Pharmacological Evaluation and Preparation of Nonsteroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit

Abstract
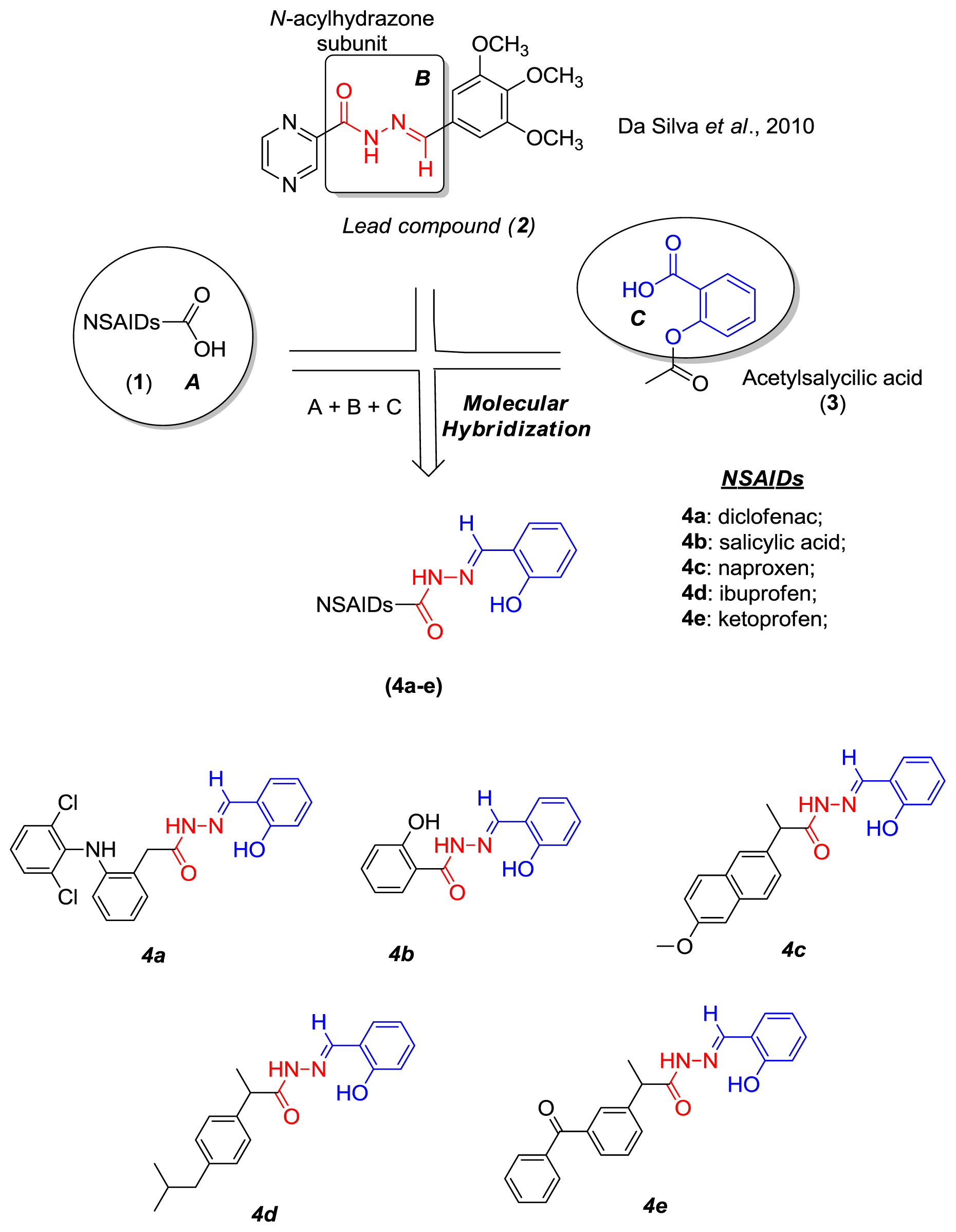
:1. Introduction
2. Results and Discussion
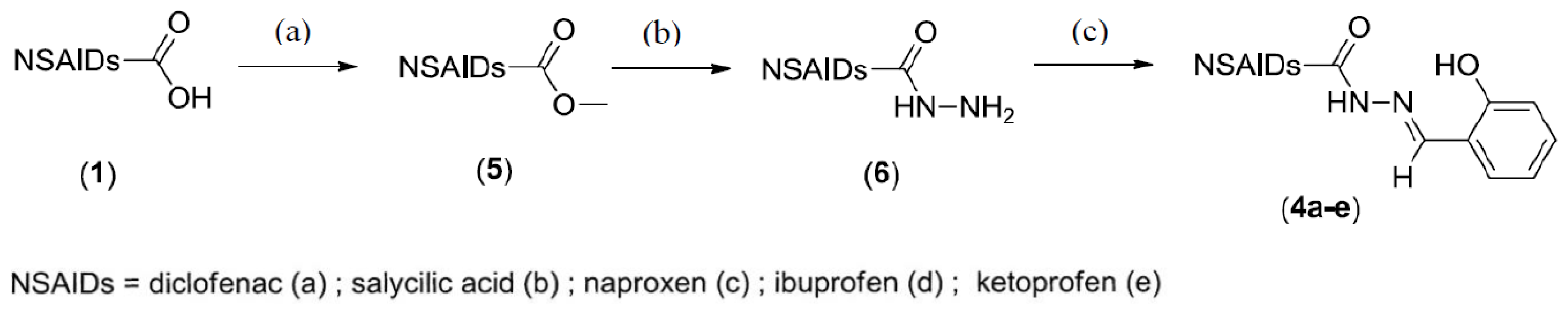
2.1. Chemistry
2.2. Anti-Inflammatory/Analgesic Activities and Ulcerogenicity Studies
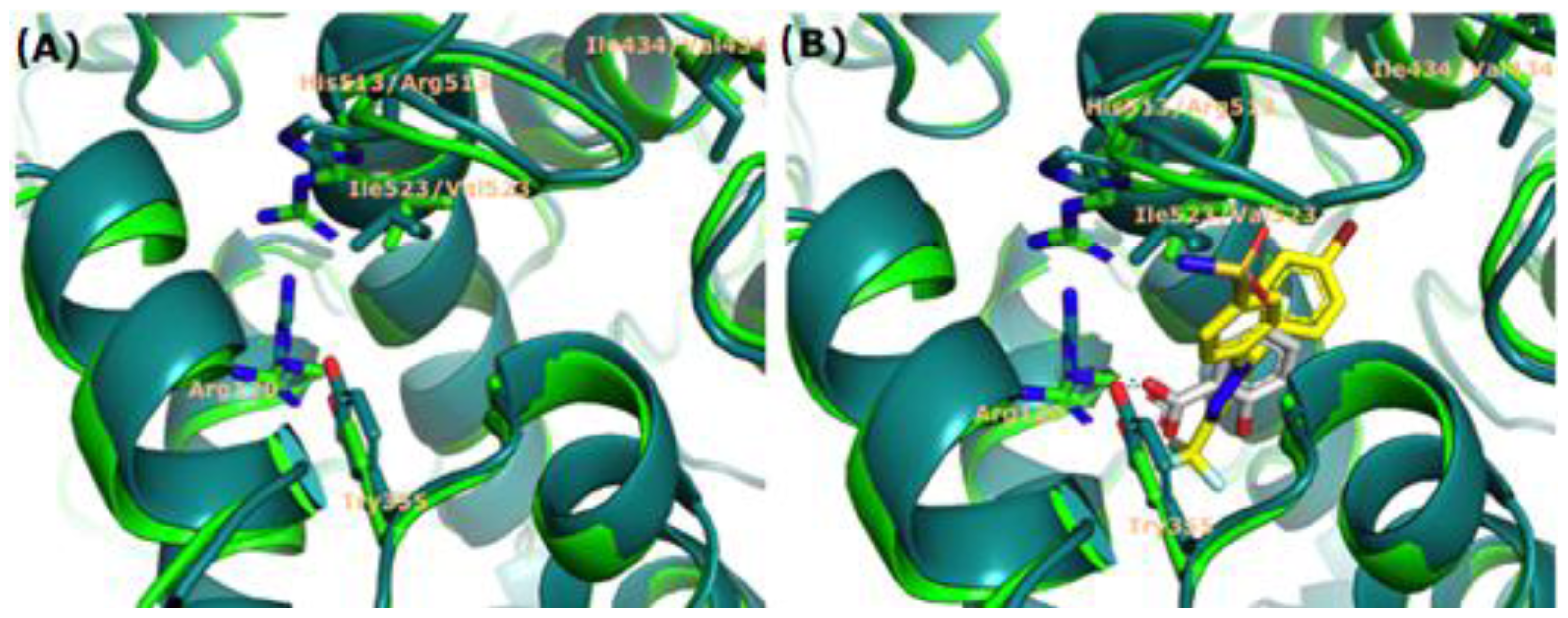
2.3. Docking Studies
3. Experimental Section
3.1. General Procedures
3.2. Preparation
3.2.1. General Procedures for the Preparation of Ester (5a–e) and Hydrazide (6a–e) Derivatives
3.2.2. General Procedures for the Preparation of N-Acyl Hydrazone Derivatives (4a–e)
3.3. Pharmacology
3.3.1. Anti-Inflammatory Activity
3.3.2. Analgesic Activity
3.3.3. Ulcerogenicity Studies
3.4. Docking Studies
4. Conclusions
Acknowledgments
Conflicts of Interest
- Author ContributionsThais Regina Ferreira de Melo and Rafael Consolin Chelucci participated in synthesis, anti-inflammatory and analgesic assays, Maria Elisa Lopes Pires participate in gastroulceration studies, Luiz Antônio Dutra participated in synthesis, purification and characterization of the compounds 4a–e, Karina Pereira Barbieri participated in analgesic study, Gustavo Henrique Trossini participated in molecular modeling studies, Priscila Longhin Bosquesi, Man Chin Chung and Jean Leandro dos Santos participated in the interpretation of the results and in manuscript writing.
References
- Rollas, S.; Küçükgüzel, S.G. Biological activities of hydrazone derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar]
- Hernández, P.; Cabrera, M.; Lavaggi, M.L.; Celano, L.; Tiscornia, I.; Rodrigues da Costa, T.; Thomson, L.; Bollati-Fogolín, M.; Miranda, A.L.; Lima, L.M.; et al. Discovery of new orally effective analgesic and anti-inflammatory hybrid furoxanyl N-acyl hydrazone derivatives. Bioorg. Med. Chem 2012, 20, 2158–2171. [Google Scholar]
- Barreiro, E.J.; Fraga, C.A.M.; Miranda, A.L.P.; Rodrigues, C.R. A química medicinal de N-acilidrazonas: Novos compostos protótipos de fármacos analgésicos, antiinflamatórios e anti-trombóticos. Quim. Nova 2002, 25, 129–148. [Google Scholar]
- Fanelli, A.; Romualdi, P.; Viganò, R.; Lora Aprile, P.; Gensini, G.; Fanelli, G. Non-selective non-steroidal anti-inflammatory drugs (NSAIDs) and cardiovascular risk. Acta Biomed 2013, 84, 5–11. [Google Scholar]
