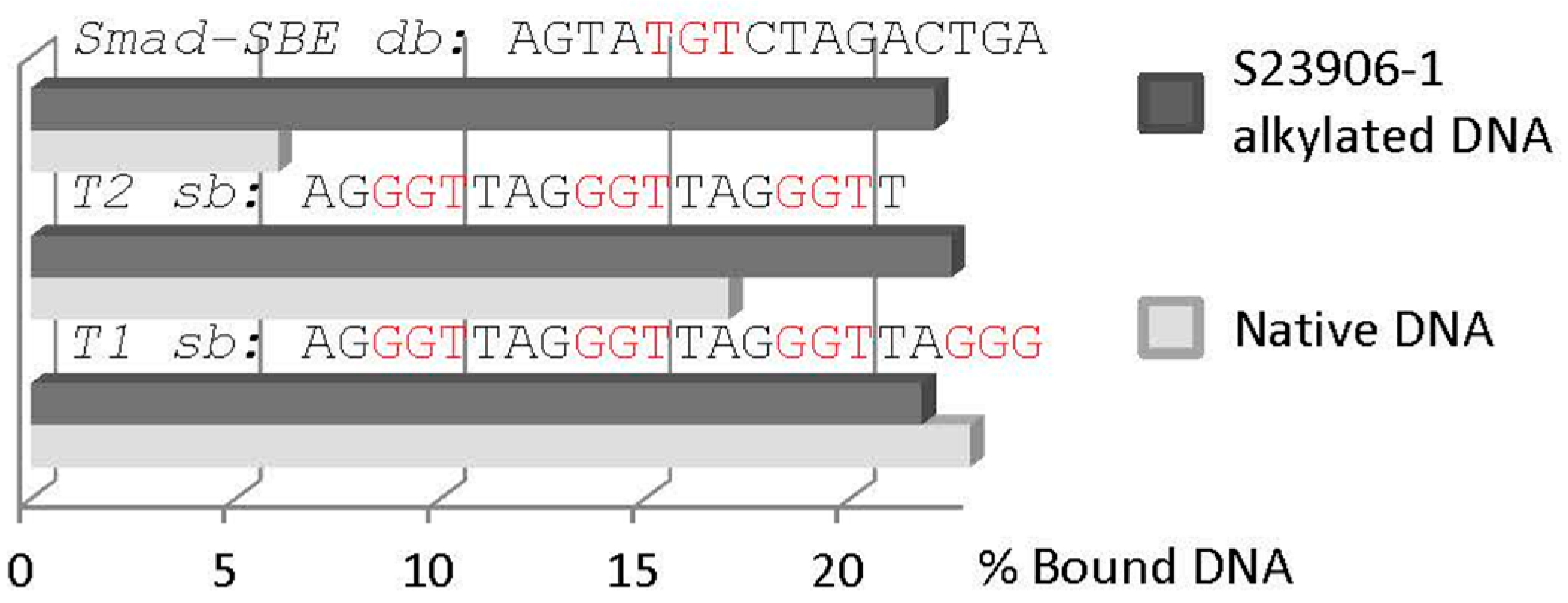


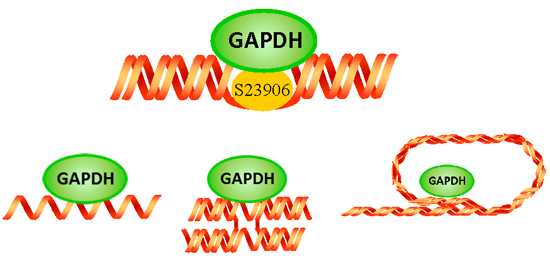{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TranSignal Membrane | Transcription Factor | GAPDH Binding | Sequence for TranSignal Oligonucleotides Containing Transcription Factor Consensus Sites |
|---|---|---|---|
| I | ARE | + | GTCTGGTACAGGGTGTTCTTTTT |
| CDP | + | TCAGAAATTGGCTAATAATCATTGGG | |
| E2F1 | ++ | ATTTAAGTTTCGCGCCCTTTCTCAA | |
| EGR(1) | + | GGATCCAGCGGGGGCGAGCGGGGGCCA | |
| GAS/ISRE | + | CGAAGTACTTTCAGTTTCATATTACTCTACAA | |
| MEF-1 | ++ | TCAGGCAGCAGGTGTTGGGGGGAT | |
| SIE | + | GTGCATTTCCCGTAAATCTTGTCTACA | |
| II | c-Rel | + | GGGGATTTCCGGGGATTTCCGGGGATTTCC |
| E4F/ATF | + | GGCTGACGTCACTGGGCTGACGTCACTG | |
| AFXH (FOXO4) | + | GTTGTTTATGGTTGTTTATGGTTGTTTATG | |
| Freac2-2 | + | TTGTTTTGTTGTTTTGTTGTTTTG | |
| HFH3 | + | GGGTGTTTGTTTAGGGTGTTTGTTTA | |
| MEF-2 (2) | ++ | GCTATTTTTAACGAGGGCTATTTTTAACGAGG | |
| PARP | ++ | ATGGGAGGGGCAATGGGAGGGGCA | |
| PAX3 | + | GATCCTGAGTCTAATTGGATCCTGAGTCTAATTG | |
| REBB2 | ++ | TGGAAATGGCGGGGGATGGTGGGGGACCGGATC | |
| RSRFC4 | + | GGTCTATTTATAGCTTGGTCTATTTATAGCTT | |
| ZIC | + | CATAGTTTCTAAAAGAGGAGGAGGTAGTTCTAG | |
| III | ADD-1 | + | TCCTAGTGTGAGCGGCCCT |
| HMG | + | CGATCTGGAACTCCGGGAATTTCCCTGGCCC | |
| HOXD-8/9/10 | + | GCGGCAGTTTTATTGTTTTATTCGC | |
| MAZ | + | GGGTTGGGGAAGTATTAGGAGGGGAGGGTT | |
| NF-4FA | + | CTCCTTTCTTTGAAGCTCCTTTCTTTGAAG | |
| TCE | ++ | GCAGAGGGCGTGGGGGAAAAGAA | |
| IV | AP-4 | + | TCAGCGCGGGTCAGCGCGGGATTC |
| ApoA1Prom | ++ | CCCTGCAAGAGCTGGCTGCTTAGAGACTGCGAGAAGGAG | |
| c-Myb (2) | + | GGACCAGGGGGTCTAGGAG | |
| CYP1A1 | ++ | GTAAGGGGGCAGAGGTCGGG | |
| EGR1 (2) | ++ | CCTCCCCCCGCCTTGCCCGGGGTTGTGG | |
| kBF-alpha | + | GGCGTTTTCGTTTTTACCCGGC | |
| LXRE-1 | + | GCTGAGGTTACTGCTGGTCATTCAAGCT | |
| MASH-1 | + | GGCTCAGGCAGCAGGTGTTGGG | |
| MBP-1 (1) | + | GTGGGAAATTCCGTGGGAAATTCC | |
| V | MDBP (1) | ++ | CTATTGGCGTTACTATGGGAACATA |
| MDBP (2) | + | GGCCATTACCTGGTGATATTACCTGGTGATGC | |
| Myb (2) | + | GCCCAGTTGTTAGCCCAGTTGTTA | |
| NCAM-BP | ++ | GCTCTGCATTTTCTTTTGGCC | |
| NF-Atp | + | TTGCATTTTCCATGGTTGCATTTTCCATGG | |
| NF-E6/CP1 | + | ACTGAGTCATGAGTCATGGTTGGCCC | |
| ODC | + | TGCGTCTCCATGACGGTCTCCATGACGAC | |
| PAX-1 | + | CACCGTTCCGCTCTAGATATCTC | |
| PBGD BP | + | TCAGTGTCCTGGAGTGTCCTGGTTACT | |
| Pit-1 (1) | + | CTAAATTATCCATTTATCCATTAGCAC | |
| PU.1 | + | AGAAAAGGAGAAGTAGGAGGC | |
| Snail | + | TGTGAACAGGTGCTTGTGAACAGGTGCT | |
| Thy-1 BP | + | GATCAGGGGTGGCAGGGGTGGAAT |
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Christakis, P. The birth of chemotherapy at Yale. Bicentennial lecture series: Surgery Grand Round. Yale J. Biol. Med. 2011, 84, 169–172. [Google Scholar] [PubMed]
- Jagetia, G.C.; Rao, S.K.; Baliga, M.S.; Babu, S.K. The evaluation of nitric oxide scavenging activity of certain herbal formulations in vitro: A preliminary study. Phytother. Res. 2004, 18, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Mishra, P.C.; Suhai, S. Reactions of DNA bases with the anti-cancer nitrogen mustard mechlorethamine: A quantum chemical study. Chem. Phys. Lett. 2007, 449, 323–328. [Google Scholar] [CrossRef]
- Millard, J.T.; Weidner, M.F.; Kirchner, J.J.; Ribeiro, S.; Hopkins, P.B. Sequence preferences of DNA interstrand crosslinking agents: Quantitation of interstrand crosslink locations in DNA duplex fragments containing multiple crosslinkable sites. Nucleic Acids Res. 1991, 9, 1885–1891. [Google Scholar] [CrossRef]
- Rink, S.M.; Hopkins, P.B. A mechlorethamine-induced DNA interstrand cross-link bends duplex DNA. Biochemistry 1995, 34, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Bellon, S.F.; Coleman, J.H.; Lippard, S.J. DNA unwinding produced by site-specific intrastrand cross-links of the antitumor drug cis-diamminedichloroplatinum(II). Biochemistry 1991, 30, 8026–8035. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Reedijk, J.; Leng, M. Sequence-dependent distortions induced in DNA by monofunctional platinum(II) binding. Biochemistry 1992, 31, 12397–12402. [Google Scholar] [CrossRef] [PubMed]
- Pilch, D.S.; Dunham, S.U.; Jamieson, E.R.; Lippard, S.J.; Breslauer, K.J. DNA sequence context modulates the impact of a cisplatin 1,2-d(GpG) intrastrand cross-link on the conformational and thermodynamic properties of duplex DNA. J. Mol. Biol. 2000, 296, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Malina, J.; Nováková, O.; Vojtiskova, M.; Natile, G.; Brabec, V. Conformation of DNA GG intrastrand cross-link of antitumor oxaliplatin and its enantiomeric analog. Biophys. J. 2007, 93, 3950–3962. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Marini, V.; Bursova, V.; Brabec, V. Biophysical studies on the stability of DNA intrastrand cross-links of transplatin. Biophys. J. 2008, 95, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Teletchéa, S.; Komeda, S.; Teuben, J.M.; Elizondo-Riojas, M.A.; Reedijk, J.; Kozelka, J. A pyrazolato-bridged dinuclear platinum(II) complex induces only minor distortions upon DNA-binding. Chemistry 2006, 12, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Nováková, O.; Chen, H.; Vrana, O.; Rodger, A.; Sadler, P.J.; Brabec, V. DNA interactions of monofunctional organometallic ruthenium(II) antitumor complexes in cell-free media. Biochemistry 2003, 42, 11544–11554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nováková, O.; Nazarov, A.A.; Hartinger, C.G.; Keppler, B.K.; Brabec, V. DNA interactions of dinuclear RuII arene antitumor complexes in cell-free media. Biochem. Pharmacol. 2009, 77, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Nováková, O.; Malina, J.; Suchankova, T.; Kasparkova, J.; Bugarcic, T.; Sadler, P.J.; Brabec, V. Energetics, conformation, and recognition of DNA duplexes modified by monodentate Ru(II) complexes containing terphenyl arenes. Chemistry 2010, 16, 5744–5754. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.E.; Khoo, A.; Fagbemi, A.F.; Schärer, O.D. The efficiencies of damage recognition and excision correlate with duplex destabilization induced by acetylaminofluorene adducts in human nucleotide excision repair. Chem. Res. Toxicol. 