The Role of Pericytes in Neurovascular Unit Remodeling in Brain Disorders

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
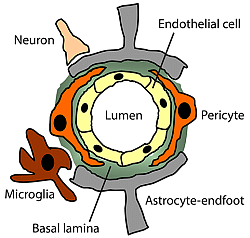
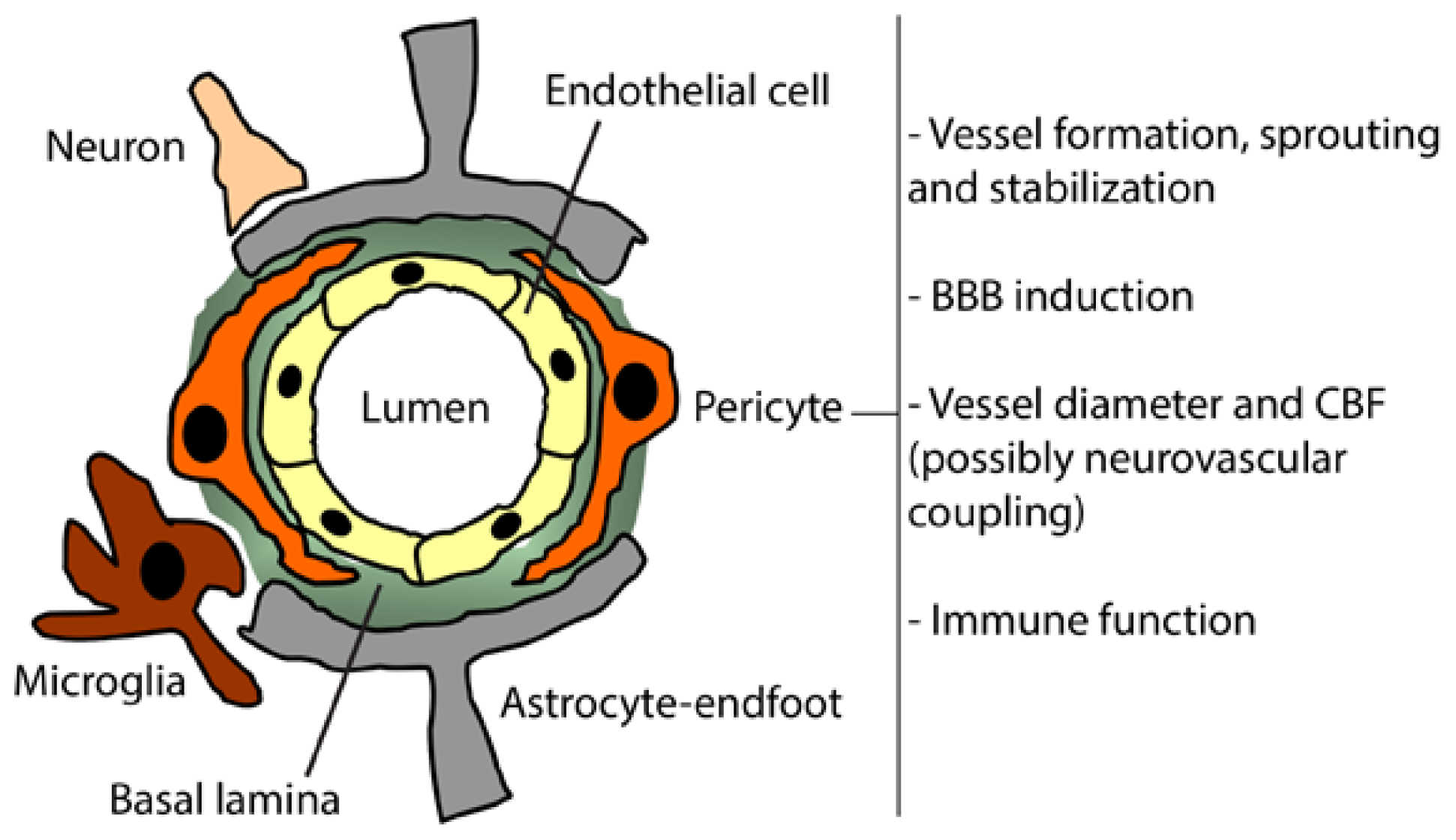
2. Pericytes
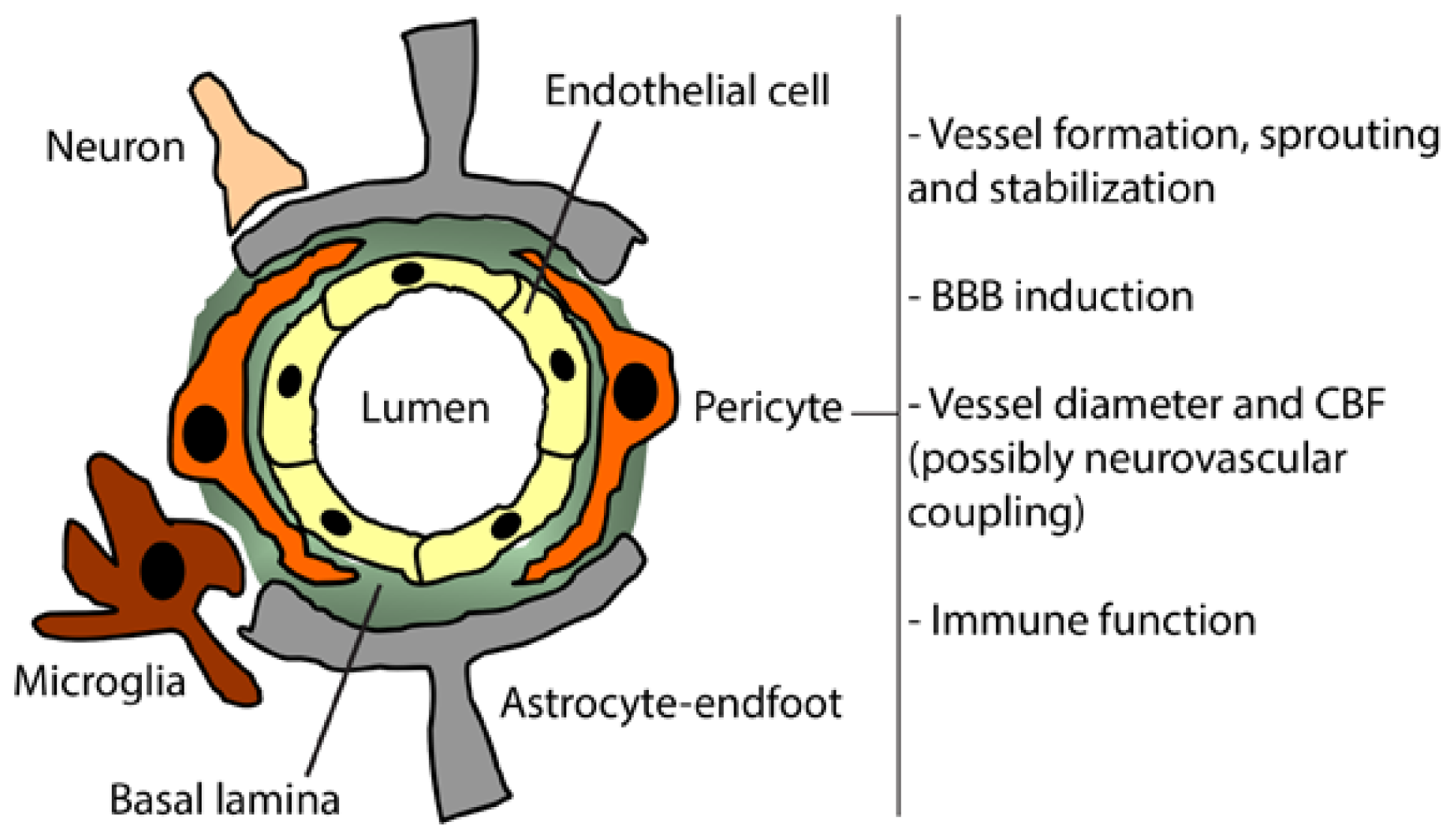
3. Pericyte Function in the Healthy Brain
3.1. Brain Vessels Sprouting
3.2. BBB Formation and Induction
3.3. Brain Vessel Diameter and CBF Regulations
3.4. Immune Function
3.5. Pluripotent Cells
4. Pericyte Interactions at the Neurovascular Unit
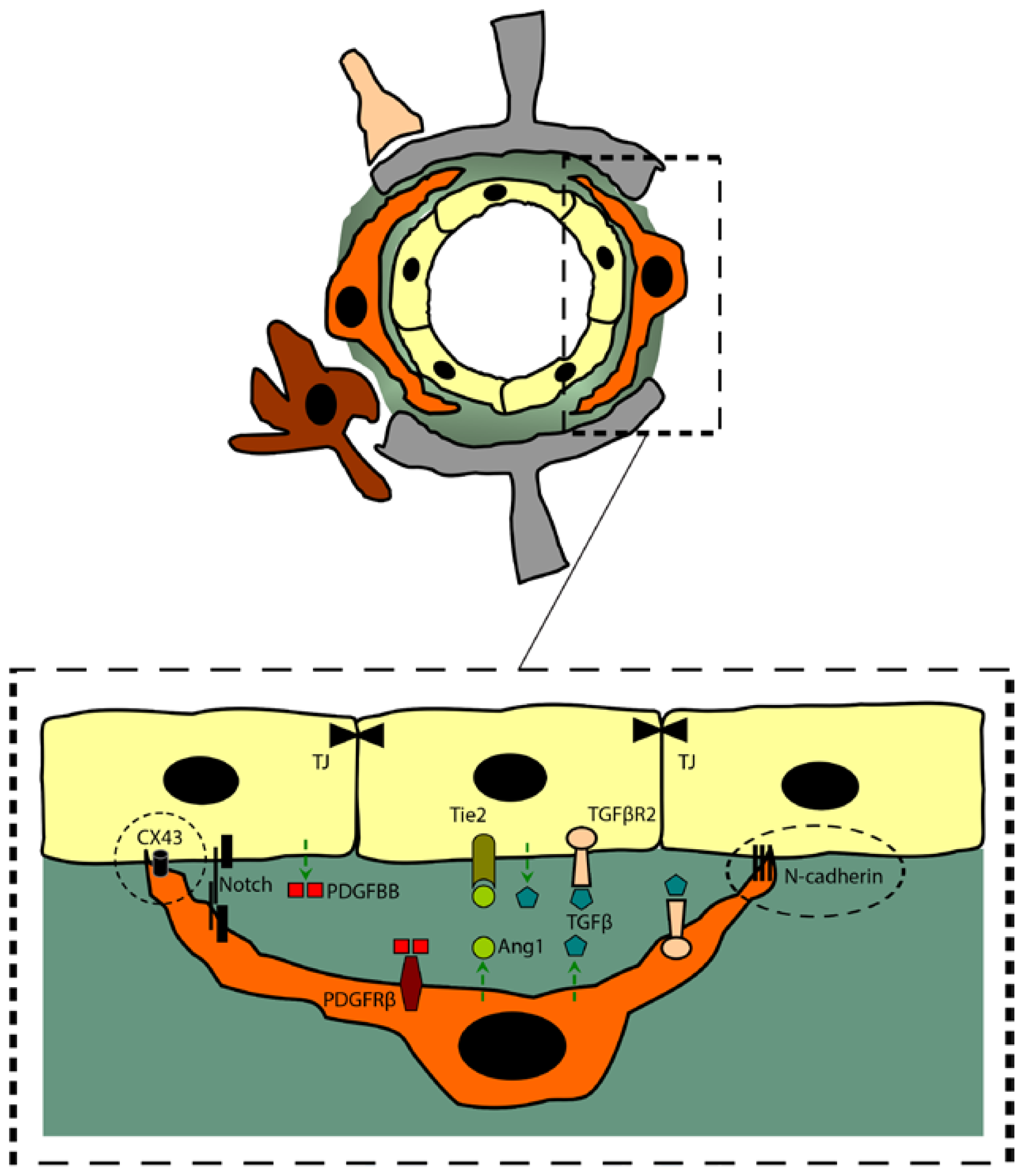
4.1. Cell-to-Cell Interactions
4.2. Signaling Pathways
- (a)
- PDGFBB/PDGFRβ: The platelet-derived growth factor B (PDGFB) signaling through PDGFRβ plays a crucial role in endothelial-to-pericyte interaction, specifically in the recruitment of brain pericytes into the abluminal side of endothelial cells [50]. In angiogenic context, endothelial cells secrete PDGFB in an active homodimer form (PDGFBB), promoting the proliferation and migration of PDGFRβ-expressing pericytes within the angiogenic endothelial sprouts [51]. PDGFRβ is a tyrosine kinase receptor that is expressed on developing and mature pericytes [11]. PDGFBB binding to PDGFRβ induces the latter’s dimerization, autophosphorylation, activating several downstream signaling cascades that include survival pathways [20,51]. Moreover, it has been shown that the recruitment and attachment of pericytes to the abluminal side of the nascent blood vessels requires the interaction between the C-terminal retention motif of the PDGFB and the heparan sulfate proteoglycans contained within the basal lamina, thus activating PDGFRβ signal transduction [20,50,52]. More importantly, it has been demonstrated that the complete deletion of PDGFB or PDGFRβ in transgenic mice results in a perinatal lethality that was partly caused by a pronounced vascular