
The Anticancer Drug Ellipticine Activated with Cytochrome P450 Mediates DNA Damage Determining Its Pharmacological Efficiencies: Studies with Rats, Hepatic Cytochrome P450 Reductase Null (HRN™) Mice and Pure Enzymes

 , , and
, , and 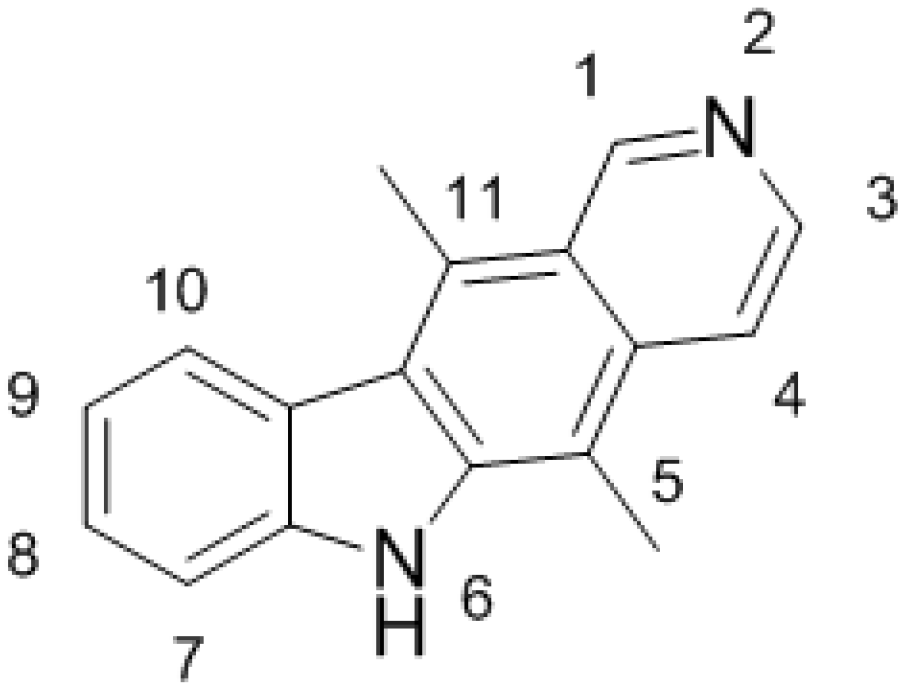{kind=link}
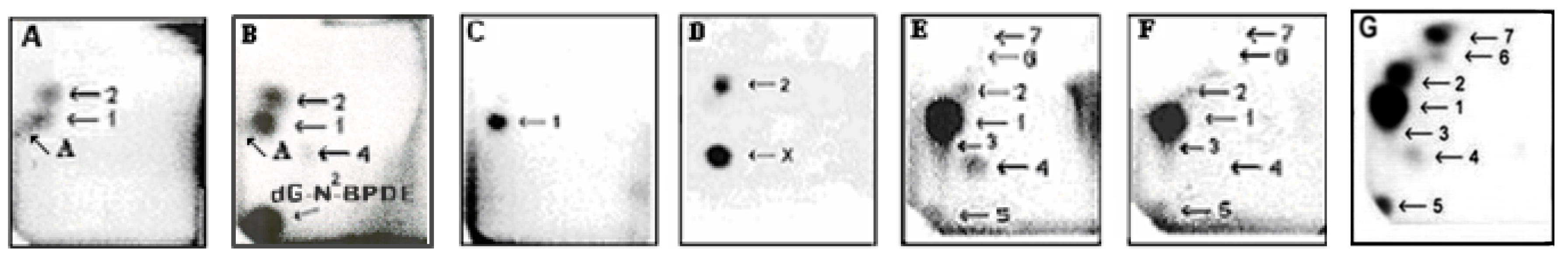{kind=link}
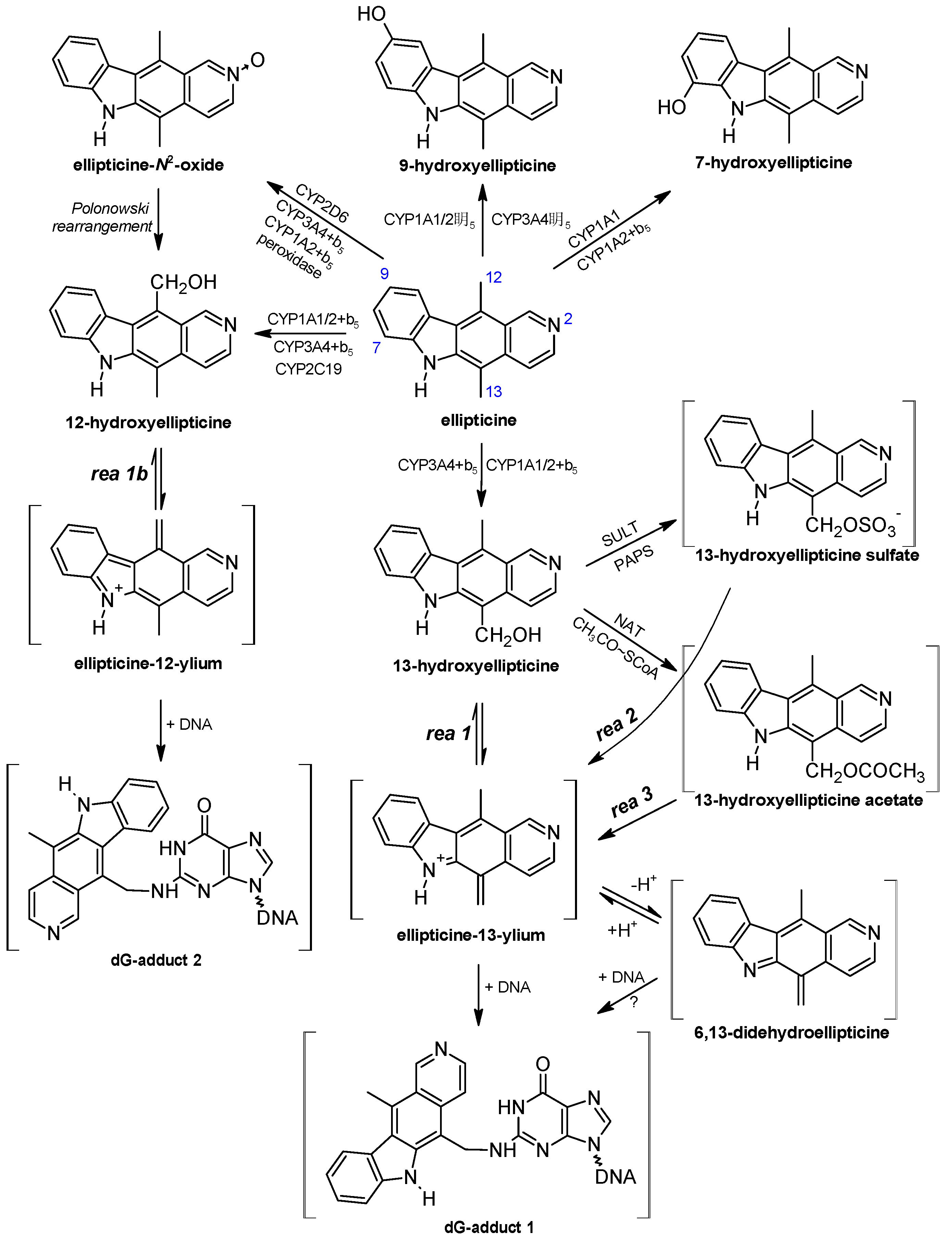{kind=link}
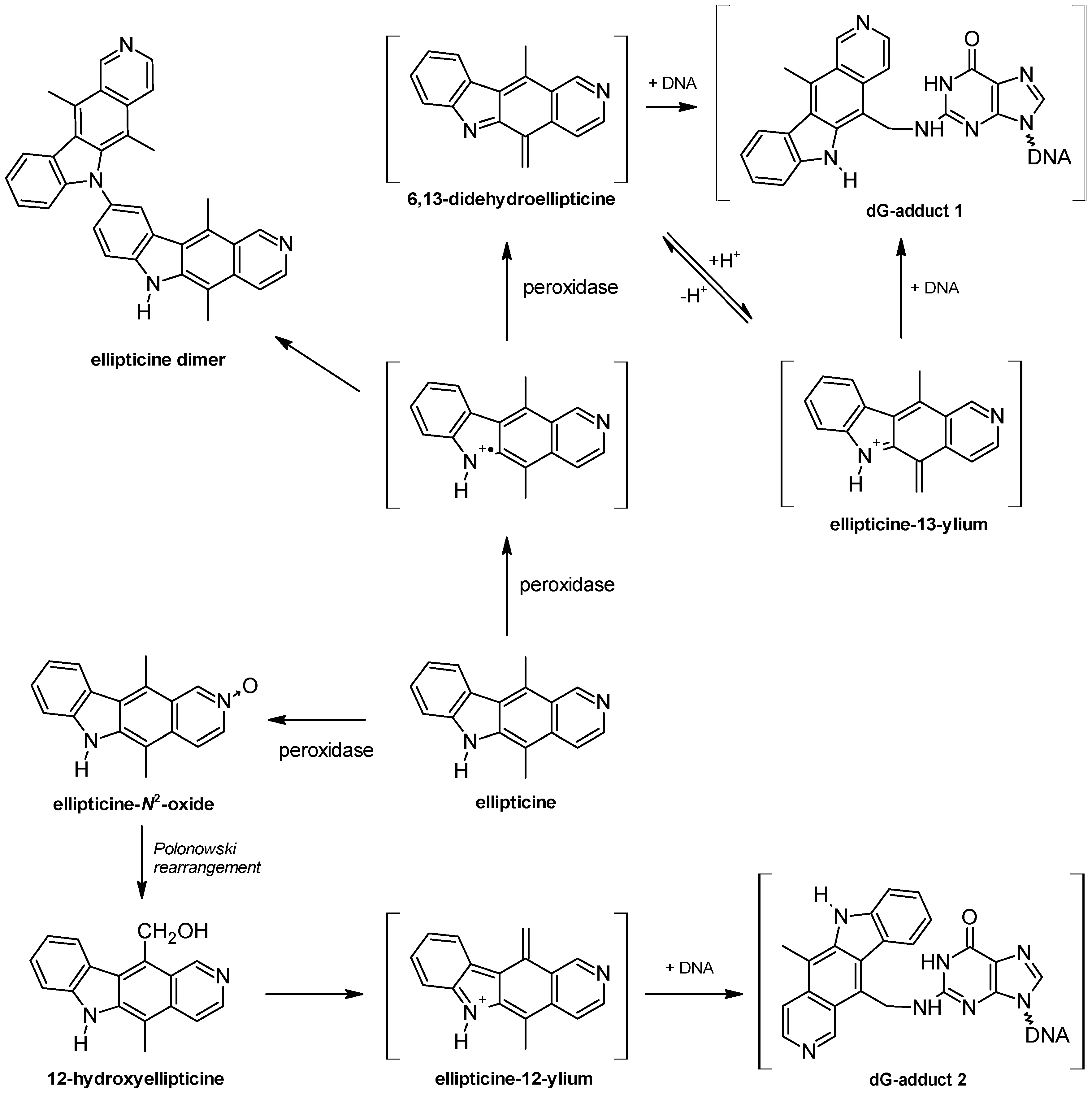{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
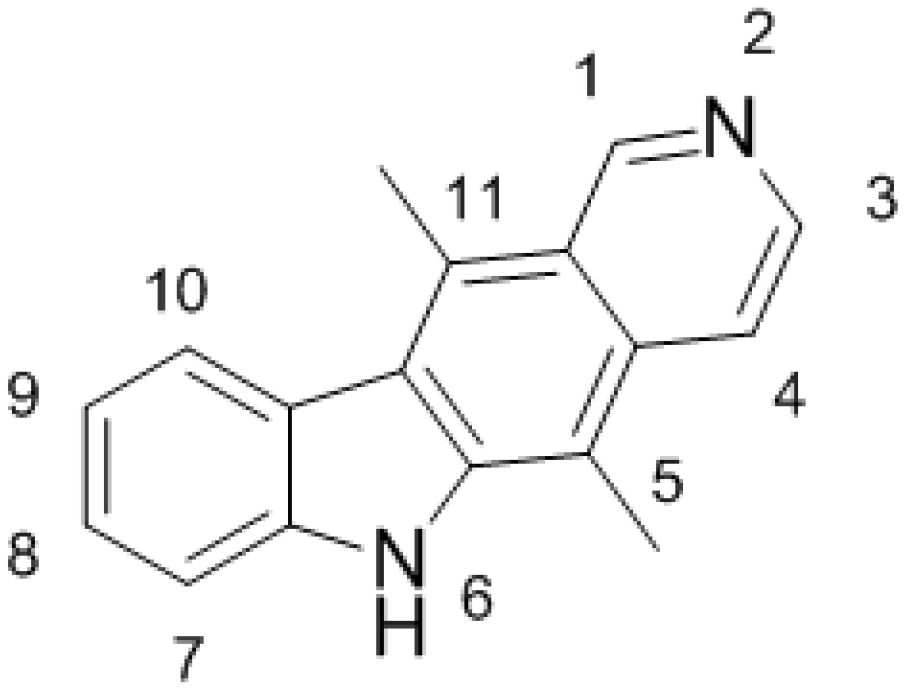
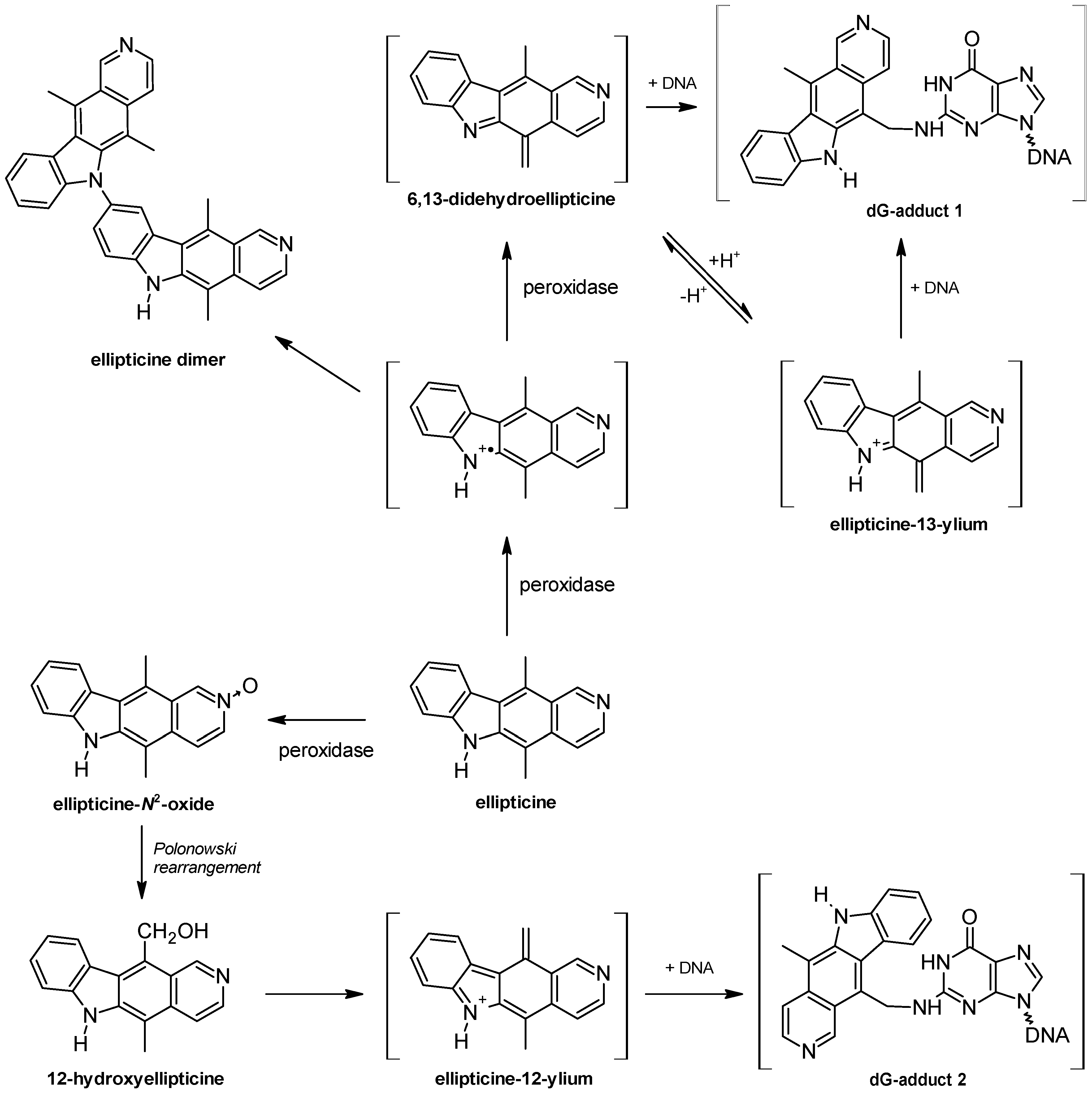
2. DNA-Damaging Mechanisms of Ellipticine Cytotoxicity to Cancer Cells
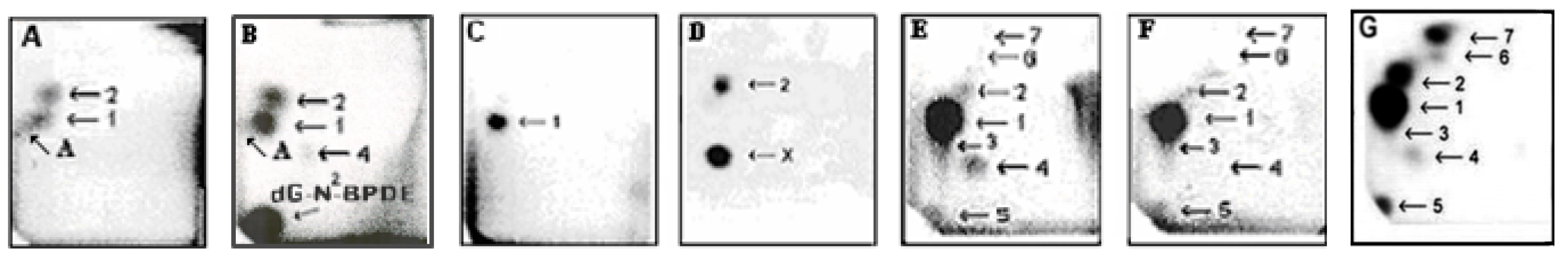
3. Metabolism of Ellipticine by Cytochromes P450 (CYPs), Peroxidases and Conjugation Enzymes in Vitro
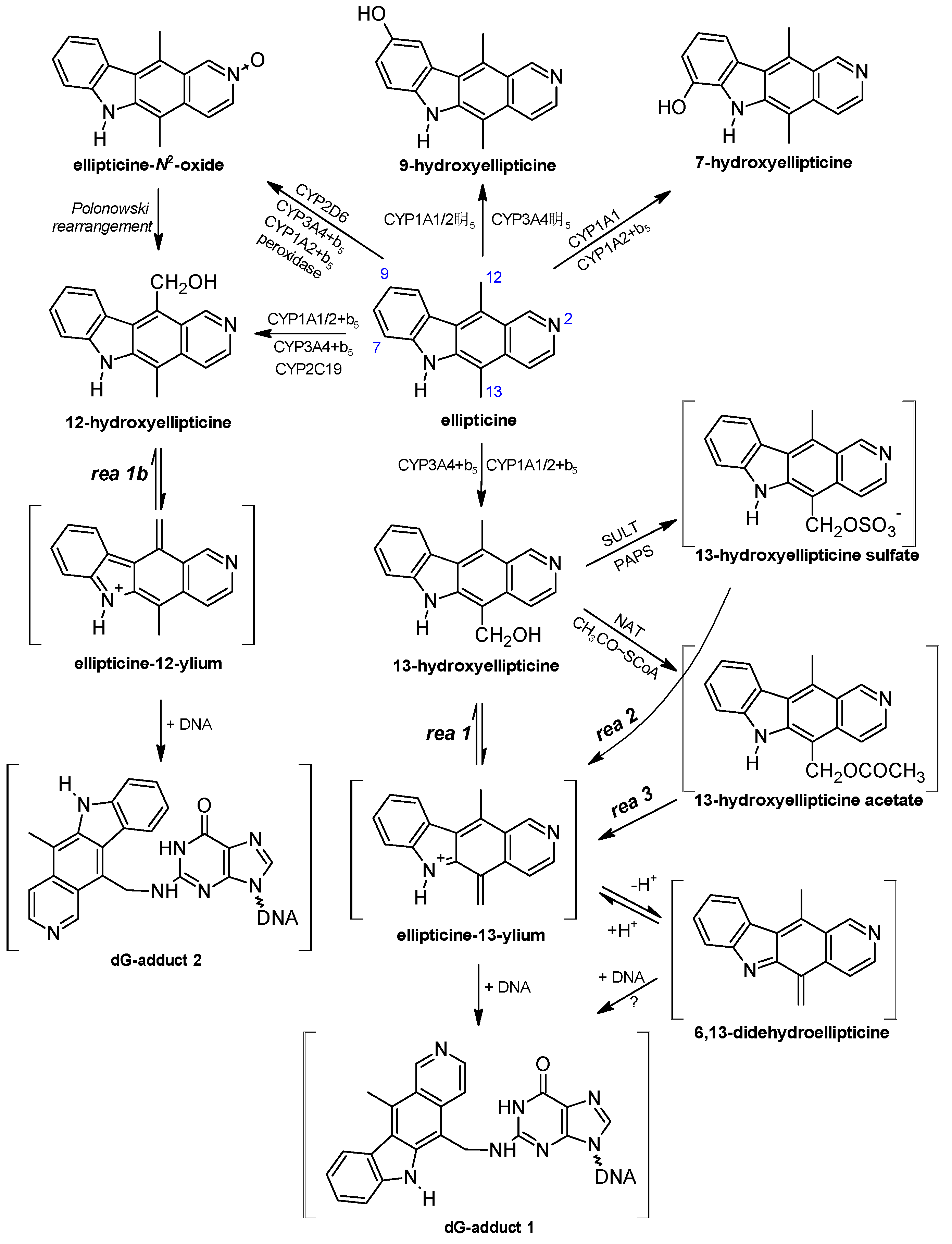
4. Oxidation of Ellipticine by CYP Enzymes in Vivo
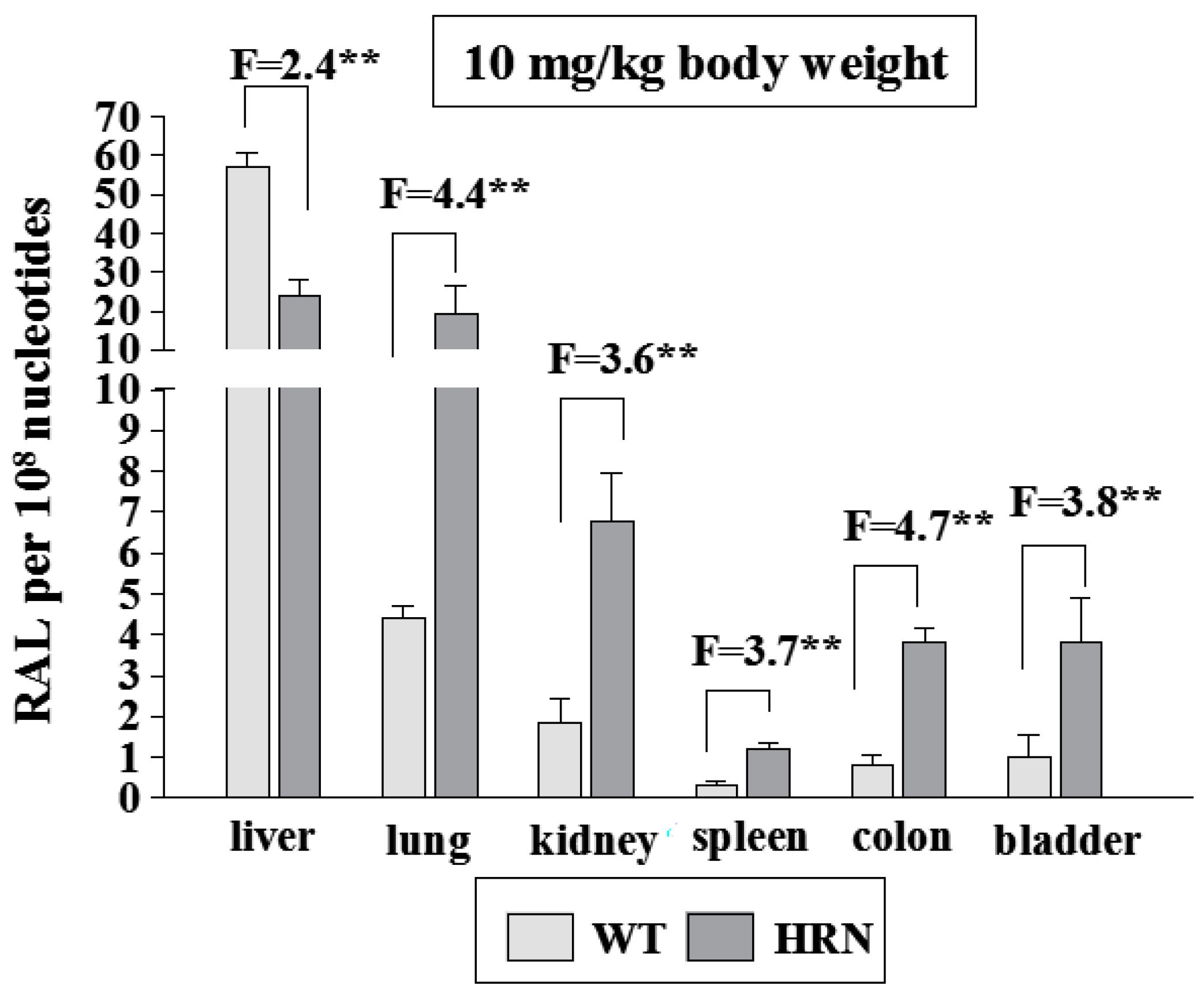
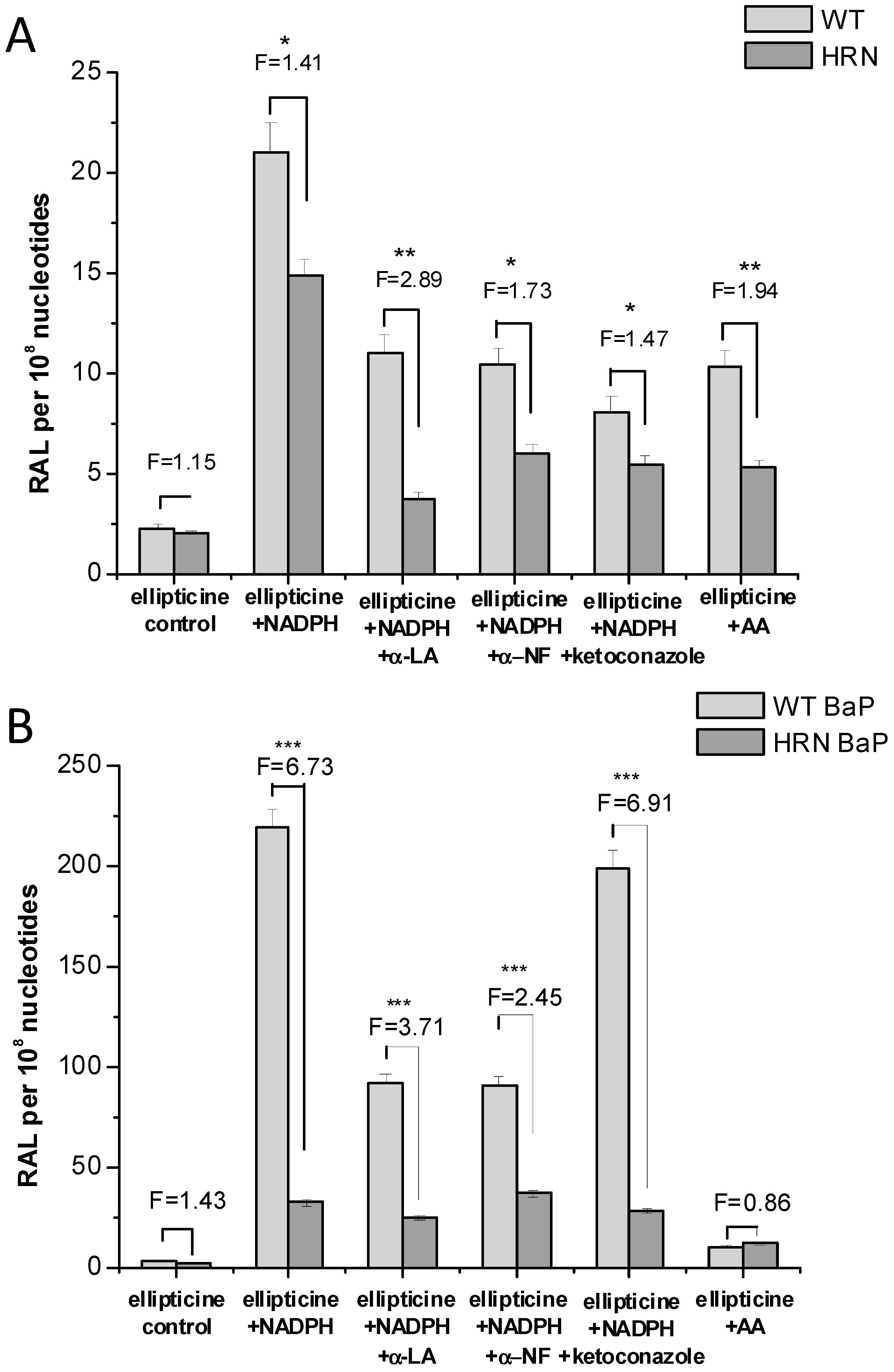
4.1. Utilization of Wild-Type (WT) and Hepatic P450 Reductase Null (HRN) Mice to Identify Enzymes Metabolizing Ellipticine in Vivo
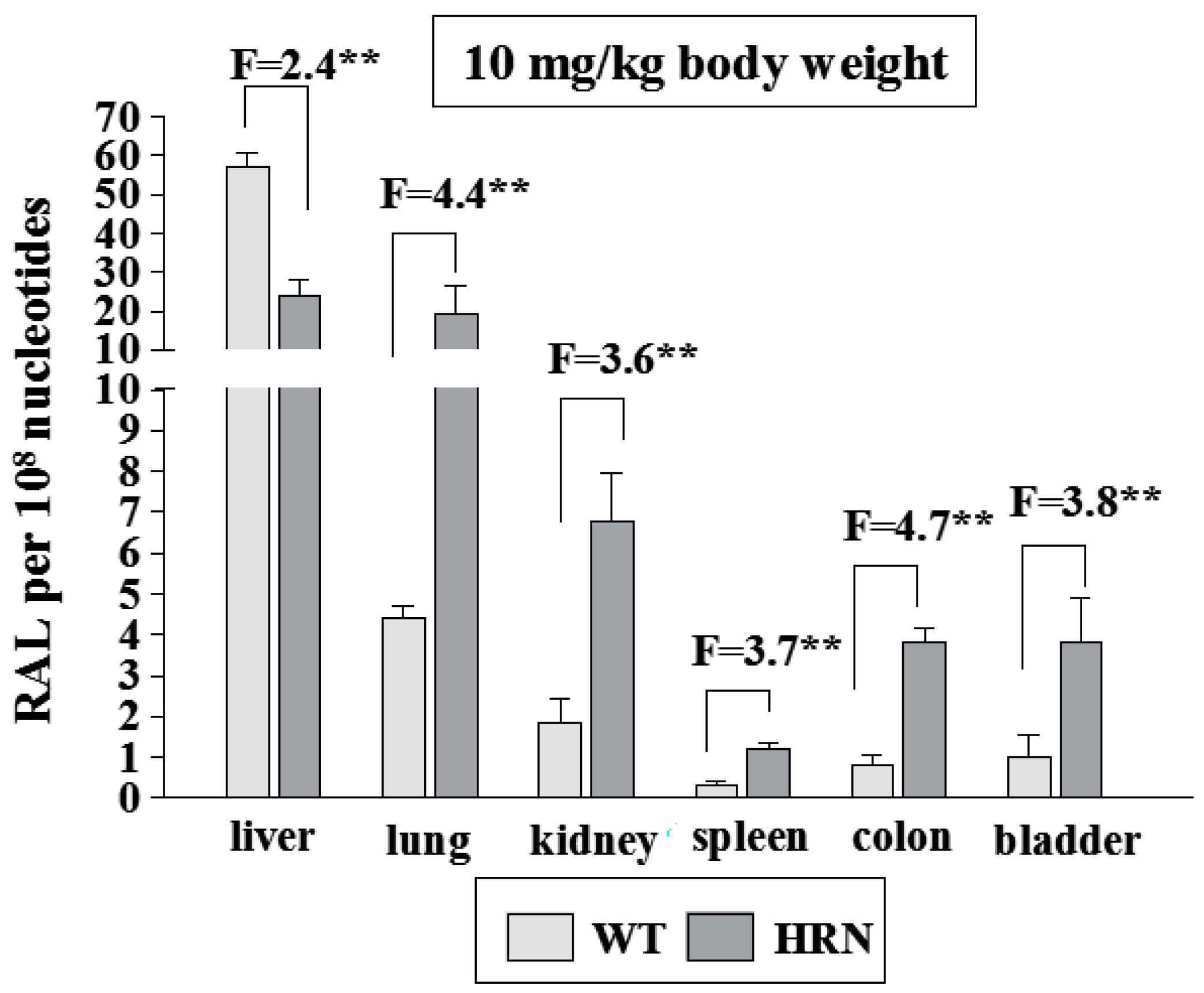
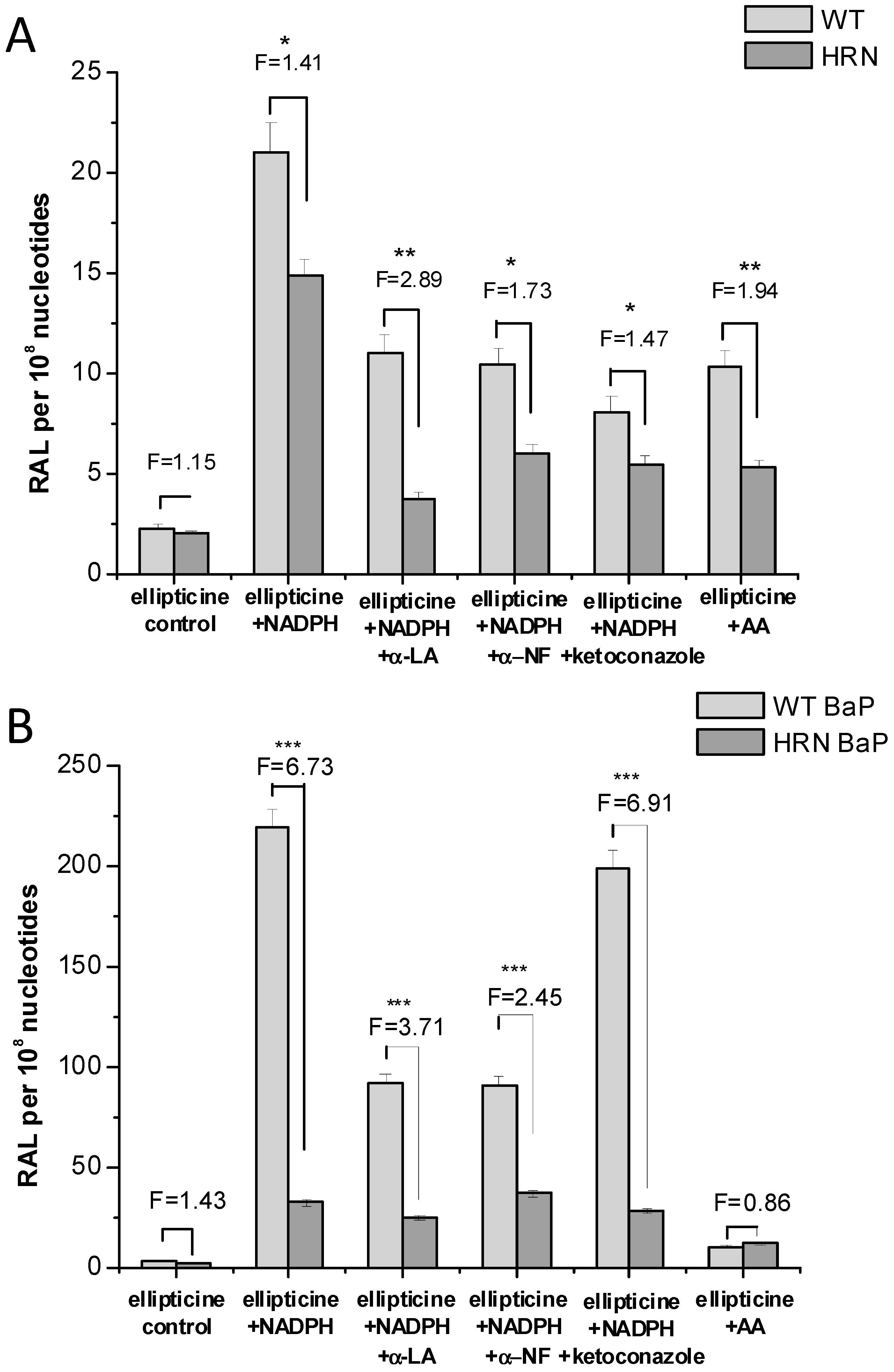

4.2. Ellipticine Metabolism in Wistar Rats
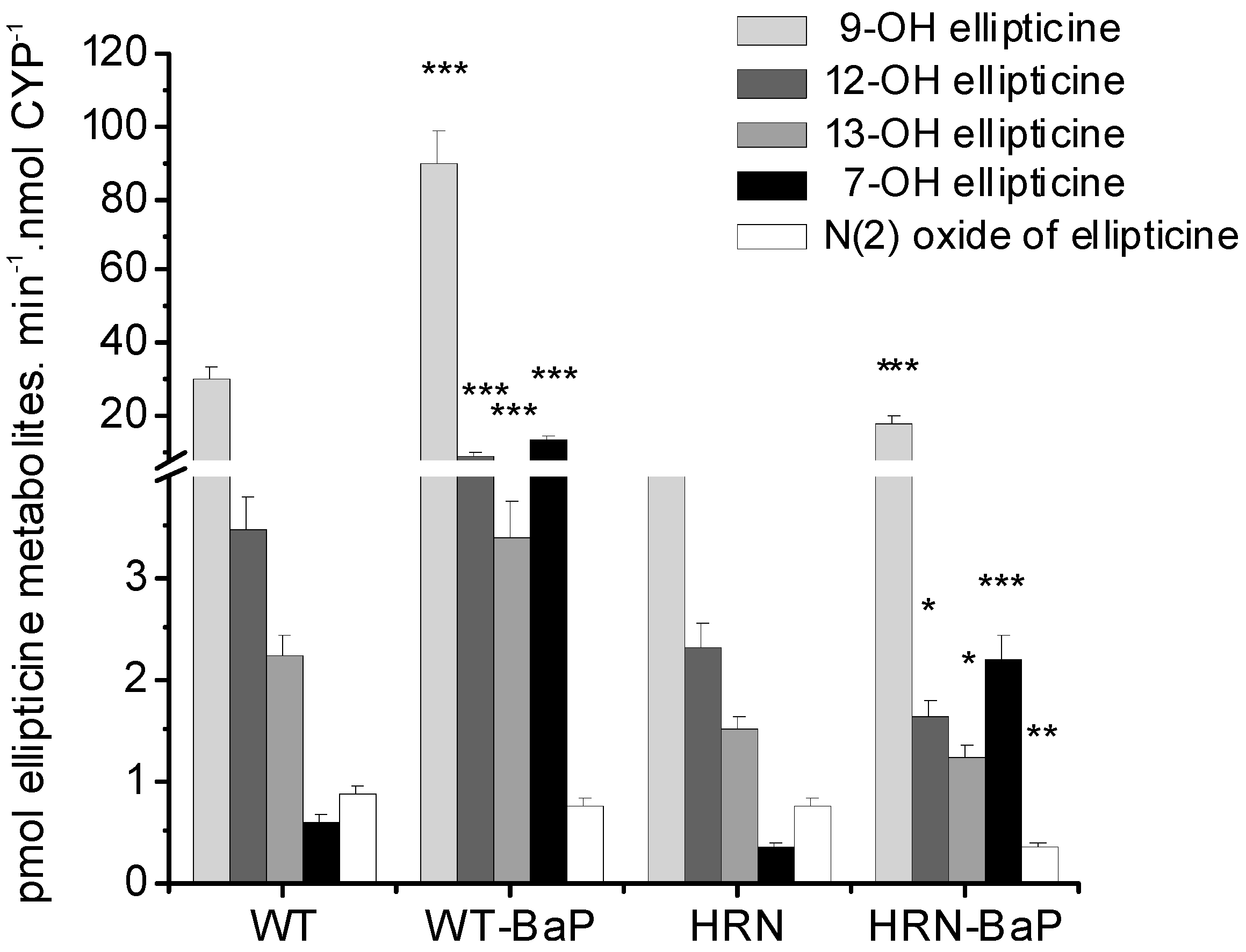
4.3. Studies with Human Microsomes and Cancer Cells
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stiborova, M.; Bieler, C.A.; Wiessler, M.; Frei, E. The anticancer agent ellipticine on activation by cytochrome P450 forms covalent DNA adducts. Biochem. Pharmacol. 2001, 62, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Rupertova, M.; Schmeiser, H.H.; Frei, E. Molecular mechanisms of antineoplastic action of an anticancer drug ellipticine. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2006, 150, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Rupertova, M.; Frei, E. Cytochrome P450- and peroxidase-mediated oxidation of anticancer alkaloid ellipticine dictates its anti-tumor efficiency. Biochim. Biophys. Acta 2011, 1814, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Kizek, R.; Adam, V.; Hrabeta, J.; Eckschlager, T.; Smutny, S.; Burda, J.V.; Frei, E.; Stiborova, M. Anthracyclines and ellipticines as DNA-damaging anticancer drugs: Recent advances. Pharmacol. Ther. 2012, 133, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Auclair, C. Multimodal action of antitumor agents on DNA: The ellipticine series. Arch. Biochem. Biophys. 1987, 259, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Garbett, N.C.; Graves, D.E. Extending nature’s leads: The anticancer agent ellipticine. Curr. Med. Chem. Anti-Cancer Agents 2004, 4, 149–172. [Google Scholar] [CrossRef]
- Kuo, P.L.; Hsu, Y.L.; Chang, C.H.; Lin, C.C. The antiproliferative inhibition of ellipticine in human breast mda-mb-231 cancer cells is through cell cycle arrest and apoptosis induction. Anti-Cancer Drugs 2005, 16, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Russell, E.G.; O’Sullivan, E.C.; Miller, C.M.; Stanicka, J.; McCarthy, F.O.; Cotter, T.G. Ellipticine derivative induces potent cytostatic effect in acute myeloid leukaemia cells. Investig. New Drugs 2014, 32, 1113–1122. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, S.G.; Chung, J.Y.; Kim, Y.J.; Park, J.E.; Koh, H.; Han, M.S.; Park, Y.C.; Yoo, Y.H.; Kim, J.M. Ellipticine induces apoptosis in human endometrial cancer cells: The potential involvement of reactive oxygen species and mitogen-activated protein kinases. Toxicology 2011, 289, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.L.; Hsu, Y.L.; Chang, C.H.; Lin, C.C. The mechanism of ellipticine-induced apoptosis and cell cycle arrest in human breast MCF-7 cancer cells. Cancer Lett. 2005, 223, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Sugikawa, E.; Nakanishi, N. Inhibition of p53 protein phosphorylation by 9-hydroxyellipticine: A possible anticancer mechanism. Jpn. J. Cancer Res. 1995, 86, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.M.; Myers, T.G.; Fan, Y.; O’Conno, R.P.M.; Paull, K.D.; Friend, S.H.; Weinstein, J.N. Mining the National Cancer Institute Anticancer Drug Discovery Database: Cluster analysis of ellipticine analogs with p53-inverse and central nervous system-selective patterns of activity. Mol. Pharmacol. 1998, 53, 241–251. [Google Scholar] [PubMed]
- Kuo, P.L.; Kuo, Y.C.; Hsu, Y.L.; Cho, C.Y.; Lin, C.C. Ellipticine induced apoptosis through p53-dependent pathway in human hepatocellular carcinoma HepG2 cells. Life Sci. 2006, 78, 2550–2557. [Google Scholar]
- Martinkova, E.; Maglott, A.; Leger, D.Y.; Bonnet, D.; Stiborova, M.; Takeda, K.; Martin, S.; Dontenwill, M. α5β1 integrin antagonists reduce chemotherapy-induced premature senescence and facilitate apoptosis in human glioblastoma cells. Int. J. Cancer 2010, 127, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Sugikawa, E.; Hosoi, T.; Yazaki, N.; Gamanuma, N.; Nakanishi, N.; Ohashi, M. Mutant p53 mediated induction of cell cycle arrest and apoptosis at G1 phase by 9-hydroxyellipticine. Anticancer Res. 1999, 19, 3099–3108. [Google Scholar] [PubMed]
- Peng, Y.; Li, C.; Chen, L.; Sebti, S.; Chen, J. Rescue of mutant p53 transcription function by ellipticine. Oncogene 2003, 22, 4478–4487. [Google Scholar] [CrossRef] [PubMed]
- Savorani, C.; Manfé, V.; Biskup, E.; Gniadecki, R. Ellipticine induces apoptosis in T-cell lymphoma via oxidative DNA damage. Leuk. Lymphoma 2014, 4, 1–9. [Google Scholar] [CrossRef]
- Fang, K.; Chen, S.P.; Lin, C.W.; Cheng, W.C.; Huang, H.T. Ellipticine-induced apoptosis depends on Akt translocation and signaling in lung epithelial cancer cells. Lung Cancer 2009, 63, 227–234. [Google Scholar] [CrossRef]
- Wang, F.; Liu, J.; Robbins, D.; Morris, K.; Sit, A.; Liu, Y.Y.; Zhao, Y. Mutant p53 exhibits trivial effects on mitochondrial functions which can be reactivated by ellipticine in lymphoma cells. Apoptosis 2011, 16, 301–310. [Google Scholar] [CrossRef]
- Miller, C.M.; McCarthy, F.O. Isolation, biological activity and synthesis of the natural product ellipticine and related pyridocarbazoles. RSC Adv. 2012, 2, 8883–8918. [Google Scholar] [CrossRef]
- Fritsche, M.; Haessler, C.; Brandner, G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene 1993, 8, 307–318. [Google Scholar] [PubMed]
- Xu, G.W.; Mawji, I.A.; Macrae, C.J.; Koch, C.A.; Datti, A.; Wrana, J.L.; Dennis, J.W.; Schimmer, A.D. A high-content chemical screen identifies ellipticine as a modulator of p53 nuclear localization. Apoptosis 2008, 13, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kohn, K.W.; Waring, M.J.; Glaubiger, D.; Friedman, C.A. Intercalative binding of ellipticine to DNA. Cancer Res. 1975, 35, 71–76. [Google Scholar] [PubMed]
- Patel, N.; Bergman, J.; Gräslund, A. 1H-NMR studies of the interaction between a self-complementary deoxyoligonucleotide duplex and indolo[2,3-b]quinoxaline derivatives active against herpes virus. Eur. J. Biochem. 1991, 197, 597–604. [Google Scholar] [PubMed]