2012, 25, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.C.; Kuper, J.; Gasteiger, K.L.; Simon, N.; Strasser, R.; Eisen, D.; Geiger, S.; Schneider, S.; Kisker, C.; Carell, T. Structural insights into the recognition of cisplatin and AAF-dG lesion by Rad14 (XPA). Proc. Natl. Acad. Sci. USA 2015, 112, 8272–8277. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Hearst, J.E. Thermostability of double-stranded deoxyribonucleic acids: Effects of covalent additions of a psoralen. Biochemistry 1986, 25, 5895–5902. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, G.; David-Cordonnier, M.-H. DNA-destabilizing agents as an alternative approach for targeting DNA: Mechanisms of action and cellular consequences. J. Nucleic Acids 2010, 2010, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, G.; Depauw, S.; Mendy-Belaiche, D.; David-Cordonnier, M.-H. DNA Helix Destabilization by Alkylating Agents: From Covalent Bonding to DNA Repair; InTech Open Access Publisher: Rijeka, Croatia, 2011; pp. 97–124. [Google Scholar]
- Lorusso, D.; Mainenti, S.; Pietragalla, A.; Ferrandina, G.; Foco, G.; Masciullo, V.; Scambia, G. Brostallicin (PNU-166196), a new minor groove DNA binder: Preclinical and clinical activity. Expert Opin. Investig. Drugs 2009, 18, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Geroni, C.; Marchini, S.; Cozzi, P.; Galliera, E.; Ragg, E.; Colombo, T.; Battaglia, R.; Howard, M.; D’Incalci, M.; Broggini, M. Brostallicin, a novel anticancer agent whose activity is enhanced upon binding to glutathione. Cancer Res. 2002, 62, 2332–2336. [Google Scholar] [PubMed]
- Gregson, S.J.; Howard, P.W.; Hartley, J.A.; Brooks, N.A.; Adams, L.J.; Jenkins, T.C.; Kelland, L.R.; Thurston, D.E. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J. Med. Chem. 2001, 44, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Hartley, J.A.; Spanswick, V.J.; Brooks, N.; Clingen, P.H.; McHugh, P.J.; Hochhauser, D.; Pedley, R.B.; Kelland, L.R.; Alley, M.C.; Schultz, R.; et al. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: Part 1: Cellular pharmacology, in vitro and initial in vivo antitumor activity. Cancer Res. 2004, 64, 6693–6699. [Google Scholar] [CrossRef] [PubMed]
- Swenson, D.H.; Li, L.H.; Hurley, L.H.; Rokem, J.S.; Petzold, G.L.; Dayton, B.D.; Wallace, T.L.; Lin, A.H.; Krueger, W.C. Mechanism of interaction of CC-1065 (NSC 298223) with DNA. Cancer Res. 1982, 42, 2821–2828. [Google Scholar] [PubMed]
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.J.; Mazumder, A.; Kohn, K.W. DNA sequence- and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.-H.; Laine, W.; Lansiaux, A.; Kouach, M.; Briand, G.; Pierré, A.; Hickman, J.A.; Bailly, C. Alkylation of guanine in DNA by S23906-1, a novel potent antitumor compound derived from the plant alkaloid acronycine. Biochemistry 2002, 41, 9911–9920. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.-H.; Laine, W.; Kouach, M.; Briand, G.; Vezin, H.; Gaslonde, T.; Michel, S.; Doan Thi Mai, H.; Tillequin, F.; Koch, M.; et al. A transesterification reaction is implicated in the covalent binding of benzo[b]acronycine anticancer agents with DNA and glutathion. Bioorg. Med. Chem. 2004, 12, 23–29. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.-H.; Laine, W.; Gaslonde, T.; Michel, S.; Tillequin, F.; Koch, M.; Léonce, S.; Pierré, A.; Bailly, C. Design of novel antitumor DNA alkylating agents: The benzoacronycine series. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 83–92. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.-H.; Laine, W.; Lansiaux, A.; Rosu, F.; Colson, P.; de Pauw, E.; Michel, S.; Tillequin, F.; Koch, M.; Hickman, J.A.; et al. Covalent binding of antitumor benzoacronycines to double-stranded DNA induces helix opening and the formation of single-stranded DNA: Unique consequences of a novel DNA-bonding mechanism. Mol. Cancer Ther. 2005, 4, 71–80. [Google Scholar] [PubMed]
- Zou, Y.; van Houten, B. Strand opening by the UvrA2B complex allows dynamic recognition of DNA damage. EMBO J. 1999, 18, 4889–4901. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Luo, C.; Geacintov, N.E. Hierarchy of DNA damage recognition in Escherichia coli nucleotide excision repair. Biochemistry 2001, 40, 2923–2931. [Google Scholar] [CrossRef] [PubMed]
- Kolbanovskiy, A.; Kuzmin, V.; Shastry, A.; Kolbanovskaya, M.; Chen, D.; Chang, M.; Bolton, J.L.; Geacintov, N.E. Base selectivity and effects of sequence and DNA secondary structure on the formation of covalent adducts derived from the equine estrogen metabolite 4-hydroxyequilenin. Chem. Res. Toxicol. 2005, 18, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Shapiro, R.; Geacintov, N.E.; Broyde, S. Equilenin-derived DNA adducts to cytosine in DNA duplexes: Structures and thermodynamics. Biochemistry 2005, 44, 14565–14576. [Google Scholar] [CrossRef] [PubMed]
- Privalov, P.L.; Dragan, A.I.; Crane-Robinson, C. The cost of DNA bending. Trends Biochem. Sci. 2009, 34, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Vojtiskova, M.; Natile, G.; Brabec, V. Unique properties of DNA interstrand cross-links of antitumor oxaliplatin and the effect of chirality of the carrier ligand. Chemistry 2008, 14, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.C.; Zamble, D.B.; Reardon, J.T.; Lippard, S.J.; Sancar, A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc. Natl. Acad. Sci. USA 1994, 91, 10394–10398. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ramanjaneyulu, A.; Ray, R.; Rajeswari, M.R. Involvement of high mobility group B proteins in cisplatin-induced cytotoxicity in squamous cell carcinoma of skin. DNA Cell Biol. 2009, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Lanuszewska, J.; Widlak, P. High mobility group 1 and 2 proteins bind preferentially to DNA that contains bulky adducts induced by benzo[a]pyrene diol epoxide and N-acetoxy-acetylaminofluorene. Cancer Lett. 2000, 158, 17–25. [Google Scholar] [CrossRef]
