
Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment

Abstract
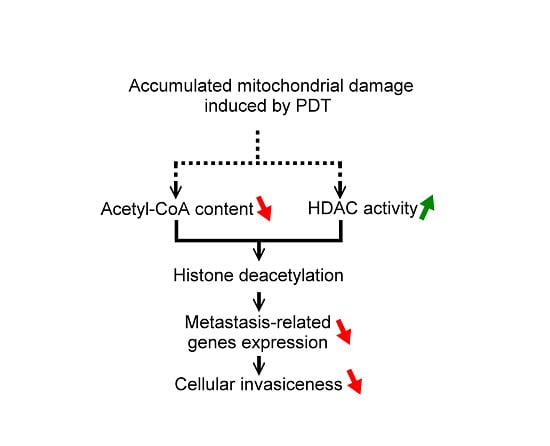
:
1. Introduction
2. Results and Discussion
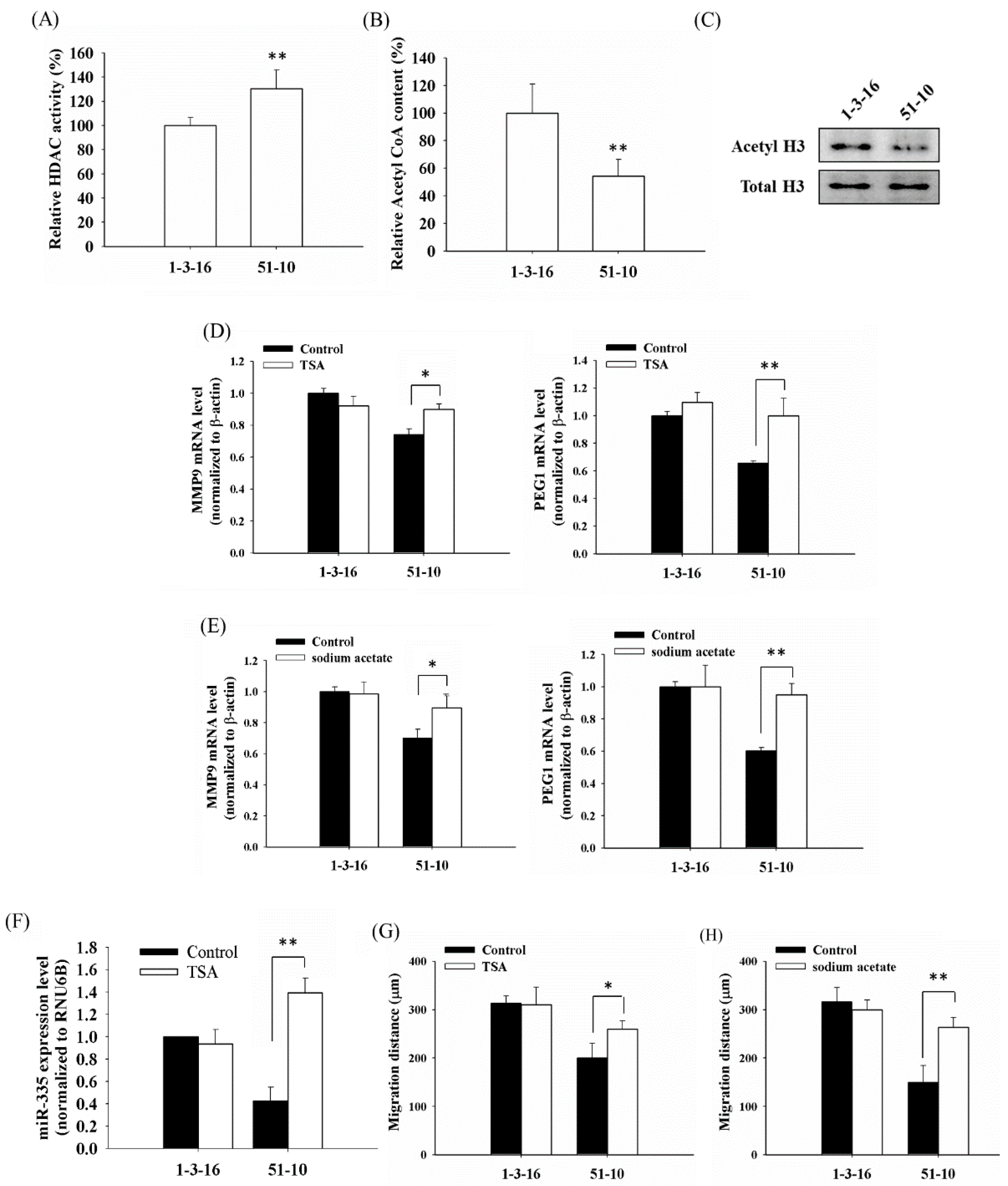
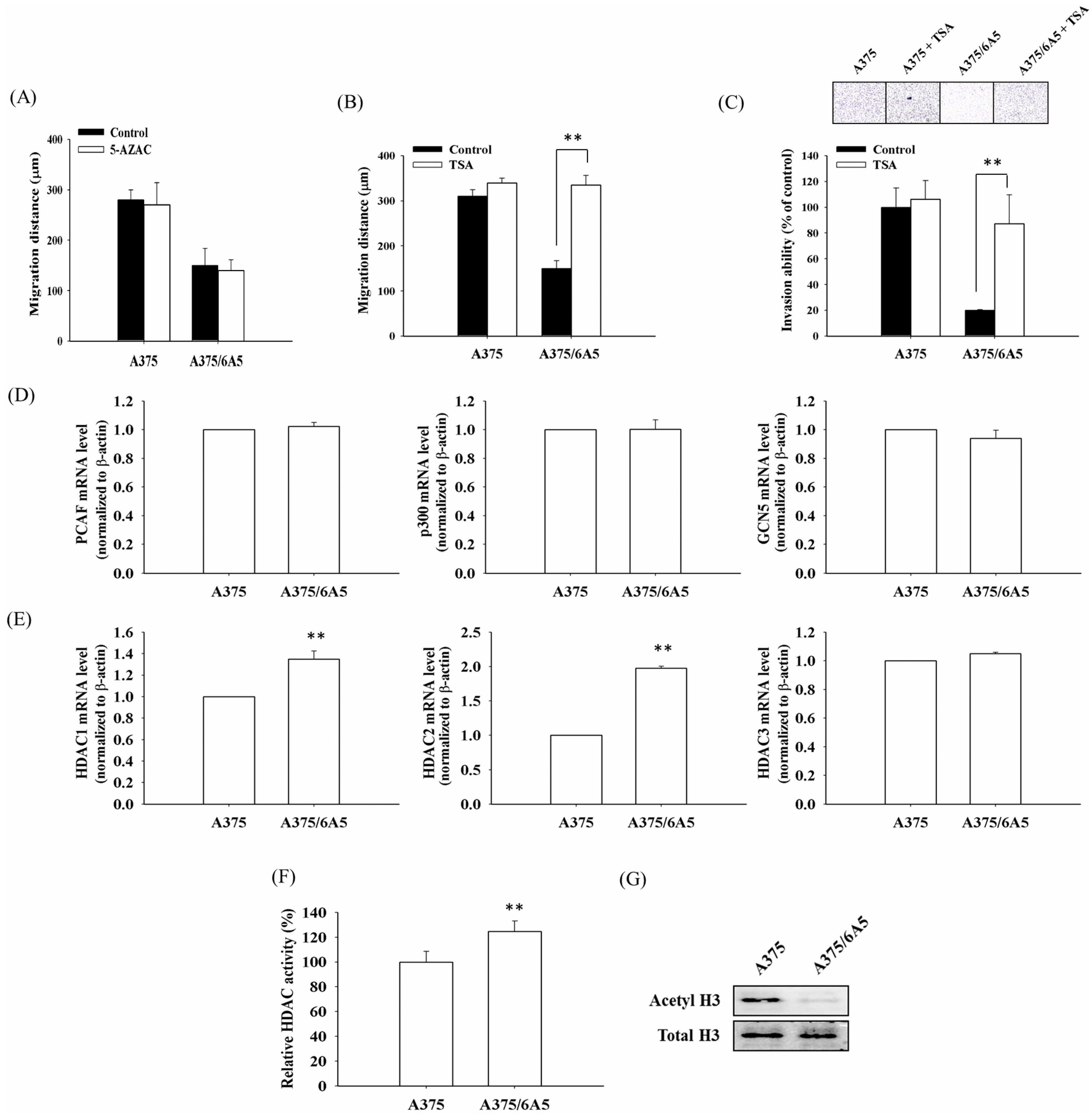
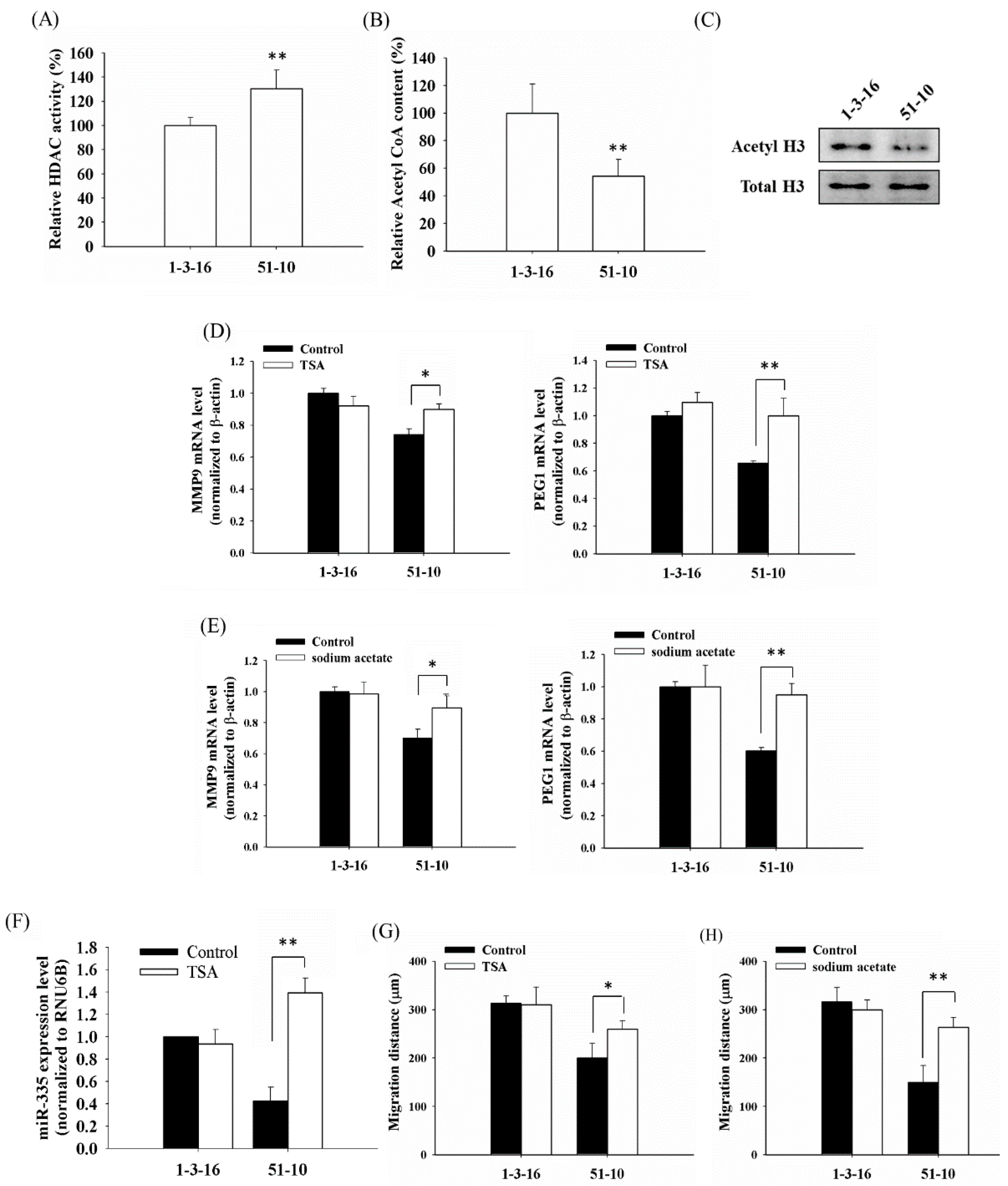
2.1. PDT-Derived A375 Variants Contain Elevated HDAC (Histone Deacetylase) Activity and Hypoacetylated Histones

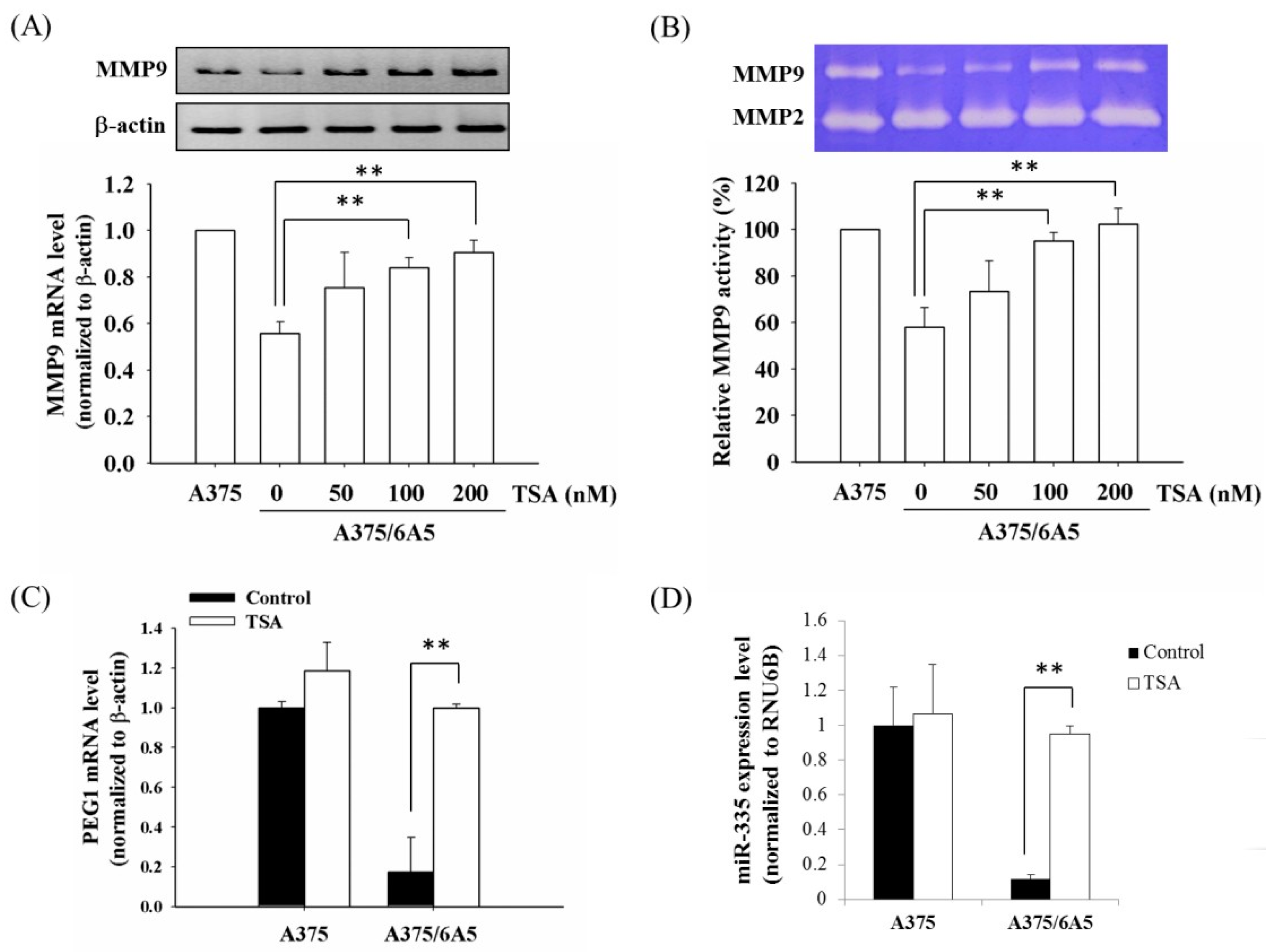
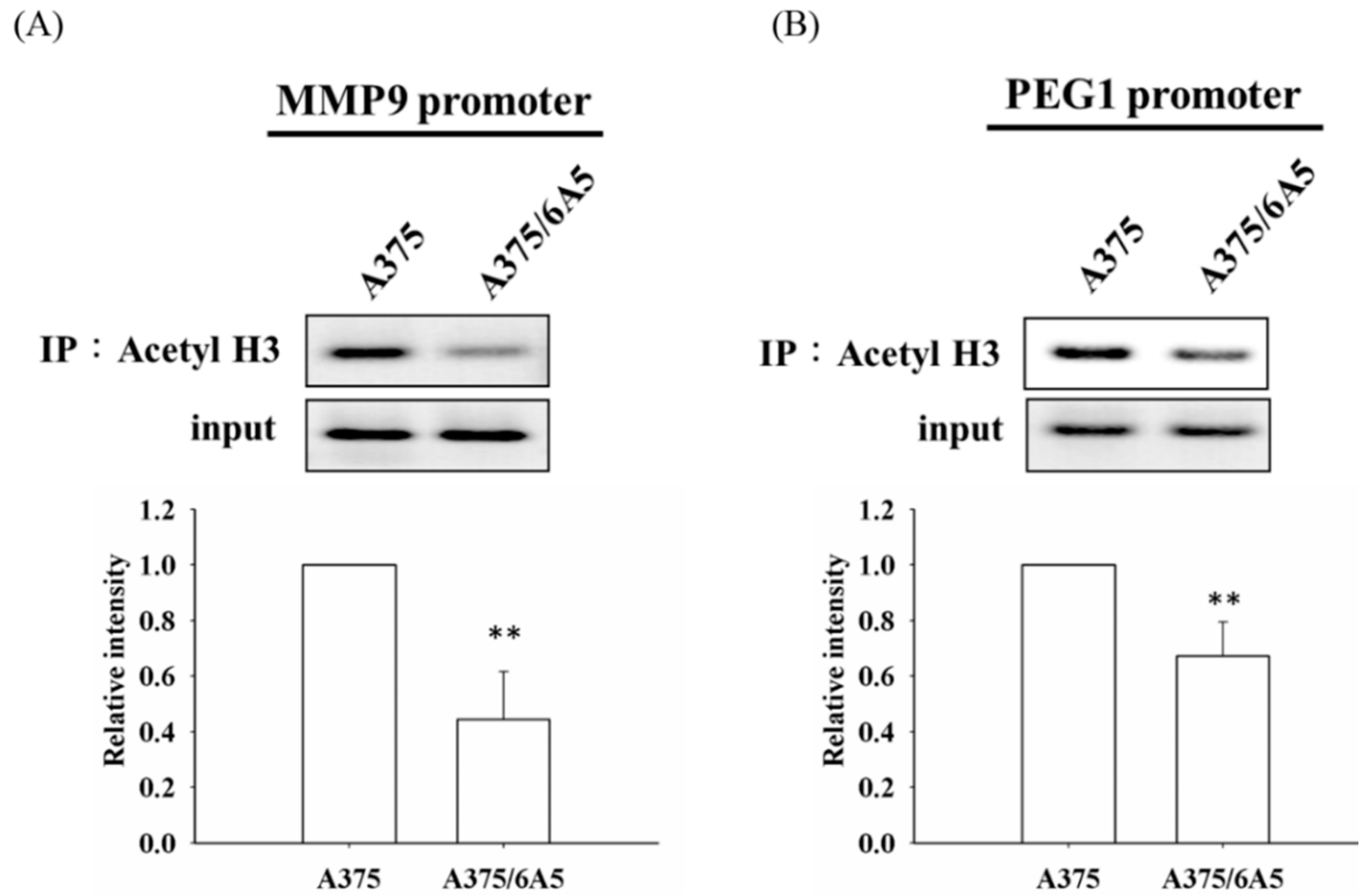
2.2. Histone Deacetylation Results in the Down-Regulation of MMP9 and PEG1 in PDT-Derived Variants

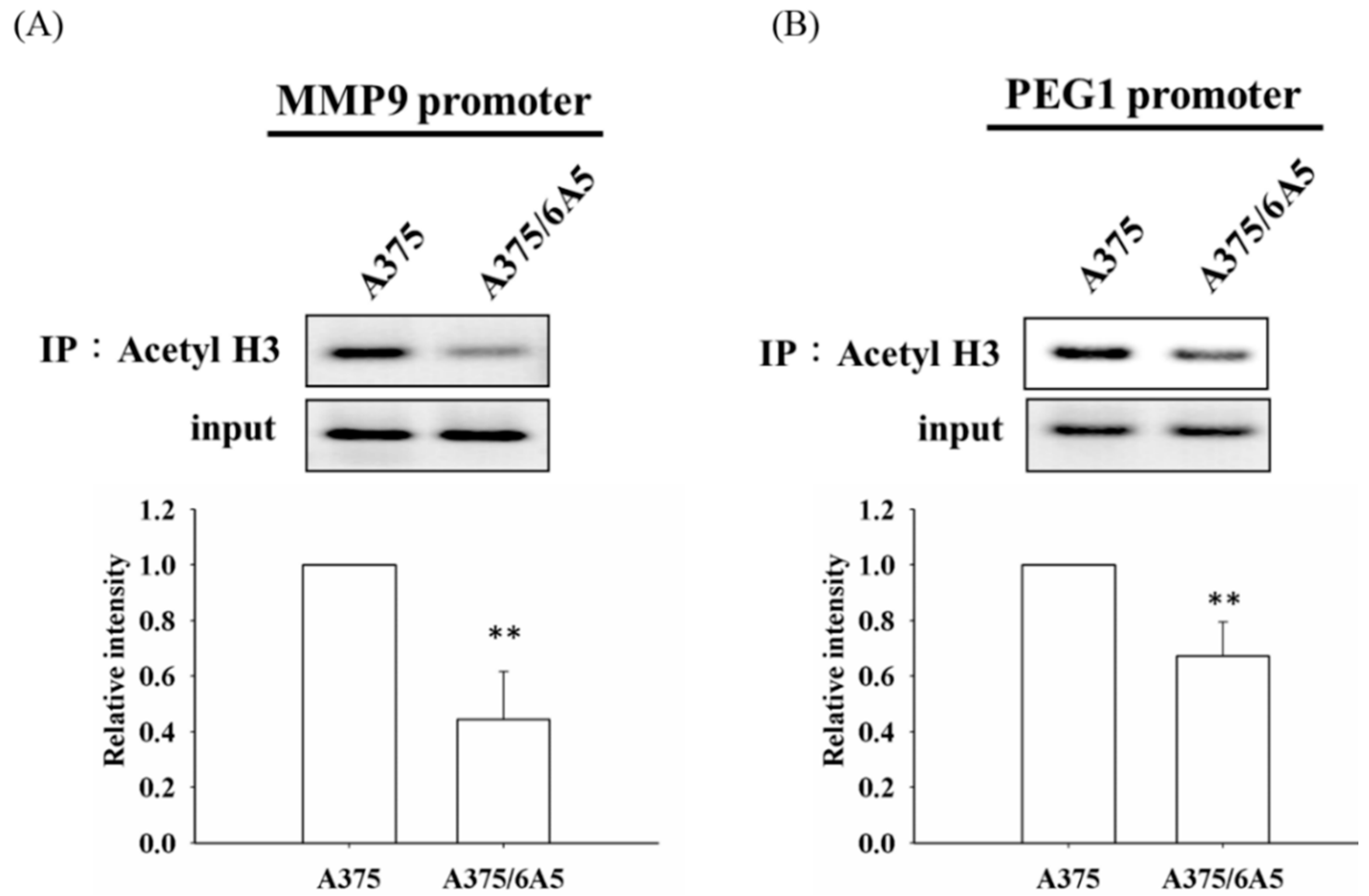
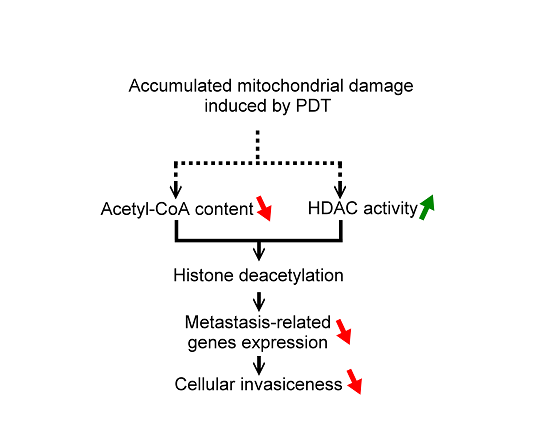
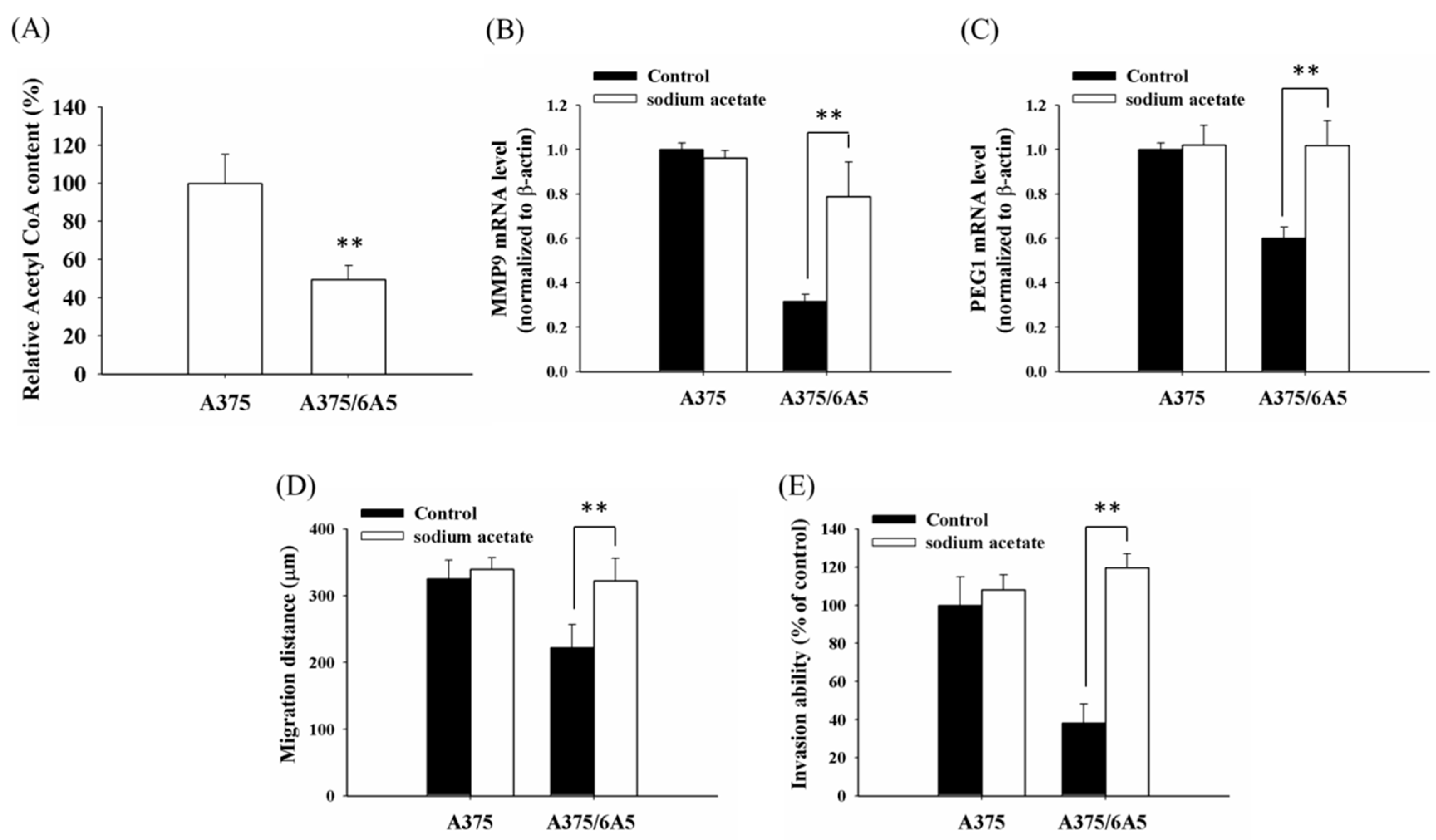
2.3. The Reduced Amount of Acetyl-CoA Also Involved in the Reduced Histone Acetylation and Cellular Migration and Invasiveness in Cells with Mitochondrial Dysfunction
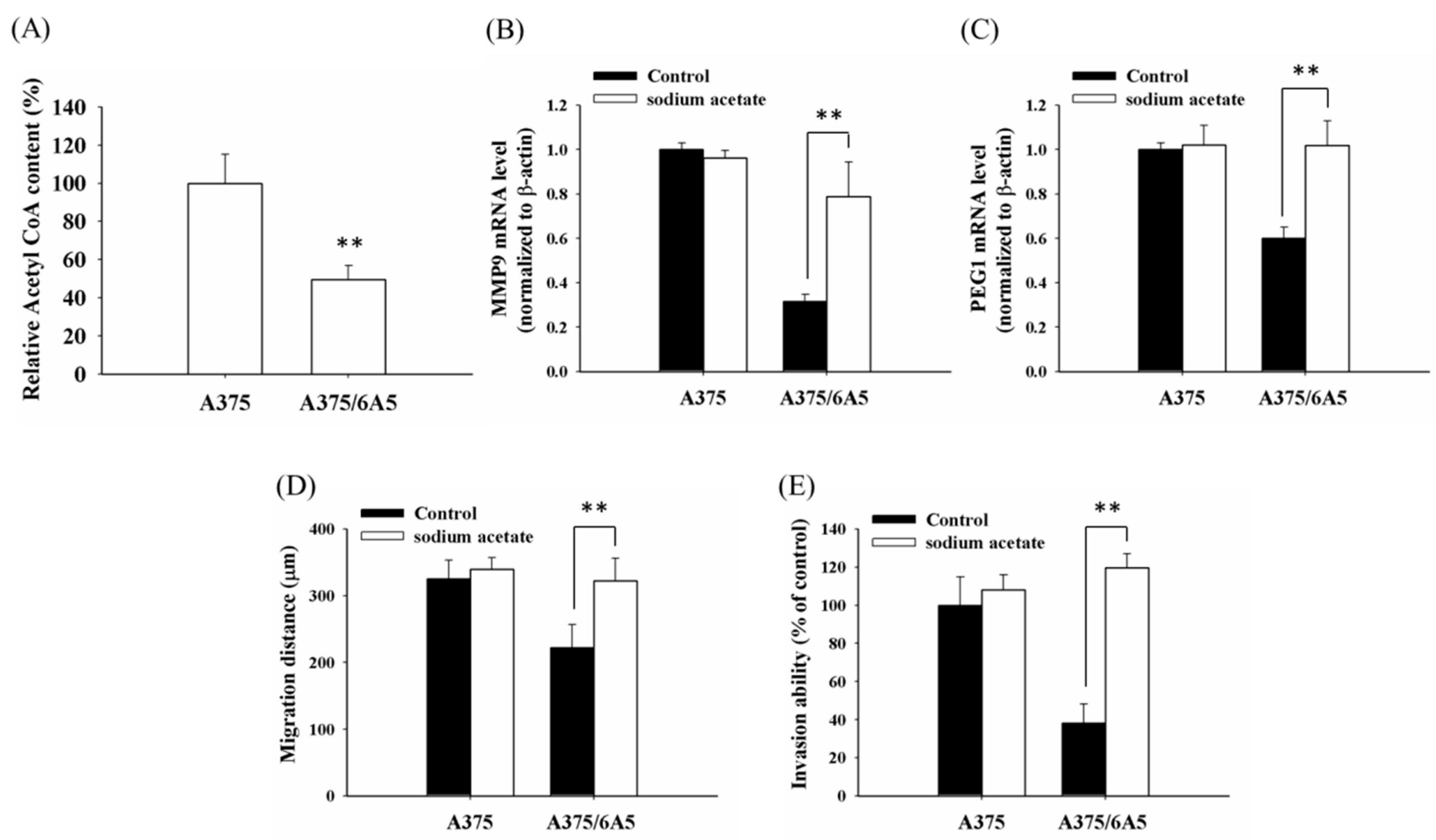
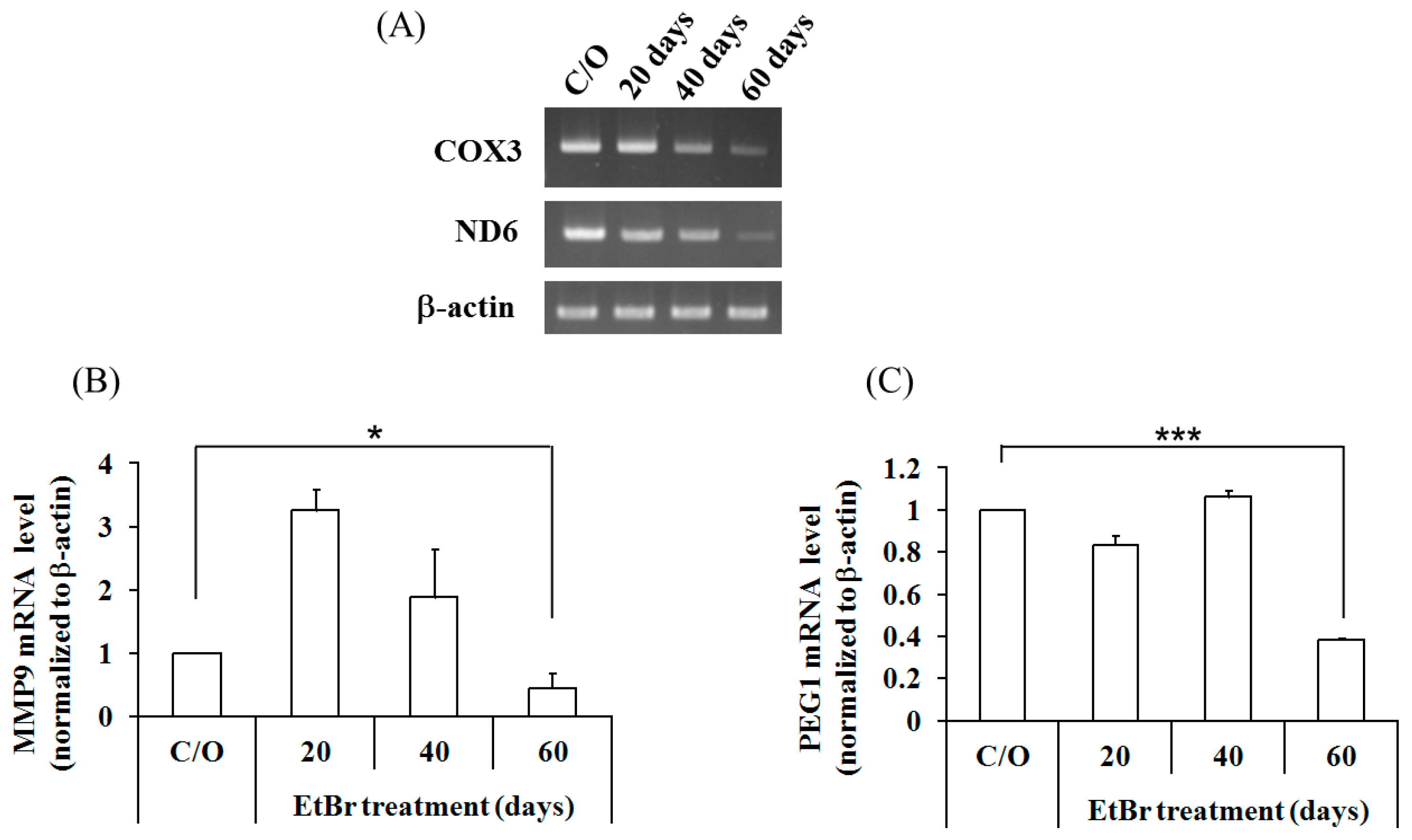
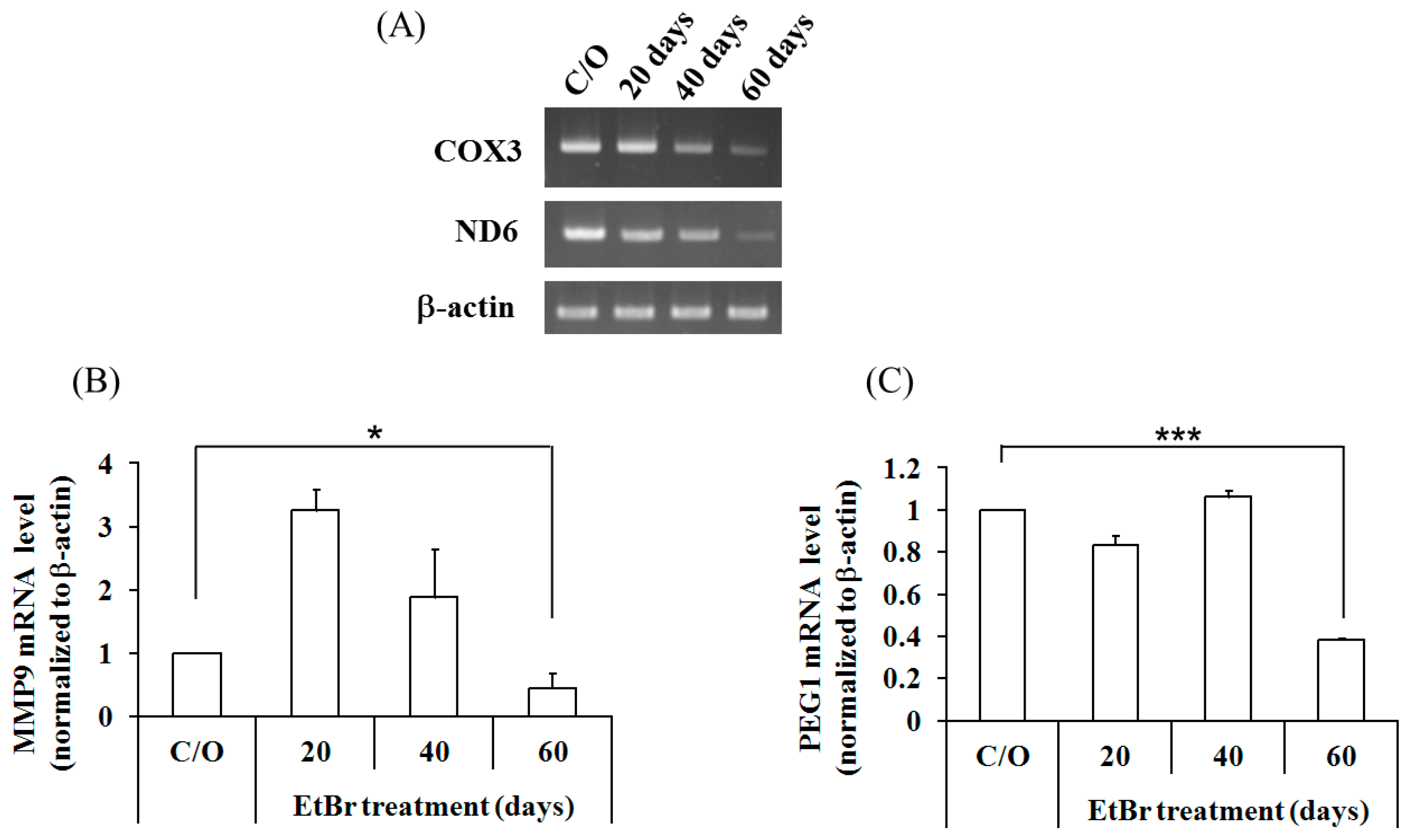
2.4. Mitochondrial Dysfunction Induced by 4977 bp Deletion of Mitochondrial DNA and Ethidium Bromide Treatment Suppressed Cellular Migration and Gene Expression of MMP9 and PEG1 via Histone Deacetylation
3. Experimental Section
3.1. Cell Culture
3.2. RNA Extraction, mRNA and miRNA Detection
3.3. Western Blot
3.4. Migration and Invasion Assays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer |
|---|---|
| PCAF (forward) | 5′-TCCTGTCGGAGTTGTAGCCA-3′ |
| PCAF (reverse) | 5′-GTTCTGGAAGAGGCTGAGAG-3′ |