- Chu, Y.; Hsu, M.T. Ellipticine increases the superhelical density of intracellular SV40 DNA by intercalation. Nucleic Acids Res. 1992, 20, 4033–4038. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.P.; Hill, G.C.; Peoch, D.; Rayner, B.; Inabach, J.L.; Lown, J.W. High-field NMR and restrained molecular modeling studies on a DNA heteroduplex containing a modified apurinic abasic site in the form of covalently linked 9-aminoellipticine. Biochemistry 1994, 33, 10271–10285. [Google Scholar] [CrossRef]
- Belehradek, J., Jr.; Fermandjian, S. DNA-drug recognition and effects on topoisomerase II-mediated cytotoxicity. A three-mode binding model for ellipticine derivatives. J. Biol. Chem. 1991, 266, 1820–1829. [Google Scholar] [PubMed]
- Froelich-Ammon, S.J.; Patchan, M.W.; Osheroff, N.; Thompson, R.B. Topoisomerase II binds to ellipticine in the absence or presence of DNA. Characterization of enzyme-drug interactions by fluorescence spectroscopy. J. Biol. Chem. 1995, 270, 14998–5004. [Google Scholar] [CrossRef] [PubMed]
- Andrews, W.J.; Panova, T.; Normand, C.; Gadal, O.; Tikhonova, I.G.; Panov, K.I. Old drug, new target: Ellipticines selectively inhibit RNA polymerase I transcription. J. Biol. Chem. 2013, 288, 4567–4582. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kar, A.; Chowdhury, S.; Dasgupta, D. Ellipticine binds to a human telomere sequence: An additional mode of action as a putative anticancer agent? Biochemistry 2013, 52, 4127–4137. [Google Scholar] [PubMed]
- Stiborová, M.; Sejbal, J.; Borek-Dohalská, L.; Aimová, D.; Poljaková, J.; Forsterová, K.; Rupertová, M.; Wiesner, J.; Hudecek, J.; Wiessler, M.; et al. The anticancer drug ellipticine forms covalent DNA adducts, mediated by human cytochromes P450, through metabolism to 13-hydroxyellipticine and ellipticine N2-oxide. Cancer Res. 2004, 64, 8374–8380. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Poljaková, J.; Ryslavá, H.; Dracínský, M.; Eckschlager, T.; Frei, E. Mammalian peroxidases activate anticancer drug ellipticine to intermediates forming deoxyguanosine adducts in DNA identical to those found in vivo and generated from 12-hydroxyellipticine and 13-hydroxyellipticine. Int. J. Cancer 2007, 120, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Rupertová, M.; Aimová, D.; Ryslavá, H.; Frei, E. Formation and persistence of DNA adducts of anticancer drug ellipticine in rats. Toxicology 2007, 236, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Indra, R.; Moserová, M.; Cerná, V.; Rupertová, M.; Martínek, V.; Eckschlager, T.; Kizek, R.; Frei, E. Cytochrome b5 increases cytochrome P450 3A4-mediated activation of anticancer drug ellipticine to 13-hydroxyellipticine whose covalent binding to DNA is elevated by sulfotransferases and N,O-acetyltransferases. Chem. Res. Toxicol. 2012, 25, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kotrbová, V.; Mrázová, B.; Moserová, M.; Martínek, V.; Hodek, P.; Hudeček, J.; Frei, E.; Stiborová, M. Cytochrome b5 shifts oxidation of the anticancer drug ellipticine by cytochromes P450 1A1 and 1A2 from its detoxication to activation, thereby modulating its pharmacological efficacy. Biochem. Pharmacol. 2011, 82, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Frei, E. Ellipticines as DNA-targeted chemotherapeutics. Curr. Med. Chem. 2014, 21, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Breuer, A.; Aimová, D.; Stiborová-Rupertová, M.; Wiessler, M.; Frei, E. DNA adduct formation by the anticancer drug ellipticine in rats determined by 32P-postlabeling. Int. J. Cancer 2003, 107, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Stiborová-Rupertová, M.; Bořek-Dohalská, L.; Wiessler, M.; Frei, E. Rat microsomes activating the anticancer drug ellipticine to species covalently binding to deoxyguanosine in DNA are a suitable model mimicking ellipticine bioactivation in humans. Chem. Res. Toxicol. 