- Trimmer, E.E.; Zamble, D.B.; Lippard, S.J.; Essigmann, J.M. Human testis-determining factor SRY binds to the major DNA adduct of cisplatin and a putative target sequence with comparable affinities. Biochemistry 1998, 37, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Chvalova, K.; Sari, M.A.; Bombard, S.; Kozelka, J. LEF-1 recognition of platinated GG sequences within double-stranded DNA. Influence of flanking bases. J. Inorg. Biochem. 2008, 102, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Buschta-Hedayat, N.; Buterin, T.; Hess, M.T.; Missura, M.; Naegeli, H. Recognition of nonhybridizing base pairs during nucleotide excision repair of DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 6090–6095. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Z.; Utzat, C.D.; Liu, Y.; Geacintov, N.E.; Basu, A.K.; Zou, Y. Interactions of human replication protein A with single-stranded DNA adducts. Biochem. J. 2005, 385, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Patrick, S.M.; Turchi, J.J. Replication protein A (RPA) binding to duplex cisplatin-damaged DNA is mediated through the generation of single-stranded DNA. J. Biol. Chem. 1999, 274, 14972–14978. [Google Scholar] [CrossRef] [PubMed]
- Patrick, S.M.; Tillison, K.; Horn, J.M. Recognition of cisplatin-DNA interstrand cross-links by replication protein A. Biochemistry 2008, 47, 10188–10196. [Google Scholar] [CrossRef] [PubMed]
- Neher, T.M.; Bodenmiller, D.; Fitch, R.W.; Jalal, S.I.; Turchi, J.J. Novel irreversible small molecule inhibitors of replication protein A display single-agent activity and synergize with cisplatin. Mol. Cancer Ther. 2011, 10, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Dormi, S.S.; Turchi, A.M.; Woods, D.S.; Turchi, J.J. Chemical inhibitor targeting the replication protein A-DNA interaction increases the efficacy of Pt-based chemotherapy in lung and ovarian cancer. Biochem. Pharmacol. 2015, 93, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Roginskaya, M.; Colis, L.C.; Basu, A.K.; Shell, S.M.; Liu, Y.; Musich, P.R.; Harris, C.M.; Harris, T.M.; Zou, Y. Specific and efficient binding of Xeroderma Pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3′- and/or 5′-ssDNA branches. Biochemistry 2006, 45, 15921–15930. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Luo, N.; Jedlovszky, P.; Simon, I.; Osman, R. Role of base flipping in specific recognition of damaged DNA by repair enzymes. J. Mol. Biol. 2002, 323, 823–834. [Google Scholar] [CrossRef]
- Yang, W. Structure and mechanism for DNA lesion recognition. Cell Res. 2008, 18, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.G.; Garcia, K.; He, C. Damage detection and base flipping in direct DNA alkylation repair. ChemBioChem 2009, 10, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Duckett, D.R.; Drummond, J.T.; Murchie, A.I.; Reardon, J.T.; Sancar, A.; Lilley, D.M.; Modrich, P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc. Natl. Acad. Sci. USA 1996, 93, 6443–6447. [Google Scholar] [CrossRef] [PubMed]
- Aebi, S.; Kurdi-Haidar, B.; Gordon, R.; Cenni, B.; Zheng, H.; Fink, D.; Christen, R.D.; Boland, C.R.; Koi, M.; Fishel, R.; et al. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 1996, 56, 3087–3090. [Google Scholar] [PubMed]
- Mello, J.A.; Acharya, S.; Fishel, R.; Essigmann, J.M. The mismatch-repair protein hMSH2 binds selectively to DNA adducts of the anticancer drug cisplatin. Chem. Biol. 1996, 3, 579–589. [Google Scholar] [CrossRef]
- Fourrier, L.; Brooks, P.; Malinge, J.M. Binding discrimination of MutS to a set of lesions and compound lesions (base damage and mismatch) reveals its potential role as a cisplatin-damaged DNA sensing protein. J. Biol. Chem. 2003, 278, 21267–21275. [Google Scholar] [CrossRef] [PubMed]
- Vooradi, V.; Romano, L.J. Effect of N-2-acetylaminofluorene and 2-aminofluorene adducts on DNA binding and synthesis by yeast DNA polymerase eta. Biochemistry 2009, 48, 4209–4216. [Google Scholar] [CrossRef] [PubMed]
- Nováková, O.; Kasparkova, J.; Bursova, V.; Hofr, C.; Vojtiskova, M.; Chen, H.; Sadler, P.J.; Brabec, V. Conformation of DNA modified by monofunctional Ru(II) arene complexes: Recognition by DNA binding proteins and repair. Relationship to cytotoxicity. Chem. Biol. 2005, 12, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Kropachev, K.; Ding, S.; Van Houten, B.; Geacintov, N.E.; Broyde, S. Exploring damage recognition models in prokaryotic nucleotide excision repair with a benzo[a]pyrene-derived lesion in UvrB. Biochemistry 2009, 48, 8948–8957. [Google Scholar] [CrossRef] [PubMed]
- Janićijević, A.; Sugasawa, K.; Shimizu, Y.; Hanaoka, F.; Wijgers, N.; Djurica, M.; Hoeijmakers, J.H.; Wyman, C. DNA bending by the human damage recognition complex XPC-HR23B. DNA Repair 2003, 2, 325–236. [Google Scholar] [CrossRef]
- Clement, F.C.; Camenisch, U.; Fei, J.; Kaczmarek, N.; Mathieu, N.; Naegeli, H. Dynamic two-stage mechanism of versatile DNA damage recognition by Xeroderma Pigmentosum group C protein. Mutat. Res. 2010, 685, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.L.; Roginskaya, M.; Zou, Y.; Altamirano, A.; Basu, A.K.; Stone, M.P. Binding of the human nucleotide excision repair proteins XPA and XPC/HR23B to the 5R-thymine glycol lesion and structure of the cis-(5R,6S) thymine glycol epimer in the 5′-GTgG-3′ sequence: Destabilization of two base pairs at the lesion site. Nucleic Acids Res. 2010, 38, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, F.A.; Cai, Y.; Lin, C.; Tang, Y.; Kolbanovskiy, A.; Amin, S.; Patel, D.J.; Broyde, S.; Geacintov, N.E. Exocyclic amino groups of flanking guanines govern sequence-dependent adduct conformations and local structural distortions for minor groove-aligned benzo[a]pyrenyl-guanine lesions in a GG mutation hotspot context. Nucleic Acids Res. 2007, 35, 1555–1568. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.H.; Zewail-Foote, M. The antitumor agent ecteinascidin 743: Characterization of its covalent DNA adducts and chemical stability. Adv. Exp. Med. Biol. 2001, 500, 289–299. [Google Scholar] [PubMed]