- Lim, Y.J.; Chun, H.J. Recent advances in NSAIDs-induced enteropathy therapeutics: New options, new challenges. Gastroenterol. Res. Pract 2013, 2013, 761060. [Google Scholar]
- Chung, M.C.; dos Santos, J.L.; Oliveira, E.V.; Blau, L.; Menegon, R.F.; Peccinini, R.G. Synthesis, ex vivo and in vitro hydrolysis study of an indoline derivative designed as an anti-inflammatory with reduced gastric ulceration properties. Molecules 2009, 14, 3187–3197. [Google Scholar]
- Khan, M.S.; Akhter, M. Synthesis, pharmacological activity and hydrolytic behavior of glyceride prodrugs of ibuprofen. Eur. J. Med. Chem 2005, 40, 371–376. [Google Scholar]
- Hamdy, N.A.; Abdel-Aziz, H.A.; Kamel, G.M.; Fakhr, I.M. Convenient synthesis, anti-inflammatory, analgesic and ulcerogenic activites of some new bis-hydrazones and pyrazole derivatives. Acta Pol. Pharm 2013, 70, 469–480. [Google Scholar]
- McGettigan, P.; Henry, D. Cardiovascular risk and inhibition of cyclooxygenase: A systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA 2006, 296, 1633–1644. [Google Scholar]
- McGettigan, P.; Henry, D. Use of non-steroidal anti-inflammatory drugs that elevate cardiovascular risk: An examination of sales and essential medicines lists in low-, middle-, and high-income countries. PLoS Med 2013, 10, e1001388. [Google Scholar]
- Bhala, N.; Emberson, J.; Merhi, A.; Abramson, S.; Arber, N.; Baron, J.A.; Bombardier, C.; Cannon, C.; Farkouh, M.E.; FitzGerald, G.A.; et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Coxib and traditional NSAID Trialists’ (CNT) Collaboration. Lancet 2013, 382, 769–779. [Google Scholar]
- Fanelli, A.; Ghisi, D.; Fanelli, G. Nonsteroideal Anti-inflammatory Drugs (NSAIDs) in clinical practice: Managing gastric and cardiovascular risks. Acta Biomed 2013, 84, 98–101. [Google Scholar]
- Mashayekhi, V.; Mohammad, Haj; Ebrahim Tehrani, K.; Amidi, S.; Kobarfard, F. Synthesis of novel indole hydrazone derivatives and evaluation of their antiplatelet aggregation activity. Chem. Pharm. Bull. (Tokyo) 2013, 61, 144–150. [Google Scholar]
- Cunha, A.C.; Figueiredo, J.M.; Tributino, J.L.; Miranda, A.L.; Castro, H.C.; Zingali, R.B.; Fraga, C.A.; de Souza, M.C.; Ferreira, V.F.; Barreiro, E.J. Antiplatelet properties of novel N-substituted-phenyl-1,2,3-triazole-4-acylhydrazone derivatives. Bioorg. Med. Chem 2003, 11, 2051–2059. [Google Scholar]