| p300 (forward) | 5′-AGCCCTGGCAGTATGTCGAT-3′ |
| p300 (reverse) | 5′-GAATCCAGCAGGCCAGATGA-3′ |
| GCN5 (forward) | 5′-GTCAGGCTTCACCATGCCAC-3′ |
| GCN5 (reverse) | 5′-TGCCGATGACATGGAACTCG-3′ |
| HDAC1 (forward) | 5′-AACCTGCCTATGCTGATGCT-3′ |
| HDAC1 (reverse) | 5′-CAGGCAATTCGTTTGTCAGA-3′ |
| HDAC2 (forward) | 5′-GGGAATACTTTCCTGGCACA-3′ |
| HDAC2 (reverse) | 5′-ACGGATTGTGTAGCCACCTC-3′ |
| HDAC3 (forward) | 5′-TGGCTTCTGCTATGTCAACG-3′ |
| HDAC3 (reverse) | 5′-GCACGTGGGTTGGTAGAAGT-3′ |
| MMP9 (forward) | 5′-CGGAGCACGGAGACGGGTAT-3′ |
| MMP9 (reverse) | 5′-TGAAGGGGAAGACGCACAGC-3′ |
| PEG1 (forward) | 5′-CAAAGATGGAGGTGTGC-3′ |
| PEG1 (reverse) | 5′-TTCCCGTCATTGTTGCG-3′ |
| β-actin (forward) | 5′-TGGACTTCGAGCAAGAGATGG-3′ |
| β-actin (reverse) | 5′-ATCTCCTTCTGCATCCTGTCG-3′ |
| ND6 (forward) | 5′-CCCGAGCAATCTCAATTACA-3′ |
| ND6 (reverse) | 5′-CCGTGCGAGAATAATGATGT-3′ |
| COX3 (forward) | 5′-AGCCATGTGATTTCACTTCC-3′ |
| COX3 (reverse) | 5′-GTTGAGCCAATAATGACGTG-3′ |
3.5. Chromatin Immunoprecipitation (ChIP) Assay
| Gene Name | Primer |
|---|---|
| MMP9 promoter (forward) | 5′-CTGAGTCAAAGAAGGCTGT-3′ |
| MMP9 promoter (reverse) | 5′-GTGATGGAAGACTCCCTGAGA-3′ |
| PEG1 promoter (forward) | 5′-TACAGACTAGAGAAGGAAGCG-3′ |
| PEG1 promoter (reverse) | 5′-CGTGCTCTCGATCCTTATATT-3′ |
| EGFR promoter (forward) | 5′-ACCCTGGCACAGATTTGG-3′ |
| EGFR promoter (reverse) | 5′-TGAGGAGTTAATTTCCGAGAGG-3′ |
3.6. MMP9 Activity
3.7. HDAC Activity
3.8. Acetyl-CoA Content