- García-Nieto, R.; Manzanares, I.; Cuevas, C.; Gago, F. Increased DNA binding specificity for antitumor ecteinascidin 743 through protein-DNA interactions. J. Med. Chem. 2000, 43, 4367–4369. [Google Scholar] [CrossRef] [PubMed]
- Aune, G.J.; Furuta, T.; Pommier, Y. Ecteinascidin 743: A novel anticancer drug with a unique mechanism of action. Anticancer Drugs 2002, 13, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.I.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Martín-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef] [PubMed]
- Bonfanti, M.; La Valle, E.; Fernandez Sousa Faro, J.M.; Faircloth, G.; Caretti, G.; Mantovani, R.; D’Incalci, M. Effect of ecteinascidin-743 on the interaction between DNA binding proteins and DNA. Anticancer Drug Des. 1999, 14, 179–186. [Google Scholar] [PubMed]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- Minuzzo, M.; Marchini, S.; Broggini, M.; Faircloth, G.; D’Incalci, M.; Mantovani, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc. Natl. Acad. Sci. USA 2000, 97, 6780–6784. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; Hu, Z.; Kolb, E.A.; Gorfajn, B.; Scotto, K.W. Ecteinascidin-743 inhibits activated but not constitutive transcription. Cancer Res. 2002, 62, 3377–3381. [Google Scholar] [PubMed]
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von Hippel-Lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of RNA polymerase II in response to trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455. [Google Scholar] [CrossRef] [PubMed]
- Massuti, B.; Cobo, M.; Camps, C.; Dómine, M.; Provencio, M.; Alberola, V.; Viñolas, N.; Rosell, R.; Tarón, M.; Gutiérrez-Calderón, V.; Lardelli, P.; Alfaro, V.; Nieto, A.; Isla, D. Trabectedin in patients with advanced non-small-cell lung cancer (NSCLC) with XPG and/or ERCC1 overexpression and BRCA1 underexpression and pretreated with platinum. Lung Cancer 2012, 76, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Ghatage, P.; Parekh, T.; Henitz, E.; Knoblauch, R.; Matos-Pita, A.S.; Nieto, A.; Park, Y.C.; Cheng, P.S.; Li, W.; et al. Effect of BRCA1 and XPG mutations on treatment response to trabectedin and pegylated liposomal doxorubicin in patients with advanced ovarian cancer: Exploratory analysis of the phase 3 OVA-301 study. Ann. Oncol. 2015, 26, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Finch, J.S.; Rosenberger, S.F.; Martinez, J.D.; Bowden, G.T. Okadaic acid induces transcription of junB through a CCAAT box and NF-Y. Gene 2001, 267, 135–144. [Google Scholar] [CrossRef]
- D’Angelo, D.; Borbone, E.; Palmieri, D.; Uboldi, S.; Esposito, F.; Frapolli, R.; Pacelli, R.; D’Incalci, M.; Fusco, A. The impairment of the High Mobility Group A (HMGA) protein function contributes to the anticancer activity of trabectedin. Eur. J. Cancer 2013, 49, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Hughes, G.K.; Lahey, F.N.; Price, J.R. Alkaloids of the Australian Rutaceae. Nature 1948, 162, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, P.L.; Robertson, A.V. The structure of acronycine. Aust. J. Chem. 1966, 19, 275–281. [Google Scholar] [CrossRef]
- Gougoutas, J.Z.; Kaski, B.A. The crystal and molecular structure of bromodihydroacronycine. Acta Cryst. B 1970, 26, 853–859. [Google Scholar] [CrossRef]
- Svoboda, G.H.; Poore, G.A.; Simpson, P.J.; Boder, G.B. Alkaloids of Acronychia baueri. Isolation of the alkaloids and study of the antitumor and other biological properties of acronycine. J. Pharmacol. Sci. 1966, 55, 758–768. [Google Scholar] [CrossRef]
- Scarffe, J.H.; Beaumont, A.R.; Crowther, D. Phase I-II evaluation of acronine in patients with multiple myeloma. Cancer Treat. Rep. 1983, 67, 93–94. [Google Scholar] [PubMed]
- Dorr, R.T.; Liddil, J.D. Development of a parental formulation of the antitumor agent acronycine. J. Drug. Dev. 1988, 1, 31–39. [Google Scholar]
- Brum-Bousquet, M.; Mitaku, S.; Skaltsounis, A.L.; Tillequin, F.; Koch, M. Acronycine epoxide: A new acridone alkaloid from several Sarcomelicope species. Planta Med. 1988, 54, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Tillequin, F. New antitumor agents in the acronycine series. Ann. Pharm. Fr. 2002, 60, 246–252. [Google Scholar] [PubMed]
- Léonce, S.; Perez, V.; Lambel, S.; Peyroulan, D.; Tillequin, F.; Michel, S.; Koch, M.; Pfeiffer, B.; Atassi, G.; Hickman, J.A.; et al. Induction of cyclin E and inhibition of DNA synthesis by the novel acronycine derivative S23906-1 precede the irreversible arrest of tumor cells in S phase leading to apoptosis. Mol. Pharmacol. 2001, 60, 1383–1391. [Google Scholar] [PubMed]
- Costes, N.; Le Deit, H.; Michel, S.; Tillequin, F.; Koch, M.; Pfeiffer, B.; Renard, P.; Léonce, S.; Guilbaud, N.; Kraus-Berthier, L.; et al. Synthesis and cytotoxic and antitumor activity of benzo[b]pyrano[3,2-h]acridin-7-one analogues of acronycine. J. Med. Chem. 2000, 43, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, N.; Kraus-Berthier, L.; Meyer-Losic, F.; Malivet, V.; Chacun, C.; Jan, M.; Tillequin, F.; Koch, M.; Pfeiffer, B.; Atassi, G.; et al. Marked antitumor activity of a new potent acronycine derivative in orthotopic models of human solid tumors. Clin. Cancer Res. 2001, 7, 2573–2580. [Google Scholar] [PubMed]
- Shieh, H.L.; Pezzuto, J.M.; Cordell, G.A. Evaluation of the cytotoxic mechanisms mediated by the broad-spectrum antitumor alkaloid acronycine and selected semisynthetic derivatives. Chem. Biol. Interact. 1992, 81, 35–55. [Google Scholar] [CrossRef]
- Depauw, S.; Gaslonde, T.; Léonce, S.; Kraus-Berthier, L.; Laine, W.; Lenglet, G.; Chiaroni, A.; Pfeiffer, B.; Bailly, C.; Michel, S.; et al. Influence of the stereoisomeric position of the reactive acetate groups of the benzo[b]acronycine derivative S23906-1 on its DNA alkylation, helix-opening, cytotoxic, and antitumor activities. Mol. Pharmacol. 2009, 76, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Lansiaux, A.; Wattez, N.; Hildebrand, M.P.; Léonce, S.; Pierré, A.; Hickman, J.A.; Bailly, C. Induction of apoptosis in HL-60 leukemia and B16 melanoma cells by the acronycine derivative S23906-1. Biochem. Pharmacol. 2003, 63, 1443–1452. [Google Scholar] [CrossRef]