- Lima, L.M.; Frattani, F.S.; dos Santos, J.L.; Castro, H.C.; Fraga, C.A.; Zingali, R.B.; Barreiro, E.J. Synthesis and anti-platelet activity of novel arylsulfonate—Acylhydrazone derivatives, designed as antithrombotic candidates. Eur. J. Med. Chem 2008, 43, 348–356. [Google Scholar]
- Chelucci, R.C.; Dutra, L.A.; Lopes Pires, M.E.; de Melo, T.R.; Bosquesi, P.L.; Chung, M.C.; Dos Santos, J.L. Antiplatelet and antithrombotic activities of non-steroidal anti-inflammatory drugs containing an N-acyl hydrazone subunit. Molecules 2014, 19, 2089–2099. [Google Scholar]
- Patrício, J.P.; Barbosa, J.P.; Ramos, R.M.; Antunes, N.F.; de Melo, P.C. Relative cardiovascular and gastrointestinal safety of non-selective non-steroidal anti-inflammatory drugs vs. cyclo-oxygenase-2 inhibitors: Implications for clinical practice. Clin. Drug Investig 2013, 33, 167–183. [Google Scholar]
- Cheng, J.W. Updates in antiplatelet agents used in cardiovascular diseases. J. Cardiovasc. Pharmacol. Ther 2013, 18, 514–524. [Google Scholar]
- Nemerovski, C.W.; Salinitri, F.D.; Morbitzer, K.A.; Moser, L.R. Aspirin for primary prevention of cardiovascular disease events. Pharmacotherapy 2012, 32, 1020–1035. [Google Scholar]
- Da Silva, Y.K.; Augusto, C.V.; de Castro Barbosa, M.L.; de Albuquerque Melo, G.M.; de Queiroz, A.C.; de Lima Matos Freire Dias, T.; Júnior, W.B.; Barreiro, E.J.; Lima, L.M.; Alexandre-Moreira, M.S. Synthesis and pharmacological evaluation of pyrazine N- acyl hydrazone derivatives designed as novel analgesic and anti-inflammatory drug candidates. Bioorg. Med. Chem 2010, 18, 5007–5015. [Google Scholar]
- Blanco, F.; Egan, B.; Caboni, L.; Elguero, J.; O’Brien, J.; McCabe, T.; Fayne, D.; Meegan, M.J.; Lloyd, D.G. Study of E/Z isomerization in a series of novel non-ligand binding pocket androgen receptor antagonists. J. Chem. Inf. Model 2012, 52, 2387–2397. [Google Scholar]
- Winter, C.; Risley, E.; Nuss, G. Carrageenin-induced edema in hind paw of the rat as an assay for anti-inflammatory drugs. Proc. Soc. Exp. Biol. Med 1962, 111, 544–547. [Google Scholar]