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.G.; Wang, Y.Y.; Yang, Y.D.; Zhang, X.C.; Gao, Y.; Yang, Y.; Zhang, J.B.; Li, G.L. Efficacy of topical ALA-PDT combined with excision in the treatment of skin malignant tumor. Photodiagn. Photodyn. Ther. 2014, 11, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Oseroff, A.R. Mitochondria-based photodynamic anti-cancer therapy. Adv. Drug Deliv. Rev. 2001, 49, 71–86. [Google Scholar] [CrossRef]
- Gibson, S.L.; Nguyen, M.L.; Havens, J.J.; Barbarin, A.; Hilf, R. Relationship of δ-aminolevulinic acid-induced protoporphyrin IX levels to mitochondrial content in neoplastic cells in vitro. Biochem. Biophys. Res. Commun. 1999, 265, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Warloe, T.; Berg, K.; Moan, J.; Kongshaug, M.; Giercksky, K.E.; Nesland, J.M. 5-aminolevulinic acid-based photodynamic therapy. Cancer 1997, 79, 2282–2308. [Google Scholar] [CrossRef]
- Hua, Z.; Gibson, S.L.; Foster, T.H.; Hilf, R. Effectiveness of δ-aminolevulinic acid-induced protoporphyrin as a photosensitizer for photodynamic therapy in vivo. Cancer Res. 1995, 55, 1723–1731. [Google Scholar] [PubMed]
- Tsai, T.; Ji, H.T.; Chiang, P.C.; Chou, R.H.; Chang, W.S.; Chen, C.T. ALA-PDT results in phenotypic changes and decreased cellular invasion in surviving cancer cells. Lasers Surg. Med. 2009, 41, 305–315. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Butow, R.A.; Avadhani, N.G. Mitochondrial signaling: The retrograde response. Mol. Cell 2004, 14, 1–15. [Google Scholar] [CrossRef]