- Leonce, S.; Kraus-Berthier, L.; Golsteyn, R.M.; David-Cordonnier, M.H.; Tardy, C.; Lansiaux, A.; Poindessous, V.; Larsen, A.K.; Pierré, A. Generation of replication-dependent double-strand breaks by the novel N2-G alkylator S23906-1. Cancer Res. 2006, 66, 7203–7210. [Google Scholar] [CrossRef] [PubMed]
- Cahuzac, N.; Studény, A.; Marshall, K.; Versteege, I.; Wetenhall, K.; Pfeiffer, B.; Léonce, S.; Hickman, J.A.; Pierré, A.; Golsteyn, R.M. An unusual DNA binding compound, S23906, induces mitotic catastrophe in cultured human cells. Cancer Lett. 2010, 289, 178–187. [Google Scholar] [CrossRef] [PubMed]
- David-Cordonnier, M.H.; Laine, W.; Joubert, A.; Tardy, C.; Goossens, J.F.; Kouach, M.; Briand, G.; Thi Mai, H.D.; Michel, S.; Tillequin, F.; et al. Covalent binding to glutathione prevents DNA alkylation by the benzoacronycine antitumor agent S23906-1. Eur. J. Biochem. 2003, 270, 2848–2859. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.J.; Poindessous, V.; Soares, D.G.; Ouadrani, K.E.; Sarasin, A.; Guérin, E.; de Gramont, A.; Henriques, J.A.; Escargueil, A.E.; Larsen, A.K. The NER proteins XPC and CSB, but not ERCC1, regulate the sensitivity to the novel DNA binder S23906: Implications for recognition and repair of antitumor alkylators. Biochem. Pharmacol. 2010, 80, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.G.; Battistella, A.; Rocca, C.J.; Matuo, R.; Henriques, J.A.; Larsen, A.K.; Escargueil, A.E. Ataxia telangiectasia mutated- and Rad3-related kinase drives both the early and the late DNA-damage response to the monofunctional antitumour alkylator S23906. Biochem. J. 2011, 437, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.J.; Soares, D.G.; Bouzid, H.; Henriques, J.A.; Larsen, A.K.; Escargueil, A.E. BRCA2 is needed for both repair and cell cycle arrest in mammalian cells exposed to S23906, an anticancer monofunctional DNA binder. Cell Cycle 2015, 14, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Caradec, J.; Sirab, N.; Revaud, D.; Keumeugni, C.; Loric, S. Is GAPDH a relevant housekeeping gene for normalisation in colorectal cancer experiments? Br. J. Cancer 2010, 103, 1475–1476. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Moothart, D.R.; Lowy, D.R.; Qian, X. The expression of glyceraldehyde-3-phosphate dehydrogenase associated cell cycle (GACC) genes correlates with cancer stage and poor survival in patients with solid tumors. PLoS ONE 2013, 8, e61262. [Google Scholar] [CrossRef] [PubMed]
- Soltany-Rezaee-Rad, M.; Mottaghi-Dastjerdi, N.; Setayesh, N.; Roshandel, G.; Ebrahimifard, F.; Sepehrizadeh, Z. Overexpression of FOXO3, MYD88, and GAPDH identified by suppression subtractive hybridization in esophageal cancer is associated with autophagy. Gastroenterol. Res. Pract. 2014, 2014, 185035. [Google Scholar] [CrossRef] [PubMed]
- Sirover, M.A. New insights into an old protein: The functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1999, 1432, 159–184. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Seidler, N.W. Basic biology of GAPDH. Adv. Exp. Med. Biol. 2013, 985, 1–36. [Google Scholar] [PubMed]
- Sirover, M.A. Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem. Cell Biol. 2014, 57, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J. Alzheimers Dis. 2010, 20, 369–393. [Google Scholar] [PubMed]
- El Kadmiri, N.; Slassi, I.; El Moutawakil, B.; Nadifi, S.; Tadevosyan, A.; Hachem, A.; Soukri, A. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease. Pathol. Biol. 2014, 62, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.I.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Green, D.R.; Ricci, J.E. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009, 16, 1573–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.F. Glyceraldehyde-3-phosphate dehydrogenase: A promising target for molecular therapy in hepatocellular carcinoma. Oncotarget 2012, 3, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liu, S.; Sun, M.Z. Novel insight into the role of GAPDH playing in tumor. Clin. Transl. Oncol. 2013, 15, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Dmitriev, A.A.; Snezhkina, A.V.; Kudryavtseva, A.V. Deregulation of glycolysis in cancer: Glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert Opin. Ther. Targets 2013, 17, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [PubMed]
- Jeffery, C. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11. [Google Scholar] [CrossRef]
- Sirover, M.A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta 2011, 1810, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Henderson, B.; Martin, A.C. Protein moonlighting: A new factor in biology and medicine. Biochem. Soc. Trans. 2014, 42, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.R.; Ward, R.D.; Oliver, C. A mutation in glyceraldehyde 3-phosphate dehydrogenase alters endocytosis in CHO cells. J. Cell Biol. 1995, 130, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Kumar, S.; Sheokand, N.; Raje, C.I.; Raje, M. The multifunctional glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a novel macrophage lactoferrin receptor. Biochem Cell Biol. 2012, 90, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Somers, M.; Engelborghs, Y.; Baert, J. Analysis of the binding of glyceraldehyde-3-phosphate dehydrogenase to microtubules, the mechanism of bundle formation and the linkage effect. Eur. J. Biochem. 1990, 193, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Landino, L.M.; Hagedorn, T.D.; Kennett, K.L. Evidence for thiol/disulfide exchange reactions between tubulin and glyceraldehyde-3-phosphate dehydrogenase. Cytoskeleton 2014, 71, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Cueille, N.; Blanc, C.T.; Riederer, I.M.; Riederer, B.M. Microtubule-associated protein 1B binds glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 2007, 6, 2640–2647. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, E.J.; Azizi, F.; Artalejo, C.R. Rab2 utilizes glyceraldehyde-3-phosphate dehydrogenase and protein kinase Cι to associate with microtubules and to recruit dynein. J. Biol. Chem. 2009, 284, 5876–5884. [Google Scholar] [CrossRef] [PubMed]