leakage due to a mural cells deficiency [8,53]. It is noteworthy here to mention that pericytic PDGFRβ signaling seems to play an important role, not only in the brain microvasculature, but also in coronary microvasculature, as the blockade of PDGFRβ signaling with tyrosine kinase inhibitor such as Sunitinib lead to pericyte loss [54], outlining the importance of this pathway in pericyte survival and function in other vascular systems.
- (b)
- TGFβ: TGFβ has a pivotal role in vascular development including the induction of pericytes differentiation and adhesion to brain microvessels, and the regulation of endothelial cell proliferation and differentiation [13,50]. Endothelial cells, neurons, glial cells and pericytes secrete the latent form of TGFβ, which is activated by thrombospondin or integrins [13,50]. In both pericytes and endothelial cells, activated TGFβ can bind to TGFβ receptor type II (TGFβR2) leading to the recruitment and the activation of the TGFβ receptor type I (TGFβR1) and activin-like kinase 1 or 5 (ALK1/5), thus inducing the activation and the nuclear translocation of Smad proteins that promotes transcriptional changes [13,55]. It has been shown that binding of endothelially secreted TGFβ to pericytic TGFβR2 inhibits their proliferation while inducing the expression of contractile proteins and promoting the production of extracellular matrix proteins [56]. In fact, genetic deletions within TGFβ signaling pathway components lead to a faulty vascular development resulting in embryonic lethality [13,50]. More precisely, specific knockout of the Smad4 gene in the brain endothelium leads to several vascular defects, such as pericyte detachment and reduced capillary coverage, increased endothelial cell proliferation, vasodilatation and intraventricular hemorrhage during the perinatal period [57]. Interestingly, these results were associated with a reduction in N-cadherin expression, which is an important adhesion molecule implicated in endothelium-pericyte interaction.