- Effenberger, K.; Breyer, S.; Ocker, M.; Schobert, R. New doxorubicin N-acyl hydrazones with improved efficacy and cell line specificity show modes of action different from the parent drug. Int. J. Clin. Pharmacol. Ther 2010, 48, 485–486. [Google Scholar]
- Seigmund, E.; Cadmus, R.; Lu, G. A method for evaluating both non-narcotic and narcotic analgesics. Proc. Soc. Exp. Biol. Med 1957, 95, 729–733. [Google Scholar]
- Fraga, C.A.; Barreiro, E.J. Medicinal chemistry of N-acylhydrazones: New lead-compounds of analgesic, antiinflammatory and antithrombotic drugs. Curr. Med. Chem 2006, 13, 167–198. [Google Scholar]
- Cioli, V.; Putzolu, S.; Rossi, V.; Corza, B.P.; Corradino, C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol. Appl. Pharmacol 1979, 50, 283–289. [Google Scholar]
- Dos Santos, J.L.; Chelucci, R.; Chiquetto, R.; Chung, M.C.; Campos, M.L.; Peccinini, R.G. Synthesis, characterization and pharmacological evaluation of 1-(2-chloro-6-fluorophenyl)-5-methylindolin-2-one: A new anti-inflammatory compound with reduced gastric ulceration properties. Molecules 2010, 15, 8039–8047. [Google Scholar]
- Sheha, M. Pharmacokinetic and ulcerogenic studies of naproxen prodrugs designed for specific brain delivery. Arch. Pharm. Res 2012, 35, 523–530. [Google Scholar]
- Velásquez, C.; Praveen Rao, P.N.; Knaus, E.E. Novel nonsteroidal antiinflammatory drugs possessing a nitric oxide donor diazen-1-ium-1,2-diolate moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem 2005, 48, 4061–4067. [Google Scholar]
- Zhao, X.; Tao, X.; Wei, D.; Song, Q. Pharmacological activity and hydrolysis behavior of novel ibuprofen glucopyranoside conjugates. Eur. J. Med. Chem 2006, 41, 1352–1358. [Google Scholar]
- Chatterjee, N.R.; Kulkarni, A.A.; Ghulekar, S.P. Synthesis, pharmacological activity and hydrolytic behavior of ethylenediamine and benzathine conjugates of ibuprofen. Eur. J. Med. Chem 2008, 43, 2819–2823. [Google Scholar]
- Shientag, L.J.; Wheeler, S.M.; Garlick, D.S.; Maranda, L.S. A therapeutic dose of ketoprofen causes acute gastrointestinal bleeding, erosions, and ulcers in rats. J. Am. Assoc. Lab. Anim. Sci 2012, 51, 832–841. [Google Scholar]
- Laine, L. Approaches to NSAID use in the high risk patient. Gastroenterology 2001, 120, 594–606. [Google Scholar]