- Oláh, J.; Tokési, N.; Vincze, O.; Horváth, I.; Lehotzky, A.; Erdei, A.; Szájli, E.; Medzihradszky, K.F.; Orosz, F.; Kovács, G.G.; Ovádi, J. Interaction of TPPP/p25 protein with glyceraldehyde-3-phosphate dehydrogenase and their co-localization in Lewy bodies. FEBS Lett. 2006, 580, 5807–5814. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Woltjer, R.L.; Cimino, P.J.; Pan, C.; Montine, K.S.; Zhang, J.; Montine, T.J. Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J. 2005, 19, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.D.; Bereiter-Hahn, J. Glyceraldehyde-3-phosphate dehydrogenase associates with actin filaments in serum deprived NIH 3T3 cells only. Cell Biol. Int. 2002, 26, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Jacquin, M.A.; Chiche, J.; Zunino, B.; Beneteau, M.; Meynet, O.; Pradelli, L.A.; Meynet, O.; Marchetti, S.; Cornille, A.; Carles, M.; et al. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013, 20, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Thangima Zannat, M.; Bhattacharjee, R.B.; Bag, J. In the absence of cellular poly (A) binding protein, the glycolytic enzyme GAPDH translocated to the cell nucleus and activated the GAPDH mediated apoptotic pathway by enhancing acetylation and serine 46 phosphorylation of p53. Biochem. Biophys. Res. Commun. 2011, 409, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Snyder, S.H. Nitric oxide-GAPDH-Siah: A novel cell death cascade. Cell. Mol. Neurobiol. 2006, 26, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Ortiz, M.A.; Moran, J.M.; Ruiz-Mesa, L.M.; Bravo-San Pedro, J.M.; Fuentes, J.M. Paraquat exposure induces nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and the activation of the nitric oxide-GAPDH-Siah cell death cascade. Toxicol. Sci. 2010, 116, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kim, C.K.; Lee, K.H.; Ahn, J.Y. S-nitrosylation of B23/ nucleophosmin by GAPDH protects cells from the SIAH1-GAPDH death cascade. J. Cell Biol. 2012, 199, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.W.; Saunders, P.A.; Wei, H.; Li, Z.; Seth, P.; Chuang, D.M. Involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and p53 in neuronal apoptosis: Evidence that GAPDH is upregulated by p53. J. Neurosci. 1999, 19, 9654–9662. [Google Scholar] [PubMed]
- Fiucci, G.; Beaucourt, S.; Duflaut, D.; Lespagnol, A.; Stumptner-Cuvelette, P.; Geant, A.; Buchwalter, G.; Tuynder, M.; Susini, L.; Lassalle, J.M.; et al. Siah-1b is a direct transcriptional target of p53: Identification of the functional p53 responsive element in the siah-1b promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 3510–3515. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Yasunaga, R.; Higashimura, Y.; Yamaji, R.; Fujimoto, K.; Moss, J.; Inui, H.; Nakano, Y. Glyceraldehyde-3-phosphate dehydrogenase enhances transcriptional activity of androgen receptor in prostate cancer cells. J. Biol. Chem. 2007, 282, 22651–22661. [Google Scholar] [CrossRef] [PubMed]
- Raje, C.I.; Kumar, S.; Harle, A.; Nanda, J.S.; Raje, M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J. Biol. Chem. 2007, 282, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011, 23, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Seidler, N.W. Compartmentation of GAPDH. Adv. Exp. Med. Biol. 2013, 985, 61–101. [Google Scholar] [PubMed]
- Singh, R.; Green, M.R. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 1993, 259, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascual, F.; Redondo-Horcajo, M.; Magán-Marchal, N.; Lagares, D.; Martínez-Ruiz, A.; Kleinert, H.; Lamas, S. Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Mol. Cell. Biol. 2008, 28, 7139–7155. [Google Scholar] [CrossRef] [PubMed]
- Backlund, M.; Paukku, K.; Daviet, L.; De Boer, R.A.; Valo, E.; Hautaniemi, S.; Kalkkinen, N.; Ehsan, A.; Kontula, K.K.; Lehtonen, J.Y.A. Posttranscriptional regulation of angiotensin II type 1 receptor expression by glyceraldehyde-3-phosphate dehydrogenase. Nucleic Acids Res. 2009, 37, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yi, X.; Stofffer, J.B.; Bonafe, N.; Gilmore-Hebert, M.; McAlpine, J.; Chambers, S.K. The multifunctional protein glyceraldehyde-3-phosphate dehydrogenase is both regulated and controls colony-stimulating factor-1 messenger RNA stability in ovarian cancer. Mol. Cancer Res. 2008, 6, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Kubota, S.; Mukudai, Y.; Nishida, T.; Yoshihama, Y.; Shirota, T.; Shintani, S.; Takigawa, M. Binding of glyceraldehyde-3-phosphate dehydrogenase to the cis-acting element of structure-anchored repression in ccn2 mRNA. Biochem. Biophys. Res. Commun. 2011, 405, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Yamaji, R.; Irie, K.; Kioka, N.; Murakami, A. Glyceraldehyde-3-phosphate dehydrogenase regulates cyclooxygenase-2 expression by targeting mRNA stability. Arch. Biochem. Biophys. 2012, 528, 141–147. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Khan, M.M.; Deredge, D.; Ross, C.R.; Quintyn, R.; Zucconi, B.E.; Wysocki, V.H.; Wintrode, P.L.; Wilson, G.M.; Garcin, E.D. A dimer interface mutation in glyceraldehyde-3-phosphate dehydrogenase regulates its binding to AU-rich RNA. J. Biol. Chem. 2015, 290, 1770–1785. [Google Scholar] [CrossRef] [PubMed]
- Ishitani, R.; Chuang, D.M. Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 9937–9941. [Google Scholar] [CrossRef] [PubMed]
- Saunders, P.A.; Chalecka-Franaszek, E.; Chuang, D.M. Subcellular distribution of glyceraldehyde-3-phosphate dehydrogenase in cerebellar granule cells undergoing cytosine arabinoside-induced apoptosis. J. Neurochem. 1997, 69, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Phadke, M.S.; Krynetskaia, N.F.; Mishra, A.K.; Krynetskiy, E. Glyceraldehyde 3-phosphate dehydrogenase depletion induces cell cycle arrest and resistance to antimetabolites in human carcinoma cell lines. J. Pharmacol. Exp. Ther. 2009, 331, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Saunders, P.A.; Chen, R.W.; Chuang, D.M. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J. Neurochem. 1999, 72, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Schlisser, A.E.; Yan, J.; Hales, B.F. Teratogen-induced oxidative stress targets glyceraldehyde-3-phosphate dehydrogenase in the organogenesis stage mouse embryo. Toxicol. Sci. 2010, 118, 686–695. [Google Scholar] [CrossRef] [PubMed]