- Jahnke, V.E.; Sabido, O.; Freyssenet, D. Control of mitochondrial biogenesis, ROS level, and cytosolic Ca2+ concentration during the cell cycle and the onset of differentiation in L6E9 myoblasts. Am. J. Physiol. Cell Physiol. 2009, 296, C1185–C1194. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia-reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.M.; Yin, P.H.; Chi, C.W.; Hsu, C.Y.; Wu, C.W.; Lee, L.M.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 2006, 45, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Yin, P.H.; Hung, W.Y.; Li, A.F.; Li, S.H.; Chi, C.W.; Wei, Y.H.; Lee, H.C. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes Chromosomes Cancer 2005, 44, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Yin, P.H.; Yu, T.N.; Chang, Y.D.; Hsu, W.C.; Kao, S.Y.; Chi, C.W.; Liu, T.Y.; Wei, Y.H. Accumulation of mitochondrial DNA deletions in human oral tissues—effects of betel quid chewing and oral cancer. Mutat. Res. 2001, 493, 67–74. [Google Scholar] [CrossRef]
- Yin, P.H.; Lee, H.C.; Chau, G.Y.; Wu, Y.T.; Li, S.H.; Lui, W.Y.; Wei, Y.H.; Liu, T.Y.; Chi, C.W. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. Br. J. Cancer 2004, 90, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Lee, H.C.; Chung, J.G.; Wei, Y.H. Mitochondrial DNA mutations in light-associated skin tumors. Anticancer Res. 2004, 24, 1753–1758. [Google Scholar] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Gregory, P.D.; Wagner, K.; Horz, W. Histone acetylation and chromatin remodeling. Exp. Cell Res. 2001, 265, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J. Biomed. Biotechnol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Minocherhomji, S.; Tollefsbol, T.O.; Singh, K.K. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012, 7, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Shaughnessy, D.T.; McAllister, K.; Worth, L.; Haugen, A.C.; Meyer, J.N.; Domann, F.E.; Van Houten, B.; Mostoslavsky, R.; Bultman, S.J.; Baccarelli, A.A.; et al. Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ. Health Perspect. 2014, 122, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Mayo, M.W.; Denlinger, C.E.; Broad, R.M.; Yeung, F.; Reilly, E.T.; Shi, Y.; Jones, D.R. Ineffectiveness of histone deacetylase inhibitors to induce apoptosis involves the transcriptional activation of NF-κB through the Akt pathway. J. Biol. Chem. 2003, 278, 18980–18989. [Google Scholar] [CrossRef] [PubMed]
- Kuljaca, S.; Liu, T.; Tee, A.E.; Haber, M.; Norris, M.D.; Dwarte, T.; Marshall, G.M. Enhancing the anti-angiogenic action of histone deacetylase inhibitors. Mol. Cancer 2007, 6. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Kohda, T.; Miyoshi, N.; Kuroiwa, Y.; Aisaka, K.; Tsutsumi, O.; Kaneko-Ishino, T.; Ishino, F. Human PEG1/MEST, an imprinted gene on chromosome 7. Hum. Mol. Genet. 1997, 6, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.S.; Dervan, P.A.; Broderick, D.; Harrison, M.; Miller, N.; Delany, E.; O’Shea, D.; Costello, P.; McGoldrick, A.; Keating, G.; et al. Frequent loss of imprinting of PEG1/MEST in invasive breast cancer. Cancer Res. 1999, 59, 5449–5451. [Google Scholar] [PubMed]