- (c)
- Ang1/Tie2: Ang1 has been shown to be predominantly expressed in perivascular mesenchymal cells, including pericytes [50]. On the other hand, the Ang1 receptor, Tie2, has been shown to be predominantly expressed on endothelial cells [50]. As such, Ang1/Tie2 signaling pathway forms a paracrine loop that has inverted orientation in comparison with PDGFB/PDGFRβ. The Ang1/Tie2 signaling pathway has been reported to play an important role in inducing endothelial cell maturation and stability, thus decreasing vascular permeability [50,58].
- (d)
- Notch: The role of Notch signaling is well defined in neurovascular development. Establishing a cell-to-cell contact is a prerequisite for an efficient Notch signal transduction, as Notch forms heterodimeric transmembrane receptors with the transmembrane ligands delta-like (DLL) and jagged (JAG) on neighboring cells. Signal transduction is induced when the receptors and ligands bind, thus triggering the sequential proteolytic cleavage and release in the intracellular space the Notch intracellular domain (NICD), which translocates to the nucleus and binds the transcription factor recombination signal binding protein Jκ (RBPJκ), leading to downstream transcriptional changes [59]. More recently, it has been suggested that Notch signaling contribute to pericyte attachment and alignment at the abluminal side of brain endothelial cells, thus enhancing endothelial cell survival [60]. Moreover, Notch signaling has been demonstrated to play an important role in the regulation of PDGFRβ in vascular smooth muscle cell (VSMC), and probably pericytes [61]. However, more work is still required to fully address and decipher Notch signaling pathway at the NVU.