- Wolfe, M.M.; Lichtenstein, D.R.; Singh, G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N. Engl. J. Med 1999, 340, 1888–1899. [Google Scholar]
- Lanas, A.; García-Rodríguez, L.A.; Arroyo, M.T.; Gomollón, F.; Feu, F.; González-Pérez, A.; Zapata, E.; Bástida, G.; Rodrigo, L.; Santolaria, S.; et al. Risk of upper gastrointestinal ulcer bleeding associated with selective cyclo-oxygenase-2 inhibitors, traditional non-aspirin non-steroidal anti-inflammatory drugs, aspirin and combinations. Gut 2006, 55, 1731–1738. [Google Scholar]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar]
- Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.; McDonald, J.J.; Stegeman, R.A.; Pak, J.Y.; Gildehaus, D.; Miyashiro, J.M.; Penning, T.D.; Seibert, K.; et al. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature 1996, 384, 644–648. [Google Scholar]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J. Clin. Investig 2006, 116, 4–15. [Google Scholar]
- Furse, K.E.; Pratt, D.A.; Schneider, C.; Brash, A.R.; Porter, N.A.; Lybrand, T.P. Molecular dynamics simulations of arachidonic acid-derived pentadienyl radical intermediate complexes with COX-1 and COX-2: Insights into oxygenation regio- and stereoselectivity. Biochemistry 2006, 45, 3206–3218. [Google Scholar]
- Gauthier, M.P.; Michaux, C.; Rolin, C.; Vastersaegher, C.; Blakumar, X.C.; de Leval, X.; Julemont, F.; Pochet, L.; Masereel, B. Synthesis, molecular modeling and enzymatic evaluation of (+/−)3,5-diphenyl-2-thioxoimidazolidin-4-ones as new cyclooxygenase inhibitors. Bioorg. Med. Chem 2006, 14, 918–927. [Google Scholar]
- Tóth, L.; Muszbek, L.; Komáromi, I. Mechanism of the irreversible inhibition of human cyclooxygenase-1 by aspirin as predited by QM/MM calculations. J. Mol. Graph. Model 2013, 40, 99–109. [Google Scholar]
- FitzGerald, G.A. COX-2 and beyond: Appproches to prostaglandin inhibition in human disease. Nat. Drug Discov. Rev 2003, 2, 879–890. [Google Scholar]
- Sutradhar, M.; Mukherjee, G.; Drew, M.G.; Ghosh, S. Synthesis, reactivity, and X-ray crystal structure of some mixed-ligand oxovanadium(V) complexes: First report of binuclear oxovanadium(V) complexes containing 4,4′-bipyridine type bridge. Inorg. Chem 2006, 45, 5150–5161. [Google Scholar]