- Pedersen, I.S.; Dervan, P.; McGoldrick, A.; Harrison, M.; Ponchel, F.; Speirs, V.; Isaacs, J.D.; Gorey, T.; McCann, A. Promoter switch: A novel mechanism causing biallelic PEG1/MEST expression in invasive breast cancer. Hum. Mol. Genet. 2002, 11, 1449–1453. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Zheng, X.; Wu, S.; Lu, H.; Leng, T.; Zhu, W.; Zhou, Y.; Ou, Y.; Lin, X.; Lin, Y.; et al. Targeting oncogenic miR-335 inhibits growth and invasion of malignant astrocytoma cells. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Xiong, Y.; Xu, W.; Gao, J.; Cheng, Y.; Wang, Z.; Chen, F.; Zheng, G. Identification of hsa-miR-335 as a prognostic signature in gastric cancer. PLoS ONE 2012, 7, e40037. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Fay, J.; Meehan, M.; Bryan, K.; Watters, K.M.; Murphy, D.M.; Stallings, R.L. MiRNA-335 suppresses neuroblastoma cell invasiveness by direct targeting of multiple genes from the non-canonical TGF-β signalling pathway. Carcinogenesis 2012, 33, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Tavazoie, S.F.; Alarcon, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human micrornas that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Scarola, M.; Schoeftner, S.; Schneider, C.; Benetti, R. MiR-335 directly targets Rb1 (pRb/p105) in a proximal connection to p53-dependent stress response. Cancer Res. 2010, 70, 6925–6933. [Google Scholar] [CrossRef] [PubMed]