2003, 16, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Poljakova, J.; Martínková, E.; Ulrichová, J.; Šimánek, V.; Dvořák, Z.; Frei, E. Ellipticine oxidation and DNA adduct formation in human hepatocytes is catalyzed by human cytochromes P450 and enhanced by cytochrome b5. Toxicology 2012, 302, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Kotrbová, V.; Aimová, D.; Březinová, A.; Janouchová, K.; Poljaková, J.; Hodek, P.; Frei, E.; Stiborová, M. Cytochromes P450 reconstituted with NADPH:P450 reductase mimic the activating and detoxicating metabolism of the anticancer drug ellipticine in microsomes. Neuro Endocrinol. Lett. 2006, 27 (Suppl. 2), 18–20. [Google Scholar]
- Stiborova, M.; Poljakova, J.; Mrizova, I.; Borek-Dohalska, L.; Eckschlager, T.; Adam, V.; Kizek, R.; Frei, E. Expression levels of enzymes metabolizing an anticancer drug ellipticine determined by electromigration assays influence its cytotoxicity to cancer cells—A comparative study. Int. J. Electrochem. Sci. 2014, 9, 5675–5689. [Google Scholar]
- Poljaková, J.; Frei, E.; Gomez, J.E.; Aimová, D.; Eckschlager, T.; Hraběta, J.; Stiborová, M. DNA adduct formation by the anticancer drug ellipticine in human leukemia HL-60 and CCRF-CEM cells. Cancer Lett. 2007, 252, 270–279. [Google Scholar] [CrossRef]
- Poljaková, J.; Eckschlager, T.; Hraběta, J.; Hřebačková, J.; Smutný, S.; Frei, E.; Martínek, V.; Kizek, R.; Stiborová, M. The mechanism of cytotoxicity and DNA adduct formation by the anticancer drug ellipticine in human neuroblastoma cells. Biochem. Pharmacol. 2009, 77, 1466–1479. [Google Scholar] [CrossRef] [PubMed]
- Poljakova, J.; Hrebackova, J.; Dvořákova, M.; Moserova, M.; Eckschlager, T.; Hrabeta, J.; Göttlicherova, M.; Kope Jtkova, B.; Frei, E.; Kizek, R.; et al. Anticancer agent ellipticine combined with histone deacetylase inhibitors, valproic acid and trichostatin A, is an effective DNA damage strategy in human neuroblastoma. Neuro Endocrinol. Lett. 2011, 32 (Suppl. 1), 101–116. [Google Scholar] [PubMed]
- Poljaková, J.; Eckschlager, T.; Kizek, R.; Frei, E.; Stiborová, M. Electrochemical determination of enzymes metabolizing ellipticine in thyroid cancer cells—A tool to explain the mechanism of ellipticine toxicity to these cells. Int. J. Electrochem. Sci. 2013, 8, 1573–1585. [Google Scholar]
- Martinkova, E.; Dontenwill, M.; Frei, E.; Stiborová, M. Cytotoxicity of and DNA adduct formation by ellipticine in human U87MG glioblastoma cancer cells. Neuro Endocrinol. Lett. 2009, 30 (Suppl. 1), 60–66. [Google Scholar] [PubMed]
- Poljaková, J.; Dračínský, M.; Frei, E.; Hudeček, J.; Stiborová, M. The effect of pH on peroxidase-mediated oxidation of and DNA-adduct formation by ellipticine. Collect. Czech Chem. Commun. 2006, 71, 1169–1185. [Google Scholar] [CrossRef]
- Stiborová, M.; Arlt, V.M.; Henderson, C.J.; Wolf, C.R.; Kotrbová, V.; Moserová, M.; Hudecek, J.; Phillips, D.H.; Frei, E. Role of hepatic cytochromes P450 in bioactivation of the anticancer drug ellipticine: Studies with the hepatic NADPH:Cytochrome P450 reductase null mouse. Toxicol. Appl. Pharmacol. 2008, 226, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Moserová, M.; Mrázová, B.; Kotrbová, V.; Frei, E. Role of cytochromes P450 and peroxidases in metabolism of the anticancer drug ellipticine: Additional evidence of their contribution to ellipticine activation in rat liver, lung and kidney. Neuro Endocrinol. Lett. 2010, 31 (Suppl. 2), 26–35. [Google Scholar]