- You, B.; Huang, S.; Qin, Q.; Yi, B.; Yuan, Y.; Xu, Z.; Sun, J. Glyceraldehyde-3-phosphate dehydrogenase interacts with proapoptotic kinase mst1 to promote cardiomyocyte apoptosis. PLoS ONE 2013, 8, e58697. [Google Scholar] [CrossRef] [PubMed]
- Steinritz, D.; Weber, J.; Balszuweit, F.; Thiermann, H.; Schmidt, A. Sulfur mustard induced nuclear translocation of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH). Chem. Biol. Interact. 2013, 206, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Tristan, C.A.; Ramos, A.; Shahani, N.; Emiliani, F.E.; Nakajima, H.; Noeh, C.C.; Kato, Y.; Takeuchi, T.; Noguchi, T.; Kadowaki, H.; et al. Role of apoptosis signal-regulating kinase 1 (ASK1) as an activator of the GAPDH-Siah1 stress-signaling cascade. J. Biol. Chem. 2015, 290, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Woo, S.R.; Shen, Y.N.; Yun, M.Y.; Shin, H.J.; Park, E.R.; Kim, S.H.; Park, J.E.; Ju, Y.J.; Hong, S.H.; et al. SIRT1 interacts with and protects glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from nuclear translocation: Implications for cell survival after irradiation. Biochem. Biophys. Res. Commun. 2012, 424, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.M.; Krynetski, E.Y.; Krynetskaia, N.F.; Grieger, D.; Mukatira, S.T.; Murti, K.G.; Slaughter, C.A.; Park, H.W.; Evans, W.E. A novel CRM1-mediated nuclear export signal governs nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase following genotoxic stress. J. Biol. Chem. 2004, 279, 5984–5992. [Google Scholar] [CrossRef] [PubMed]
- Schlisser, A.E.; Hales, B.F. Deprenyl enhances the teratogenicity of hydroxyurea in organogenesis stage mouse embryos. Toxicol. Sci. 2013, 134, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Long, C.A.; Boulom, V.; Albadawi, H.; Tsai, S.; Yoo, H.J.; Oklu, R.; Goldman, M.H.; Watkins, M.T. Poly-ADP-ribose-polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann. Surg. 2013, 258, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Feng, J.J.; Wu, Y.P.; Zhang, G.Y. Cerebral ischemia-reperfusion induces GAPDH S-nitrosylation and nuclear translocation. Biochemistry 2012, 77, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Inadomi, C.; Murata, H.; Ihara, Y.; Goto, S.; Urata, Y.; Yodoi, J.; Kondo, T.; Sumikawa, K. Overexpression of glutaredoxin protects cardiomyocytes against nitric oxide-induced apoptosis with suppressing the S-nitrosylation of proteins and nuclear translocation of GAPDH. Biochem. Biophys. Res. Commun. 2012, 425, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Leisner, T.M.; Moran, C.; Holly, S.P.; Parise, L.V. CIB1 prevents nuclear GAPDH accumulation and non-apoptotic tumor cell death via AKT and ERK signaling. Oncogene 2013, 32, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Fiorini, C.; Pozza, E.D.; Padroni, C.; Costanzo, C.; Palmieri, M.; Donadelli, M. UCP2 inhibition triggers ROS-dependent nuclear translocation of GAPDH and autophagic cell death in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2013, 1833, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Hara, M.R.; Ahmad, A.S.; Cascio, M.B.; Kamiya, A.; Ehmsen, J.T.; Agrawal, N.; Hester, L.; Doré, S.; Snyder, S.H.; et al. GOSPEL: A neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron 2009, 63, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation. J. Biol. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Rhim, J.H.; Jang, I.S.; Kim, G.E.; Park, S.C.; Yeo, E.J. Activation of AMP-activated protein kinase stimulates the nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in human diploid fibroblasts. Exp. Mol. Med. 2010, 42, 254–269. [Google Scholar] [CrossRef] [PubMed]
- Morgenegg, G.; Winkler, G.C.; Hübscher, U.; Heizmann, C.W.; Mous, J.; Kuenzle, C.C. Glyceraldehyde-3-phosphate dehydrogenase is a nonhistone protein and a possible activator of transcription in neurons. J. Neurochem. 1986, 47, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Kim, J. Regulation of oncogenic transcription factor hTAF(II)68-TEC activity by human glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Biochem. J. 2007, 404, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [PubMed]
- Zheng, L.; Roeder, R.G.; Luo, Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 2003, 114, 255–266. [Google Scholar] [CrossRef]
- Dai, R.P.; Yu, F.X.; Goh, S.R.; Chng, H.W.; Tan, Y.L.; Fu, J.L.; Zheng, L.; Luo, Y. Histone 2B (H2B) expression is confined to a proper NAD+/NADH redox status. J. Biol. Chem. 2008, 283, 26894–26901. [Google Scholar] [CrossRef] [PubMed]
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 2014, 27, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Arutyunova, E.I.; Danshina, P.V.; Domnina, L.V.; Pleten, A.P.; Muronetz, V.I. Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids. Biochem. Biophys. Res. Commun. 2003, 307, 547–552. [Google Scholar] [CrossRef]
- Holtgrefe, S.; Gohlke, J.; Starmann, J.; Druce, S.; Klocke, S.; Altmann, B.; Wojtera, J.; Lindermayr, C.; Scheibe, R. Regulation of plant cytosolic glyceraldehyde 3-phosphate dehydrogenase isoforms by thiol modifications. Physiol. Plant 2008, 133, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Loecken, E.M.; Guengerich, F.P. Reactions of glyceraldehyde 3-phosphate dehydrogenase sulfhydryl groups with bis-electrophiles produce DNA-protein cross-links but not mutations. Chem. Res. Toxicol. 2008, 21, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Sundararaj, K.P.; Wood, R.E.; Ponnusamy, S.; Salas, A.M.; Szulc, Z.; Bielawska, A.; Obeid, L.M.; Hannun, Y.A.; Ogretmen, B. Rapid shortening of telomere length in response to ceramide involves the inhibition of telomere binding activity of nuclear glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2004, 279, 6152–6162. [Google Scholar] [CrossRef] [PubMed]