5. Pericytes in Brain Diseases
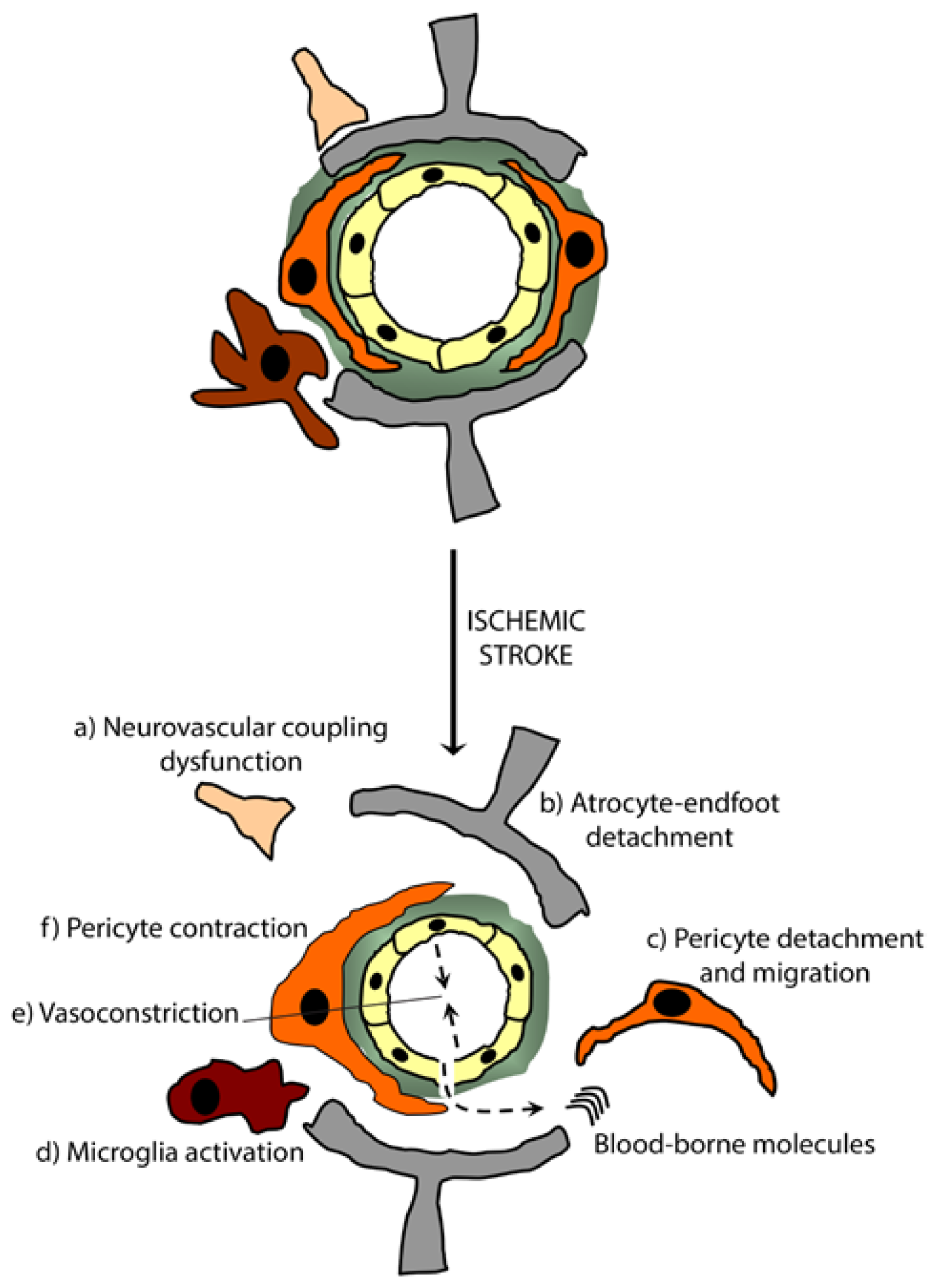
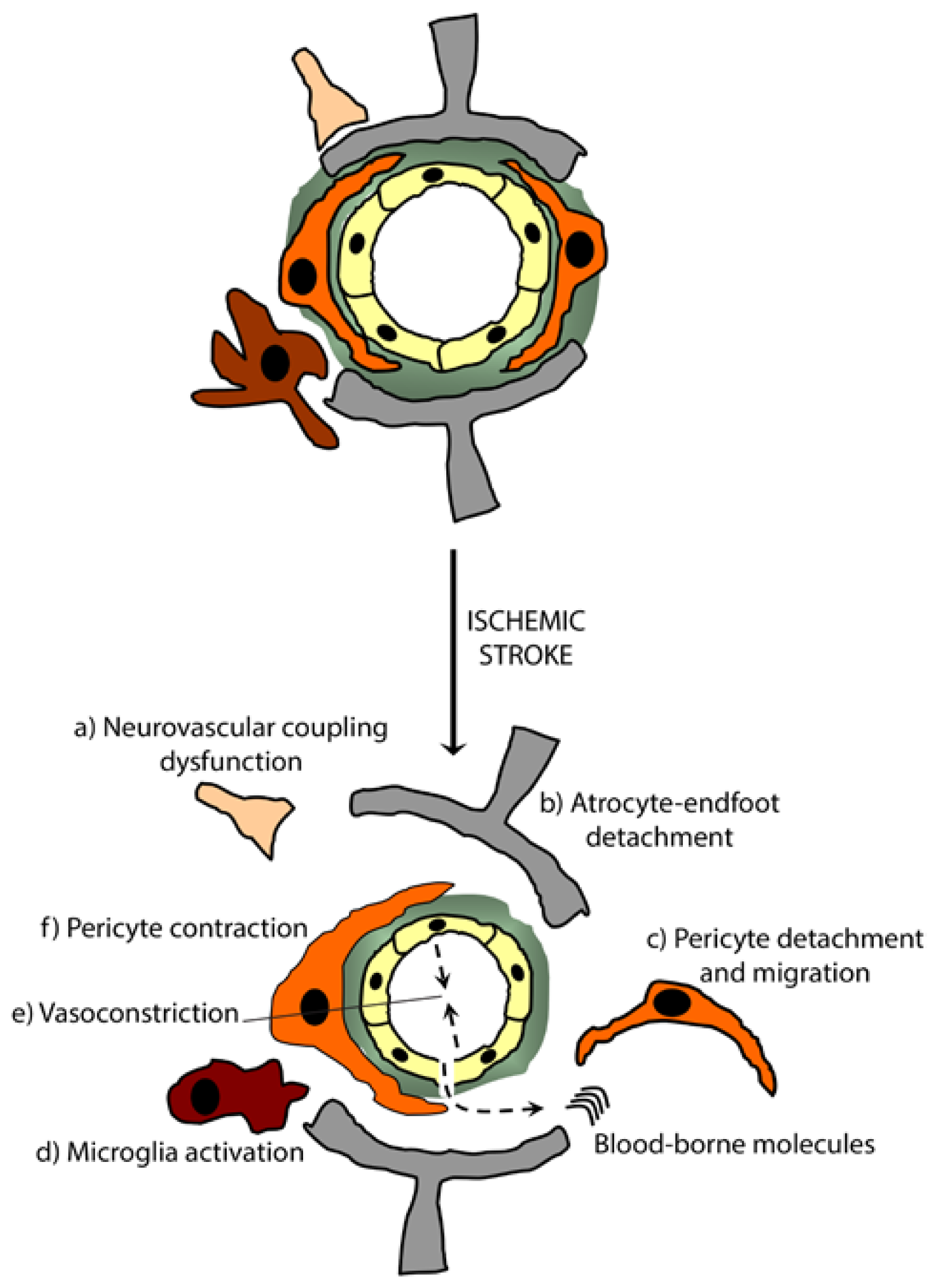
5.1. Ischemic Stroke
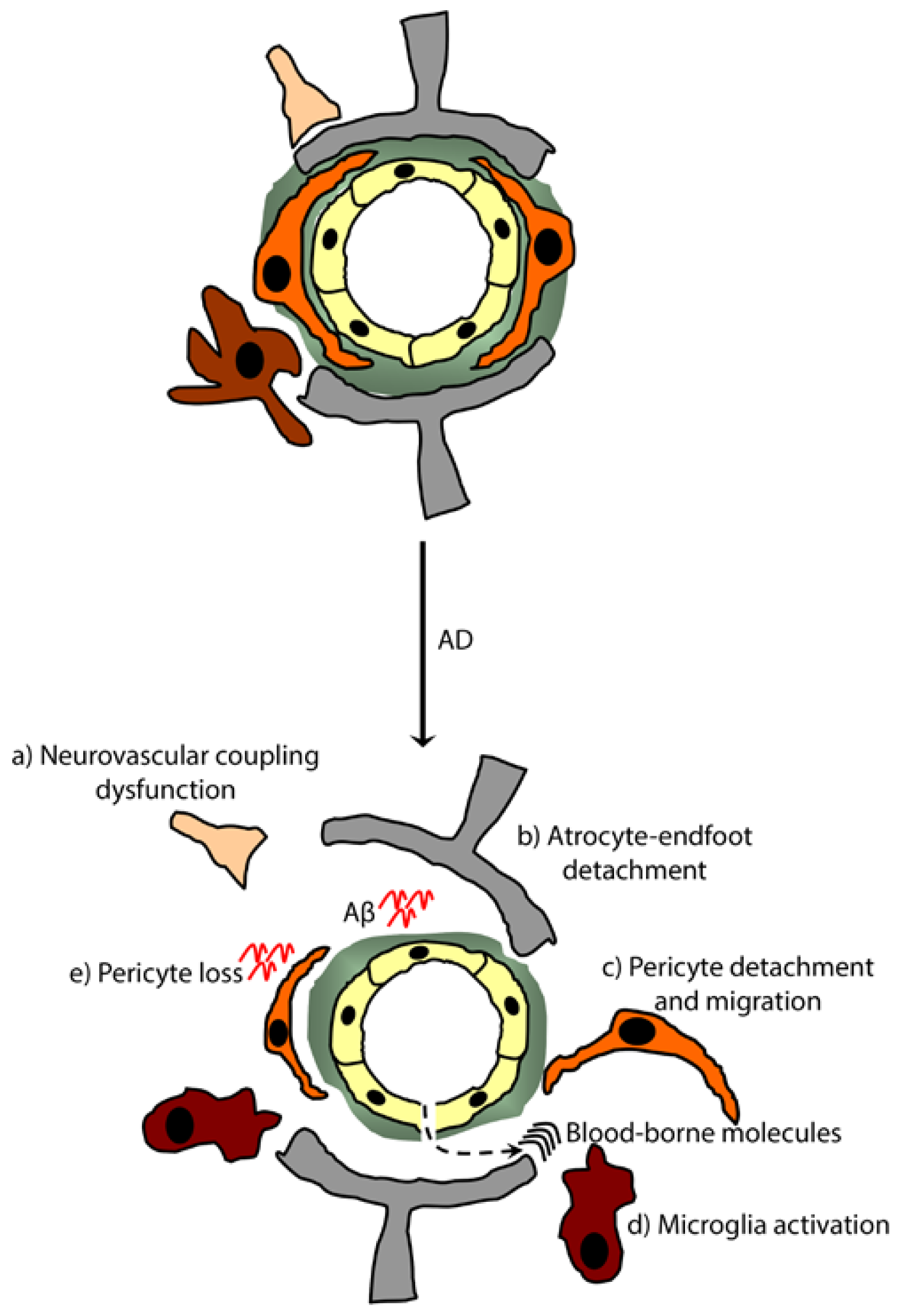
5.2. Alzheimer’s Disease (AD)
6. Pericytes in Neurovascular Unit Repair: Therapeutic Implications and Perspectives
Acknowledgments
Conflicts of Interest
- Author ContributionsA.E.A., P.T. and S.R. contributed to writing and finalizing the review.