- Stiborová, M.; Eckschlager, T.; Poljaková, J.; Hraběta, J.; Adam, V.; Kizek, R.; Frei, E. The synergistic effects of DNA-targeted chemotherapeutics and histone deacetylase inhibitors as therapeutic strategies for cancer treatment. Curr. Med. Chem. 2012, 19, 4218–4238. [Google Scholar] [CrossRef] [PubMed]
- Vranová, I.; Moserová, M.; Hodek, P.; Kizek, R.; Frei, E.; Stiborová, M. The anticancer drug ellipticine induces cytochromes P450 1A1, 1A2 and 3A, cytochrome b5 and NADPH:Cytochrome P450 in rat liver, kidney and lung. Int. J. Electrochem. Sci. 2013, 8, 1586–1597. [Google Scholar]
- Chadwick, M.; Silveira, D.M.; Platz, B.R.; Hayes, D. Comparative physiological disposition of ellipticine in several animal species after intravenous administration. Drug Metab. Dispos. 1978, 6, 528–541. [Google Scholar] [PubMed]
- Branfam, A.R.; Bruni, R.J.; Reihold, V.N.; Silveira, D.M.; Chadwick, M.; Yesair, D.W. Characterization of metabolites of ellipticine in rat bile. Drug Metab. Dispos. 1978, 6, 542–548. [Google Scholar] [PubMed]
- Ismail, M.A.; Sanders, K.J.; Fennell, G.C.; Latham, H.C.; Wormell, P.; Rodger, A. Spectroscopic studies of 9-hydroxyellipticine binding to DNA. Biopolymers 1998, 46, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Fossé, P.; René, B.; le Bret, M.; Paoletti, C.; Saucier, J.M. Sequence requirements for mammalian topoisomerase II mediated DNA cleavage stimulated by an ellipticine derivative. Nucleic Acids Res. 1991, 19, 2861–2868. [Google Scholar] [CrossRef] [PubMed]
- Fossé, P.; René, B.; Charra, M.; Paoletti, C.; Saucier, J.M. Stimulation of topoisomerase II-mediated DNA cleavage by ellipticine derivatives: Structure-activity relationships. Mol. Pharmacol. 1992, 42, 590–595. [Google Scholar] [PubMed]
- Hofle, G.; Glase, N.; Leibold, T.; Sefkow, M. Epothilone A–D and their thiazole-modified analogs as novel anticancer agents. Pure Appl. Chem. 1999, 71, 2019–2024. [Google Scholar] [CrossRef]
- Moserova, M.; Kotrbova, V.; Rupertova, M.; Naiman, K.; Hudecek, J.; Hodek, P.; Frei, E.; Stiborova, M. Isolation and partial characterization of the adduct formed by 13-hydroxyellipticine with deoxyguanosine in DNA. Neuro Endocrinol. Lett. 2008, 29, 728–732. [Google Scholar] [PubMed]
- Martínek, V.; Sklenár, J.; Dracínsky, M.; Sulc, M.; Hofbauerová, K.; Bezouska, K.; Frei, E.; Stiborová, M. Glycosylation protects proteins against free radicals generated from toxic xenobiotics. Toxicol. Sci. 2010, 117, 59–74. [Google Scholar]
- Stiborová, M.; Borek-Dohalská, L.; Aimová, D.; Kotrbová, V.; Kukacková, K.; Janouchová, K.; Rupertová, M.; Ryslavá, H.; Hudecek, J.; Frei, E. Oxidation pattern of the anticancer drug ellipticine by hepatic microsomes—Similarity between human and rat systems. Gen. Physiol. Biophys. 2006, 25, 245–261. [Google Scholar]
- Stiborova, M.; Cerna, V.; Moserova, M.; Arlt, V.M.; Frei, E. The effect of benzo[a]pyrene on metabolic activation of anticancer drug ellipticine in mice. Neuro Endocrinol. Lett. 2013, 34 (Suppl. 2), 43–54. [Google Scholar]
- Henderson, C.J.; Otto, D.M.; Carrie, D.; Magnuson, M.A.; McLaren, A.W.; Rosewell, I.; Wolf, C.R. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J. Biol. Chem. 2003, 278, 13480–13486. [Google Scholar] [CrossRef]
- Arlt, V.M.; Stiborova, M.; Henderson, C.J.; Osborne, M.R.; Bieler, C.A.; Frei, E.; Martinek, V.; Sopko, B.; Wolf, C.R.; Schmeiser, H.H.; et al. Environmental pollutant and potent mutagen 3-nitrobenzanthrone forms DNA adducts after reduction by NAD(P)H:quinone oxidoreductase and conjugation by acetyltransferases and sulfotransferases in human hepatic cytosols. Cancer Res. 2005, 65, 2644–2652. [Google Scholar] [CrossRef]