- Ronchetti, D.; Lionetti, M.; Mosca, L.; Agnelli, L.; Andronache, A.; Fabris, S.; Deliliers, G.L.; Neri, A. An integrative genomic approach reveals coordinated expression of intronic miR-335, miR-342, and miR-561 with deregulated host genes in multiple myeloma. BMC Med. Genom. 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Dohi, O.; Yasui, K.; Gen, Y.; Takada, H.; Endo, M.; Tsuji, K.; Konishi, C.; Yamada, N.; Mitsuyoshi, H.; Yagi, N.; et al. Epigenetic silencing of MiR-335 and its host gene mest in hepatocellular carcinoma. Int. J. Oncol. 2013, 42, 411–418. [Google Scholar] [PubMed]
- Kaneko-Ishino, T.; Kuroiwa, Y.; Miyoshi, N.; Kohda, T.; Suzuki, R.; Yokoyama, M.; Viville, S.; Barton, S.C.; Ishino, F.; Surani, M.A. PEG1/MEST imprinted gene on chromosome 6 identified by cdna subtraction hybridization. Nat. Genet. 1995, 11, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, L.; Viville, S.; Barton, S.C.; Ishino, F.; Keverne, E.B.; Surani, M.A. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene mest. Nat. Genet. 1998, 20, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, H.; Park, H.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. MiR-326-histone deacetylase-3 feedback loop regulates the invasion and tumorigenic and angiogenic response to anti-cancer drugs. J. Biol. Chem. 2014, 289, 28019–28039. [Google Scholar] [CrossRef] [PubMed]
- Galdieri, L.; Vancura, A. Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 2012, 287, 23865–23876. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Drago, I.; Pizzo, P.; Pozzan, T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ. 2007, 14, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [PubMed]