References
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar]
- Ruitenberg, A.; den Heijer, T.; Bakker, S.L.; van Swieten, J.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam study. Ann. Neurol 2005, 57, 789–794. [Google Scholar]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci 2011, 12, 723–738. [Google Scholar]
- Hermann, D.M.; ElAli, A. The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci. Signal 2012. [Google Scholar] [CrossRef]
- Park, L.; Gallo, E.F.; Anrather, J.; Wang, G.; Norris, E.H.; Paul, J.; Strickland, S.; Iadecola, C. Key role of tissue plasminogen activator in neurovascular coupling. Proc. Natl. Acad. Sci. USA 2008, 105, 1073–1078. [Google Scholar]
- ElAli, A.; Hermann, D.M. Apolipoprotein E controls ATP-binding cassette transporters in the ischemic brain. Sci. Signal 2010. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. Beta-Amyloid efflux mediated by p-glycoprotein. J. Neurochem 2001, 76, 1121–1128. [Google Scholar]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol 2001, 153, 543–553. [Google Scholar]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar]
- Winkler, E.A.; Sengillo, J.D.; Bell, R.D.; Wang, J.; Zlokovic, B.V. Blood-spinal cord barrier pericyte reductions contribute to increased capillary permeability. J. Cereb. Blood Flow Metab 2012, 32, 1841–1852. [Google Scholar]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener 2010, 5, 32:1–32:11. [Google Scholar]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res 2005, 97, 512–523. [Google Scholar]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar]
- Fernández-Klett, F.; Offenhauser, N.; Dirnagl, U.; Priller, J.; Lindauer, U. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 22290–22295. [Google Scholar]
- Dehouck, M.P.; Vigne, P.; Torpier, G.; Breittmayer, J.P.; Cecchelli, R.; Frelin, C. Endothelin-1 as a mediator of endothelial cell-pericyte interactions in bovine brain capillaries. J. Cereb. Blood Flow Metab 1997, 17, 464–469. [Google Scholar]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar]
- Krueger, M.; Bechmann, I. CNS pericytes: Concepts, misconceptions, and a way out. Glia 2010, 58, 1–10. [Google Scholar]
- Birbrair, A.; Zhang, T.; Wang, Z.M.; Messi, M.L.; Enikolopov, G.N.; Mintz, A.; Delbono, O. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res 2013, 10, 67–84. [Google Scholar]
- Zehendner, C.M.; Wedler, H.E.; Luhmann, H.J. A novel in vitro model to study pericytes in the neurovascular unit of the developing cortex. PLoS One 2013, 8, e81637. [Google Scholar]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci 2011, 14, 1398–1405. [Google Scholar]
- Bautch, V.L.; James, J.M. Neurovascular development: The beginning of a beautiful friendship. Cell Adh. Migr 2009, 3, 199–204. [Google Scholar]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci 2006, 7, 41–53. [Google Scholar]
- Balabanov, R.; Dore-Duffy, P. Role of the CNS microvascular pericyte in the blood-brain barrier. J. Neurosci. Res 1998, 53, 637–644. [Google Scholar]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar]
- Girouard, H.; Iadecola, C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J. Appl. Physiol 2006, 100, 328–335. [Google Scholar]
- Dalkara, T.; Gursoy-Ozdemir, Y.; Yemisci, M. Brain microvascular pericytes in health and disease. Acta Neuropathol 2011, 122, 1–9. [Google Scholar]
- Yemisci, M.; Gursoy-Ozdemir, Y.; Vural, A.; Can, A.; Topalkara, K.; Dalkara, T. Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med 2009, 15, 1031–1037. [Google Scholar]
- Chen, Q.; Anderson, D.R. Effect of CO2 on intracellular pH and contraction of retinal capillary pericytes. Investig. Ophthalmol. Vis. Sci 1997, 38, 643–651. [Google Scholar]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar]
- Edelman, D.A.; Jiang, Y.; Tyburski, J.G.; Wilson, R.F.; Steffes, C.P. Lipopolysaccharide activation of pericyte’s Toll-like receptor-4 regulates co-culture permeability. Am. J. Surg 2007, 193, 730–735. [Google Scholar]
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain microvascular pericytes are immunoactive in culture: Cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflammation 2011, 8, 139:1–139:9. [Google Scholar]
- Pieper, C.; Marek, J.J.; Unterberg, M.; Schwerdtle, T.; Galla, H.J. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014, 1550, 1–8. [Google Scholar]
- Dohgu, S.; Takata, F.; Yamauchi, A.; Nakagawa, S.; Egawa, T.; Naito, M.; Tsuruo, T.; Sawada, Y.; Niwa, M.; Kataoka, Y. Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res 2005, 1038, 208–215. [Google Scholar]
- Balabanov, R.; Washington, R.; Wagnerova, J.; Dore-Duffy, P. CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvasc. Res 1996, 52, 127–142. [Google Scholar]
- Mato, M.; Ookawara, S.; Saito-Taki, T. Serological determinants of fluorescent granular perithelial cells along small cerebral blood vessels in rodent. Acta Neuropathol 1986, 72, 117–123. [Google Scholar]
- Mato, M.; Ookawara, S.; Sakamoto, A.; Aikawa, E.; Ogawa, T.; Mitsuhashi, U.; Masuzawa, T.; Suzuki, H.; Honda, M.; Yazaki, Y.; et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 3269–3274. [Google Scholar]
- Bechmann, I.; Priller, J.; Kovac, A.; Böntert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci 2001, 14, 1651–1658. [Google Scholar]
- Thomas, W.E. Brain macrophages: On the role of pericytes and perivascular cells. Brain Res. Brain Res. Rev 1999, 31, 42–57. [Google Scholar]