- Arlt, V.M.; Henderson, C.J.; Wolf, C.R.; Schmeiser, H.H.; Phillips, D.H.; Stiborova, M. Bioactivation of 3-aminobenzanthrone, a human metabolite of the environmental pollutant 3-nitrobenzanthrone: Evidence for DNA adduct formation mediated by cytochrome P450 enzymes and peroxidases. Cancer Lett. 2006, 234, 220–2231. [Google Scholar] [CrossRef]
- Arlt, V.M.; Stiborová, M.; Henderson, C.J.; Thiemann, M.; Frei, E.; Aimová, D.; Singh, R.; Gamboa da Costa, G.; Schmitz, O.J.; Farmer, P.B.; et al. Metabolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo: Experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis 2008, 29, 656–665. [Google Scholar] [CrossRef]
- Arlt, V.M.; Poirier, M.C.; Sykes, S.E.; John, K.; Moserova, M.; Stiborova, M.; Wolf, C.R.; Henderson, C.J.; Phillips, D.H. Exposure to benzo[a]pyrene of Hepatic Cytochrome P450 Reductase Null (HRN) and P450 Reductase Conditional Null (RCN) mice: Detection of benzo[a]pyrene diol epoxide-DNA adducts by immunohistochemistry and 32P-postlabelling. Toxicol. Lett. 2012, 213, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Pass, G.J.; Carrie, D.; Boylan, M.; Lorimore, S.; Wright, E.; Houston, B.; Henderson, C.J.; Wolf, C.R. Role of hepatic cytochrome P450s in the pharmacokinetics and toxicity of cyclophosphamide: Studies with the hepatic cytochrome P450 reductase null mouse. Cancer Res. 2005, 65, 4211–4217. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ge, M.; Xue, X.; Wang, H.; Wu, X.; Li, L.; Liu, L.; Qi, X.; Zhang, Y.; Li, Y.; et al. Detoxication role of hepatic cytochrome P450s in the kidney toxicity induced by aristolochic acid. Kidney Int. 2008, 73, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Levová, K.; Moserová, M.; Kotrbová, V.; Šulc, M.; Henderson, C.J.; Wolf, C.R.; Phillips, D.H.; Frei, E.; Schmeiser, H.H.; Mareš, J.; et al. Role of cytochromes P450 1A1/2 in detoxication and activation of carcinogenic aristolochic acid I: Studies with the hepatic NADPH:cytochrome P450 reductase null (HRN) mouse model. Toxicol. Sci. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Eling, T.E.; Thompson, D.C.; Foureman, G.L.; Curtis, J.F.; Hughes, M.F. Prostaglandin H synthase and xenobiotic oxidation. Annu. Rev. Pharmacol. Toxicol. 1990, 30, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Eling, T.E.; Curtis, J.F. Xenobiotic metabolism by prostaglandin H synthase. Pharm. Ther. 1992, 53, 261–273. [Google Scholar] [CrossRef]
- Stiborova, M.; Frei, E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human hepatic and renal microsomes, cytochromes P450 1A1/2, NADPH:cytochrome P450 reductase and prostaglandin H synthase mediate the formation of aristolochic acid-DNA adducts found in patients with urothelial cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Downie, D.; McFadyen, M.C.; Rooney, P.H.; Cruickshank, M.E.; Parkin, D.E.; Miller, I.D.; Telfer, C.; Melvin, W.T.; Murray, G.I. Profiling cytochrome P450 expression in ovarian cancer: Identification of prognostic markers. Clin. Cancer Res. 2005, 11, 7369–7735. [Google Scholar] [CrossRef] [PubMed]