- Dinda, R.; Sengupta, P.; Ghosh, S.; Mak, T.C. Valence delocalization in a mixed-oxidation divanadium (IV, V) complex electrogenerated from its structurally characterized divanadium (V) analogue with a tridentate (ONO) ligand. Inorg. Chem 2002, 41, 1684–1688. [Google Scholar]
- Sriram, D.; Yogeeswari, P.; Devakaram, R.V. Synthesis, in vitro and in vivo antimycobacterial activities of diclofenac acid hydrazones and amides. Bioorg. Med. Chem 2006, 14, 3113–3118. [Google Scholar]
- Abdel-Azeem, A.Z.; Abdel-Hafez, A.A.; El-Karamany, G.S.; Farag, H.H. Chlorzoxazone esters of some non-steroidal anti-inflammatory (NSAI) carboxylic acids as mutual prodrugs: Design, synthesis, pharmacological investigations and docking studies. Bioorg. Med. Chem 2009, 17, 3665–3670. [Google Scholar]
- Institute of Laboratory Animal Research, Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; p. 220.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | N | % Inhibition mean ± S.E.M. | |||||
|---|---|---|---|---|---|---|---|
| 60 min | 120 min | 180 min | 240 min | 300 min | 360 min | ||
| Indomethacin | 6 | 66.7 ± 1.3 | 71.8 ± 0.96 | 75.7 ± 1.32 | 78.8 ± 2.13 | 76.6 ± 1.93 | 75.7 ± 0.98 |
| Diclofenac | 6 | 59.3 ± 0.27 * | 64.7 ± 1.21 * | 66.6 ± 1.48 * | 69.2 ± 1.71 * | 68.2 ± 0.98 * | 75.1 ± 2.11 * |
| 4a | 6 | 0 † | 0 † | 0 † | 0 † | 0 † | 0 † |
| Salicylic acid | 6 | 63.4 ± 2.31 * | 58.9 ± 1.83 * | 73.3 ± 2.44 * | 73.0 ± 1.17 * | 72.7 ± 1.76 * | 74.1 ± 2.10 * |
| 4b | 6 | 7.6 ± 0.10 † | 14.9 ± 0.08 † | 11.3 ± 0.07 † | 12.5 ± 0.09 † | 12.2 ± 0.08 † | 32.1 ± 0.05 † |
| Naproxen | 6 | 65.8 ± 1.32 * | 72.7 ± 1.32 * | 69.2 ± 1.61 * | 58.8 ± 0.91 | 53.9 ± 1.77 | 41.9 ± 1.87 |
| 4c | 6 | 8.0 ± 0.10 † | 35.9 ± 0.11 † | 35.0 ± 0.11 † | 52.8 ± 0.07 † | 67.5 ± 0.04 †,* | 81.5 ± 0.04 * |
| Ibuprofen | 6 | 64.8 ± 1.86 * | 42.1 ± 2.18 * | 46.7 ± 1.73 * | 42.3 ± 1.6 * | 45.5 ± 1.55 * | 62.5 ± 2.09 * |
| 4d | 6 | 22.7 ± 0.07 † | 25.7 ± 0.05 † | 9.7 ± 0.09 † | 10.6 ± 0.08 † | 22.9 ± 0.08 † | 45 ± 0.07 † |
| Ketoprofen | 6 | 71.8 ± 1.68 * | 64.7 ± 1.54 * | 63.8 ± 2.21 * | 66.9 ± 1.72 * | 72.7 ± 1.31 * | 75.1 ± 1.84 * |
| 4e | 6 | 11.3 ± 0.08 † | 15.7 ± 0.04 † | 16.1 ± 0.05 † | 18.5 ± 0.07 † | 29.4 ± 0.1 † | 34.7 ± 0.12 † |
| Compounds | % Protection |
|---|---|
| Dypirone | 37.2 ± 1.4 |
| Diclofenac | 28.3 ± 2.2 |
| 4a | 42.0 ± 1.1 †,* |