- Soliman, M.L.; Rosenberger, T.A. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol. Cell. Biochem. 2011, 352, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; Lee, C.F.; Lee, H.C.; Ma, Y.S.; Wang, C.W.; Lu, C.Y.; Pang, C.Y. Increases of mitochondrial mass and mitochondrial genome in association with enhanced oxidative stress in human cells harboring 4977 bp-deleted mitochondrial DNA. Ann. N. Y. Acad. Sci. 2001, 928, 97–112. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.-T.; Tsai, Y.-J.; Lee, M.-J.; Chen, C.-T. Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment. Int. J. Mol. Sci. 2015, 16, 23994-24010. https://doi.org/10.3390/ijms161023994
Li P-T, Tsai Y-J, Lee M-J, Chen C-T. Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment. International Journal of Molecular Sciences. 2015; 16(10):23994-24010. https://doi.org/10.3390/ijms161023994
Chicago/Turabian StyleLi, Pei-Tzu, Yi-Jane Tsai, Ming-Jen Lee, and Chin-Tin Chen. 2015. "Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment" International Journal of Molecular Sciences 16, no. 10: 23994-24010. https://doi.org/10.3390/ijms161023994
APA StyleLi, P.-T., Tsai, Y.-J., Lee, M.-J., & Chen, C.-T. (2015). Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment. International Journal of Molecular Sciences, 16(10), 23994-24010. https://doi.org/10.3390/ijms161023994