- Demarse, N.A.; Ponnusamy, S.; Spicer, E.K.; Apohan, E.; Baatz, J.E.; Ogretmen, B.; Davies, C. Direct binding of glyceraldehyde 3-phosphate dehydrogenase to telomeric DNA protects telomeres against chemotherapy-induced rapid degradation. J. Mol. Biol. 2009, 394, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, G.; Depauw, S.; Mendy, D.; David-Cordonnier, M.H. Protein recognition of the S23906-1-DNA adduct by nuclear proteins: Direct involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Biochem. J. 2013, 452, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Pariona-Llanos, R.; Pavani, R.S.; Reis, M.; Noël, V.; Silber, A.M.; Armelin, H.A.; Cano, M.I.; Elias, M.C. Glyceraldehyde 3-phosphate dehydrogenase-telomere association correlates with redox status in Trypanosoma cruzi. PLoS ONE 2015, 10, e0120896. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.; Pinto, A.R.; Li, H.; Li, L.; Wang, L.; Simpson, R.; Liu, J.P. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) induces cancer cell senescence by interacting with telomerase RNA component. Proc. Natl. Acad. Sci. USA 2012, 109, 13308–13313. [Google Scholar] [CrossRef] [PubMed]
- Ringel, A.E.; Ryznar, R.; Picariello, H.; Huang, K.L.; Lazarus, A.G.; Holmes, S.G. Yeast Tdh3 (glyceraldehyde 3-phosphate dehydrogenase) is a Sir2-interacting factor that regulates transcriptional silencing and rDNA recombination. PLoS Genet. 2013, 9, e1003871. [Google Scholar] [CrossRef] [PubMed]
- Plowright, A.T.; Schaus, S.E.; Myers, A.G. Transcriptional response pathways in a yeast strain sensitive to saframycin a and a more potent analog: Evidence for a common basis of activity. Chem. Biol. 2002, 9, 607–618. [Google Scholar] [CrossRef]
- Xing, C.; LaPorte, J.R.; Barbay, J.K.; Myers, A.G. Identification of GAPDH as a protein target of the saframycin antiproliferative agents. Proc. Natl. Acad. Sci. USA 2004, 101, 5862–5866. [Google Scholar] [CrossRef] [PubMed]
- Kosova, A.A.; Khodyreva, S.N.; Lavrik, O.I. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) interacts with apurinic/apyrimidinic sites in DNA. Mutat. Res. 2015, 779, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Grosse, F.; Nasheuer, H.P.; Scholtissek, S.; Schomburg, U. Lactate dehydrogenase and glyceraldehyde-phosphate dehydrogenase are single-stranded DNA-binding proteins that affect the DNA-polymerase-alpha-primase complex. Eur. J. Biochem. 1986, 160, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.R.; Yim, S.H.; Kim, Y.M.; Jeong, J.; Song, E.J.; Lee, Y.; Lee, J.H.; Choi, S.; Lee, K.J. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 2009, 423, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Madhusudan, S. Human AP endonuclease 1 (APE1): From mechanistic insights to druggable target in cancer. Cancer Treat. Rev. 2010, 36, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Siegler, K.; Mauro, D.J.; Seal, G.; Wurzer, J.; deRiel, J.K.; Sirover, M.A. A human nuclear uracil DNA glycosylase is the 37-kDa subunit of glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. USA 1991, 88, 8460–8464. [Google Scholar] [CrossRef] [PubMed]
- Vollberg, T.M.; Cool, B.L.; Sirover, M.A. Biosynthesis of the human base excision repair enzyme uracil-DNA glycosylase. Cancer Res. 1987, 47, 123–128. [Google Scholar] [PubMed]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, A.; Prudovsky, I.; Suzuki, H.; Dal Pra, I.; Kisselev, L. Opposite effects of cell differentiation and apoptosis on Ap3A/Ap4A ratio in human cell cultures. FEBS Lett. 1997, 415, 160–162. [Google Scholar] [CrossRef]
- Baxi, M.D.; Vishwanatha, J.K. Uracil DNA-glycosylase/glyceraldehyde-3-phosphate dehydrogenase is an Ap4A binding protein. Biochemistry 1995, 34, 9700–9707. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Krynetski, E.Y.; Krynetskaia, N.F.; Gallo, A.E.; Murti, K.G.; Evans, W.E. A novel protein complex distinct from mismatch repair binds thioguanylated DNA. Mol. Pharmacol. 2001, 59, 367–374. [Google Scholar] [PubMed]
- Krynetski, E.Y.; Krynetskaia, N.F.; Bianchi, M.E.; Evans, W.E. A nuclear protein complex containing high mobility group proteins B1 and B2, heat shock cognate protein 70, ERp60, and glyceraldehyde-3-phosphate dehydrogenase is involved in the cytotoxic response to DNA modified by incorporation of anticancer nucleoside analogues. Cancer Res. 2003, 63, 100–106. [Google Scholar] [PubMed]
- Krynetskaia, N.F.; Phadke, M.S.; Jadhav, S.H.; Krynetskiy, E.Y. Chromatin-associated proteins HMGB1/2 and PDIA3 trigger cellular response chemotherapy-induced DNA damage. Mol. Cancer Ther. 2009, 8, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Buskin, J.N.; Hauschka, S.D. Identification of a myocyte nuclear factor that binds to the muscle-specific enhancer of the mouse muscle creatine kinase gene. Mol. Cell. Biol. 1989, 9, 2627–2640. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yu, Y.; Kang, R.; Yang, M.; Xie, M.; Wang, Z.; Tang, D.; Zhao, M.; Liu, L.; Zhang, H.; Cao, L. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk. Lymphoma 2012, 53, 315–322. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savreux-Lenglet, G.; Depauw, S.; David-Cordonnier, M.-H. Protein Recognition in Drug-Induced DNA Alkylation: When the Moonlight Protein GAPDH Meets S23906-1/DNA Minor Groove Adducts. Int. J. Mol. Sci. 2015, 16, 26555-26581. https://doi.org/10.3390/ijms161125971
Savreux-Lenglet G, Depauw S, David-Cordonnier M-H. Protein Recognition in Drug-Induced DNA Alkylation: When the Moonlight Protein GAPDH Meets S23906-1/DNA Minor Groove Adducts. International Journal of Molecular Sciences. 2015; 16(11):26555-26581. https://doi.org/10.3390/ijms161125971
Chicago/Turabian StyleSavreux-Lenglet, Gaëlle, Sabine Depauw, and Marie-Hélène David-Cordonnier. 2015. "Protein Recognition in Drug-Induced DNA Alkylation: When the Moonlight Protein GAPDH Meets S23906-1/DNA Minor Groove Adducts" International Journal of Molecular Sciences 16, no. 11: 26555-26581. https://doi.org/10.3390/ijms161125971