| Salicylic acid | 30.2 ± 1.5 |
| 4b | 36.8 ± 2.1 * |
| Naproxen | 36.8 ± 0.8 |
| 4c | 36.4 ± 1.7 |
| Ibuprofen | 25.0 ± 0.7 |
| 4d | 28.1 ± 1.0 † |
| Ketoprofen | 22.3 ± 0.9 |
| 4e | 25.6 ± 2.2 † |
| Compounds | Number of ulcers | <1 mm | 1–2 mm | >2 mm |
|---|---|---|---|---|
| Celecoxib | 11 ± 3.1 | 7.5 ± 2.7 (68.2%) | 3.5 ± 1.4 (31.8%) | - |
| Diclofenac | 72 ± 6.9 | 60 ± 7.7 (83.3%) | 8.1 ± 5.3 (11.3%) | 3.9 ± 3.1 (5.4%) |
| 4a | 26 ± 7.7 †,* | 20.8 ± 4.8 (80%) | 5.2 ± 2.1 (20%) | - |
| Salicylic acid | 69.8 ± 3.4 | 52.56 ± 5.2 (75.2%) | 9.21 ± 3.6 (13.2%) | 8.03 ± 2.7 (11.6%) |
| 4b | 17.5 ± 3.3 * | 14.3 ± 3.9 (81.7%) | 3.2 ± 1.1 (18.3%) | - |
| Naproxen | 57.8 ± 4.2 | 47.97 ± 2.7 (83%) | 9.82 ± 0.8 (17%) | - |
| 4c | 7.1 ± 2.8 * | 6.5 ± 2.8 (91.5%) | 0.6 ± 0.2 (8.5%) | - |
| Ibuprofen | 70.7 ± 4.4 | 52.31 ± 6.1 (73.9) | 16.54 ± 2.8 (23.4%) | 1.83 ± 0.8 (2.7%) |
| 4d | 14.1 ± 3.5 * | 13 ± 3.6 (92.2%) | 1.1 ± 1.2 (7.8%) | - |
| Ketoprofen | 68.9 ± 3.3 | 59.46 (86.3%) | 8.33 ± 1.7 (12.1%) | 1.10 ± 0.4 (1.6%) |
| 4e | 8.2 ± 2.2 * | 7.4 ± 2.9 (90.2%) | 0.8 ± 0.4 (9.8%) | - |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Melo, T.R.F.; Chelucci, R.C.; Pires, M.E.L.; Dutra, L.A.; Barbieri, K.P.; Bosquesi, P.L.; Trossini, G.H.G.; Chung, M.C.; Dos Santos, J.L. Pharmacological Evaluation and Preparation of Nonsteroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit. Int. J. Mol. Sci. 2014, 15, 5821-5837. https://doi.org/10.3390/ijms15045821
De Melo TRF, Chelucci RC, Pires MEL, Dutra LA, Barbieri KP, Bosquesi PL, Trossini GHG, Chung MC, Dos Santos JL. Pharmacological Evaluation and Preparation of Nonsteroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit. International Journal of Molecular Sciences. 2014; 15(4):5821-5837. https://doi.org/10.3390/ijms15045821
Chicago/Turabian StyleDe Melo, Thais Regina Ferreira, Rafael Consolin Chelucci, Maria Elisa Lopes Pires, Luiz Antonio Dutra, Karina Pereira Barbieri, Priscila Longhin Bosquesi, Gustavo Henrique Goulart Trossini, Man Chin Chung, and Jean Leandro Dos Santos. 2014. "Pharmacological Evaluation and Preparation of Nonsteroidal Anti-Inflammatory Drugs Containing an N-Acyl Hydrazone Subunit" International Journal of Molecular Sciences 15, no. 4: 5821-5837. https://doi.org/10.3390/ijms15045821