- Balabanov, R.; Beaumont, T.; Dore-Duffy, P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J. Neurosci. Res 1999, 55, 578–587. [Google Scholar]
- Verbeek, M.M.; Westphal, J.R.; Ruiter, D.J.; de Waal, R.M. T lymphocyte adhesion to human brain pericytes is mediated via very late antigen-4/vascular cell adhesion molecule-1 interactions. J. Immunol 1995, 154, 5876–5884. [Google Scholar]
- Proebstl, D.; Voisin, M.B.; Woodfin, A.; Whiteford, J.; D’Acquisto, F.; Jones, G.E.; Rowe, D.; Nourshargh, S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J. Exp. Med. 2012, 209, 1219–1234. [Google Scholar]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Brühl, M.L.; Gärtner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R.; et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat. Immunol 2013, 14, 41–51. [Google Scholar]
- Dore-Duffy, P.; Katychev, A.; Wang, X.; van Buren, E. CNS microvascular pericytes exhibit multipotential stem cell activity. J. Cereb. Blood Flow Metab 2006, 26, 613–624. [Google Scholar]
- Dore-Duffy, P.; Mehedi, A.; Wang, X.; Bradley, M.; Trotter, R.; Gow, A. Immortalized CNS pericytes are quiescent smooth muscle actin-negative and pluripotent. Microvasc. Res 2011, 82, 18–27. [Google Scholar]
- Karow, M.; Sánchez, R.; Schichor, C.; Masserdotti, G.; Ortega, F.; Heinrich, C.; Gascón, S.; Khan, M.A.; Lie, D.C.; Dellavalle, A.; et al. Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 2012, 11, 471–476. [Google Scholar]
- Cai, X.; Lin, Y.; Friedrich, C.C.; Neville, C.; Pomerantseva, I.; Sundback, C.A.; Zhang, Z.; Vacanti, J.P.; Hauschka, P.V.; Grottkau, B.E. Bone marrow derived pluripotent cells are pericytes which contribute to vascularization. Stem Cell Rev 2009, 5, 437–445. [Google Scholar]
- Paul, G.; Özen, I.; Christophersen, N.S.; Reinbothe, T.; Bengzon, J.; Visse, E.; Jansson, K.; Dannaeus, K.; Henriques-Oliveira, C.; Roybon, L.; et al. The adult human brain harbors multipotent perivascular mesenchymal stem cells. PLoS One 2012, 7, e35577. [Google Scholar]
- Gerhardt, H.; Wolburg, H.; Redies, C. N-cadherin mediates pericytic-endothelial interaction during brain angiogenesis in the chicken. Dev. Dyn 2000, 218, 472–479. [Google Scholar]
- Bobbie, M.W.; Roy, S.; Trudeau, K.; Munger, S.J.; Simon, A.M.; Roy, S. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Investig. Ophthalmol. Vis. Sci 2010, 51, 3758–3763. [Google Scholar]
- Gaengel, K.; Genové, G.; Armulik, A.; Betsholtz, C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol 2009, 29, 630–638. [Google Scholar]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar]
- Lindblom, P.; Gerhardt, H.; Liebner, S.; Abramsson, A.; Enge, M.; Hellström, M.; Backstrom, G.; Fredriksson, S.; Landegren, U.; Nystrom, H.C.; et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 2003, 17, 1835–1840. [Google Scholar]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Chintalgattu, V.; Rees, M.L.; Culver, J.C.; Goel, A.; Jiffar, T.; Zhang, J.; Dunner, K., Jr.; Pati, S.; Bankson, J.A.; Pasqualini, R.; et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci. Transl. Med 2013. [Google Scholar] [CrossRef]
- Lebrin, F.; Deckers, M.; Bertolino, P.; Ten Dijke, P. TGF-β receptor function in the endothelium. Cardiovasc. Res 2005, 65, 599–608. [Google Scholar]
- Sieczkiewicz, G.J.; Herman, I.M. TGF-beta 1 signaling controls retinal pericyte contractile protein expression. Microvasc. Res 2003, 66, 190–196. [Google Scholar]
- Li, F.; Lan, Y.; Wang, Y.; Wang, J.; Yang, G.; Meng, F.; Han, H.; Meng, A.; Wang, Y.; Yang, X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 2011, 20, 291–302. [Google Scholar]
- Thurston, G.; Suri, C.; Smith, K.; McClain, J.; Sato, T.N.; Yancopoulos, G.D.; McDonald, D.M. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 1999, 286, 2511–2514. [Google Scholar]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch signaling in blood vessels: Who is talking to whom about what? Circ. Res 2007, 100, 1556–1568. [Google Scholar]
- Liu, H.; Kennard, S.; Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res 2009, 104, 466–475. [Google Scholar]
- Jin, S.; Hansson, E.M.; Tikka, S.; Lanner, F.; Sahlgren, C.; Farnebo, F.; Baumann, M.; Kalimo, H.; Lendahl, U. Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circ. Res 2008, 102, 1483–1491. [Google Scholar]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci 1999, 22, 391–397. [Google Scholar]
- Charo, I.F.; Ransohoff, R.M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med 2006, 354, 610–621. [Google Scholar]
- Kamouchi, M.; Kitazono, T.; Ago, T.; Wakisaka, M.; Kuroda, J.; Nakamura, K.; Hagiwara, N.; Ooboshi, H.; Ibayashi, S.; Iida, M. Hydrogen peroxide-induced Ca2+ responses in CNS pericytes. Neurosci. Lett 2007, 416, 12–16. [Google Scholar]
- Del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab 2003, 23, 879–894. [Google Scholar]
- Fernández-Klett, F.; Potas, J.R.; Hilpert, D.; Blazej, K.; Radke, J.; Huck, J.; Engel, O.; Stenzel, W.; Genové, G.; Priller, J. Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. J. Cereb. Blood Flow Metab 2013, 33, 428–439. [Google Scholar]
- Dore-Duffy, P.; Owen, C.; Balabanov, R.; Murphy, S.; Beaumont, T.; Rafols, J.A. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res 2000, 60, 55–69. [Google Scholar]
- Hermann, D.M.; Zechariah, A. Implications of vascular endothelial growth factor for postischemic neurovascular remodeling. J. Cereb. Blood Flow Metab 2009, 29, 1620–1643. [Google Scholar]