- Saarikoski, T.; Rivera, S.P.; Hankinson, O.; Husgafvel-Pursiainen, K. CYP2S1: A short review. Toxicol. Appl. Pharmacol. 2005, 207, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Bui, P.H.; Hankinson, O. Functional characterization of human cytochrome P450 2S1 using a synthetic gene-expressed protein in Escherichia coli. Mol. Pharmacol. 2009, 76, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Bui, P.H.; Hsu, E.L.; Hankinson, O. Fatty acid hydroperoxides support cytochrome P450 2S1-mediated bioactivation of benzo[a]pyrene-7,8-dihydrodiol. Mol. Pharmacol. 2009, 76, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Mrizová, I.; Moserová, M.; Milichovsky, J.; Šulc, M.; Guengerich, F.P.; Stiborová, M. Heterologous expression of cytochrome P450 2S1 in Escherichia coli. Interdisc. Toxicol. 2014, 7 (Suppl. 1), 66. [Google Scholar]
- Stiborova, M.; Moserova, M.; Černá, V.; Indra, R.; Dračínský, M.; Šulc, M.; Henderson, C.J.; Wolf, C.R.; Schmeiser, H.H.; Phillips, D.H.; et al. Cytochrome b5 and epoxide hydrolase contribute to benzo[a]pyrene-DNA adduct formation catalyzed by cytochrome P450 1A1 under low NADPH:P450 oxidoreductase conditions. Toxicology 2014, 318, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; DiCarlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–480. [Google Scholar] [CrossRef] [PubMed]
- Ueng, Y.-F.; Kuwabara, T.; Chun, Y.-J.; Guengerich, F.P. Cooperativity in oxidation catalyzed by cytochrome P450 3A4. Biochemistry 1997, 36, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Patterson, L.H.; McKeown, S.R.; Robson, T.; Gallagher, R.; Raleigh, S.M.; Orr, S. Antitumor prodrug development using cytochrome P450 (CYP) mediated activation. Anti-Cancer Drug Des. 1999, 14, 473–486. [Google Scholar]
- Murray, G.I.; Melvin, W.T.; Burke, M. Cytochrome P450 expression in tumors. J. Pathol. 1995, 176, 323–324. [Google Scholar]
- El-Rayes, B.F.; Ali, S.; Heilbrun, L.K.; Lababidi, S.; Bouwman, S.; Vischer, D.; Philip, P.A. Cytochrome P450 and glutathione transferase expression in human breast cancer. Clin. Cancer Res. 2003, 9, 1705–1709. [Google Scholar]
- Godschalk, R.W.L.; Moonen, E.J.C.; Schilderman, P.A.E.L.; Broekmans, W.M.R.; Kleinjans, J.C.S.; van Schooten, F.J. Exposure-route-dependent DNA adduct formation by polycyclic aromatic hydrocarbons. Carcinogenesis 2000, 21, 87–92. [Google Scholar] [PubMed]
- Poirier, M.C. Chemical-induced DNA damage and human cancer risk. Nat. Rev. 2004, 4, 630–637. [Google Scholar] [CrossRef]
- Randerath, K.; Haglund, R.E.; Phillips, D.H.; Reddy, M.V. 32P-post-labelling analysis of DNA adducts formed in the livers of animals treated with safrole, estragole and other naturally-occurring alkenylbenzenes. I. Adult female CD-1 mice. Carcinogenesis 1984, 5, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Nelson, G.B.; Wilson, K.H.; Rabinowitz, J.R.; Galati, A.; Stoner, G.D.; Nesnow, S.; Mass, M.J. Adenomas induced by polycyclic aromatic hydrocarbons in strain A/J mouse lung correlate with time-integrated DNA adduct levels. Cancer Res. 1995, 55, 1039–1044. [Google Scholar] [PubMed]
- Smith, B.A.; Fullerton, N.F.; Heflich, R.H.; Beland, F.A. DNA adduct formation and T-lymphocyte mutation induction in F344 rats implanted with tumorigenic doses of 1,6-dinitropyrene. Cancer Res. 1995, 55, 2316–2324. [Google Scholar] [PubMed]
- Yamazaki, H.; Gillam, E.M.; Dong, M.S.; Johnson, W.W.; Guengerich, F.P.; Shimada, T. Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: Effects of cytochrome P450–P450 and cytochrome P450–b5 interactions. Arch. Biochem. Biophys. 1997, 342, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Shimada, T.; Martin, M.V.; Guengerich, F.P. Stimulation of cytochrome P450 reactions by apo-cytochrome b5: Evidence against transfer of heme from cytochrome P450 3A4 to apo-cytochrome b5 or heme oxygenase. J. Biol. Chem. 2001, 276, 30885–30891. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, J.B.; Jansson, I. The many roles of cytochrome b5. Pharmacol. Ther. 2003, 97, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Myshkin, E.; Waskell, L. Role of cytochrome b5 in catalysis by cytochrome P450 2B4. Biochem. Biophys. Res. Commun. 2005, 338, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Im, S.C.; Waskell, L. Cytochrome b5 increases the rate of product formation by cytochrome P450 2B4 and competes with cytochrome P450 reductase for a binding site on cytochrome P450 2B4. J. Biol. Chem. 2007, 282, 29766–29776. [Google Scholar] [CrossRef] [PubMed]
- Kotrbová, V.; Aimová, D.; Ingr, M.; Borek-Dohalská, L.; Martínek, V.; Stiborová, M. Preparation of a biologically active apo-cytochrome b5 via heterologous expression in Escherichia coli. Protein Expr. Purif. 2009, 66, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; Guengerich, F.P. Contributions of human enzymes in carcinogen metabolism. Chem. Res. Toxicol. 2012, 25, 1316–1383. [Google Scholar] [CrossRef] [PubMed]
- Aimová, D.; Svobodová, L.; Kotrbová, V.; Mrázová, B.; Hodek, P.; Hudeček, J.; Václavíková, R.; Frei, E.; Stiborová, M. The anticancer drug ellipticine is a potent inducer of rat cytochromes P450 1A1 and 1A2, thereby modulating its own metabolism. Drug Metab. Dispos. 2007, 35, 1926–1934. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; McLaughlin, L.A.; Ronseaux, S.; Rosewell, I.; Houston, J.B.; Henderson, C.J.; Wolf, C.R. Defining the in vivo role for cytochrome b5 in cytochrome P450 function through the conditional hepatic deletion of microsomal cytochrome b5. J. Biol. Chem. 2008, 283, 31385–31393. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.J.; McLaughlin, L.A.; Wolf, C.R. Evidence that cytochrome b5 and cytochrome b5 reductase can act as sole electron donors to the hepatic cytochrome P450 system. Mol. Pharmacol. 2013, 83, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Waxman, D.J. Collaboration between hepatic and intratumoral prodrug activation in a P450 prodrug-activation gene therapy model for cancer treatment. Mol. Cancer Ther. 2007, 6, 2879–2890. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, C.S.; Waxman, D.J. Potentiation of methoxymorpholinyl doxorubicin antitumor activity by P450 3A4 gene transfer. Cancer Gene Ther. 2009, 16, 393–404. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiborová, M.; Černá, V.; Moserová, M.; Mrízová, I.; Arlt, V.M.; Frei, E. The Anticancer Drug Ellipticine Activated with Cytochrome P450 Mediates DNA Damage Determining Its Pharmacological Efficiencies: Studies with Rats, Hepatic Cytochrome P450 Reductase Null (HRN™) Mice and Pure Enzymes. Int. J. Mol. Sci. 2015, 16, 284-306. https://doi.org/10.3390/ijms16010284
Stiborová M, Černá V, Moserová M, Mrízová I, Arlt VM, Frei E. The Anticancer Drug Ellipticine Activated with Cytochrome P450 Mediates DNA Damage Determining Its Pharmacological Efficiencies: Studies with Rats, Hepatic Cytochrome P450 Reductase Null (HRN™) Mice and Pure Enzymes. International Journal of Molecular Sciences. 2015; 16(1):284-306. https://doi.org/10.3390/ijms16010284
Chicago/Turabian StyleStiborová, Marie, Věra Černá, Michaela Moserová, Iveta Mrízová, Volker M. Arlt, and Eva Frei. 2015. "The Anticancer Drug Ellipticine Activated with Cytochrome P450 Mediates DNA Damage Determining Its Pharmacological Efficiencies: Studies with Rats, Hepatic Cytochrome P450 Reductase Null (HRN™) Mice and Pure Enzymes" International Journal of Molecular Sciences 16, no. 1: 284-306. https://doi.org/10.3390/ijms16010284