- Manoonkitiwongsa, P.S.; Jackson-Friedman, C.; McMillan, P.J.; Schultz, R.L.; Lyden, P.D. Angiogenesis after stroke is correlated with increased numbers of macrophages: The clean-up hypothesis. J. Cereb. Blood Flow Metab 2001, 21, 1223–1231. [Google Scholar]
- Arimura, K.; Ago, T.; Kamouchi, M.; Nakamura, K.; Ishitsuka, K.; Kuroda, J.; Sugimori, H.; Ooboshi, H.; Sasaki, T.; Kitazono, T. PDGF receptor β signaling in pericytes following ischemic brain injury. Curr. Neurovasc. Res 2012, 9, 1–9. [Google Scholar]
- Zacharek, A.; Chen, J.; Zhang, C.; Cui, X.; Roberts, C.; Jiang, H.; Teng, H.; Chopp, M. Nitric oxide regulates Angiopoietin1/Tie2 expression after stroke. Neurosci. Lett 2006, 404, 28–32. [Google Scholar]
- Iurlaro, M.; Scatena, M.; Zhu, W.H.; Fogel, E.; Wieting, S.L.; Nicosia, R.F. Rat aorta-derived mural precursor cells express the Tie2 receptor and respond directly to stimulation by angiopoietins. J. Cell Sci 2003, 116, 3635–3643. [Google Scholar]
- Hori, S.; Ohtsuki, S.; Hosoya, K.I.; Nakashima, E.; Terasaki, T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J. Neurochem. 2004, 89, 503–513. [Google Scholar]
- Arumugam, T.V.; Chan, S.L.; Jo, D.G.; Yilmaz, G.; Tang, S.C.; Cheng, A.; Gleichmann, M.; Okun, E.; Dixit, V.D.; Chigurupati, S.; et al. Gamma secretase-mediated Notch signaling worsens brain damage and functional outcome in ischemic stroke. Nat. Med 2006, 12, 621–623. [Google Scholar]
- Wang, Y.; Pan, L.; Moens, C.B.; Appel, B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2014, 141, 307–317. [Google Scholar]
- Arboleda-Velasquez, J.F.; Zhou, Z.; Shin, H.K.; Louvi, A.; Kim, H.H.; Savitz, S.I.; Liao, J.K.; Salomone, S.; Ayata, C.; Moskowitz, M.A.; et al. Linking Notch signaling to ischemic stroke. Proc. Natl. Acad. Sci. USA 2008, 105, 4856–4861. [Google Scholar]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar]
- Iadecola, C.; Zhang, F.; Niwa, K.; Eckman, C.; Turner, S.K.; Fischer, E.; Younkin, S.; Borchelt, D.R.; Hsiao, K.K.; Carlson, G.A. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci 1999, 2, 157–161. [Google Scholar]
- Claudio, L. Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol 1996, 91, 6–14. [Google Scholar]
- Farkas, E.; Luiten, P.G. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog. Neurobiol 2001, 64, 575–611. [Google Scholar]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun 2013. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol 2013, 23, 303–310. [Google Scholar]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar]
- Niwa, K.; Kazama, K.; Younkin, S.G.; Carlson, G.A.; Iadecola, C. Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol. Dis 2002, 9, 61–68. [Google Scholar]
- Pimentel-Coelho, P.M.; Michaud, J.P.; Rivest, S. Effects of mild chronic cerebral hypoperfusion and early amyloid pathology on spatial learning and the cellular innate immune response in mice. Neurobiol. Aging 2013, 34, 679–693. [Google Scholar]
- Wu, Z.; Guo, H.; Chow, N.; Sallstrom, J.; Bell, R.D.; Deane, R.; Brooks, A.I.; Kanagala, S.; Rubio, A.; Sagare, A.; et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat. Med 2005, 11, 959–965. [Google Scholar]
- Verbeek, M.M.; de Waal, R.M.; Schipper, J.J.; van Nostrand, W.E. Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J. Neurochem 1997, 68, 1135–1141. [Google Scholar]
- Veszelka, S.; Tóth, A.E.; Walter, F.R.; Datki, Z.; Mózes, E.; Fülöp, L.; Bozsó, Z.; Hellinger, E.; Vastag, M.; Orsolits, B.; et al. Docosahexaenoic acid reduces amyloid-β induced toxicity in cells of the neurovascular unit. J. Alzheimers Dis 2013, 36, 487–501. [Google Scholar]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss1–40 peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig 2000, 106, 1489–1499. [Google Scholar]
- Wilhelmus, M.M.; Otte-Höller, I.; van Triel, J.J.; Veerhuis, R.; Maat-Schieman, M.L.; Bu, G.; de Waal, R.M.; Verbeek, M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am. J. Pathol 2007, 171, 1989–1999. [Google Scholar]
- Verbeek, M.M.; van Nostrand, W.E.; Otte-Höller, I.; Wesseling, P.; de Waal, R.M. Amyloid-β-induced degeneration of human brain pericytes is dependent on the apolipoprotein E genotype. Ann. N. Y. Acad. Sci 2000, 903, 187–199. [Google Scholar]
- Zechariah, A.; ElAli, A.; Doeppner, T.R.; Jin, F.; Hasan, M.R.; Helfrich, I.; Mies, G.; Hermann, D.M. Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 2013, 44, 1690–1697. [Google Scholar]
- Cui, X.; Chopp, M.; Zacharek, A.; Cui, Y.; Roberts, C.; Chen, J. The neurorestorative benefit of GW3965 treatment of stroke in mice. Stroke 2013, 44, 153–161. [Google Scholar]
- Hill, W.D.; Hess, D.C.; Martin-Studdard, A.; Carothers, J.J.; Zheng, J.; Hale, D.; Maeda, M.; Fagan, S.C.; Carroll, J.E.; Conway, S.J. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J. Neuropathol. Exp. Neurol 2004, 63, 84–96. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
ElAli, A.; Thériault, P.; Rivest, S. The Role of Pericytes in Neurovascular Unit Remodeling in Brain Disorders. Int. J. Mol. Sci. 2014, 15, 6453-6474. https://doi.org/10.3390/ijms15046453
ElAli A, Thériault P, Rivest S. The Role of Pericytes in Neurovascular Unit Remodeling in Brain Disorders. International Journal of Molecular Sciences. 2014; 15(4):6453-6474. https://doi.org/10.3390/ijms15046453
Chicago/Turabian StyleElAli, Ayman, Peter Thériault, and Serge Rivest. 2014. "The Role of Pericytes in Neurovascular Unit Remodeling in Brain Disorders" International Journal of Molecular Sciences 15, no. 4: 6453-6474. https://doi.org/10.3390/ijms15046453