Comparative Molecular Dynamics Simulations of Mitogen-Activated Protein Kinase-Activated Protein Kinase 5


Abstract
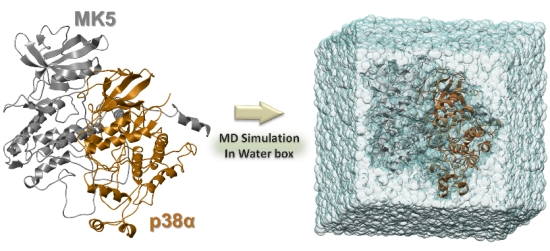
:
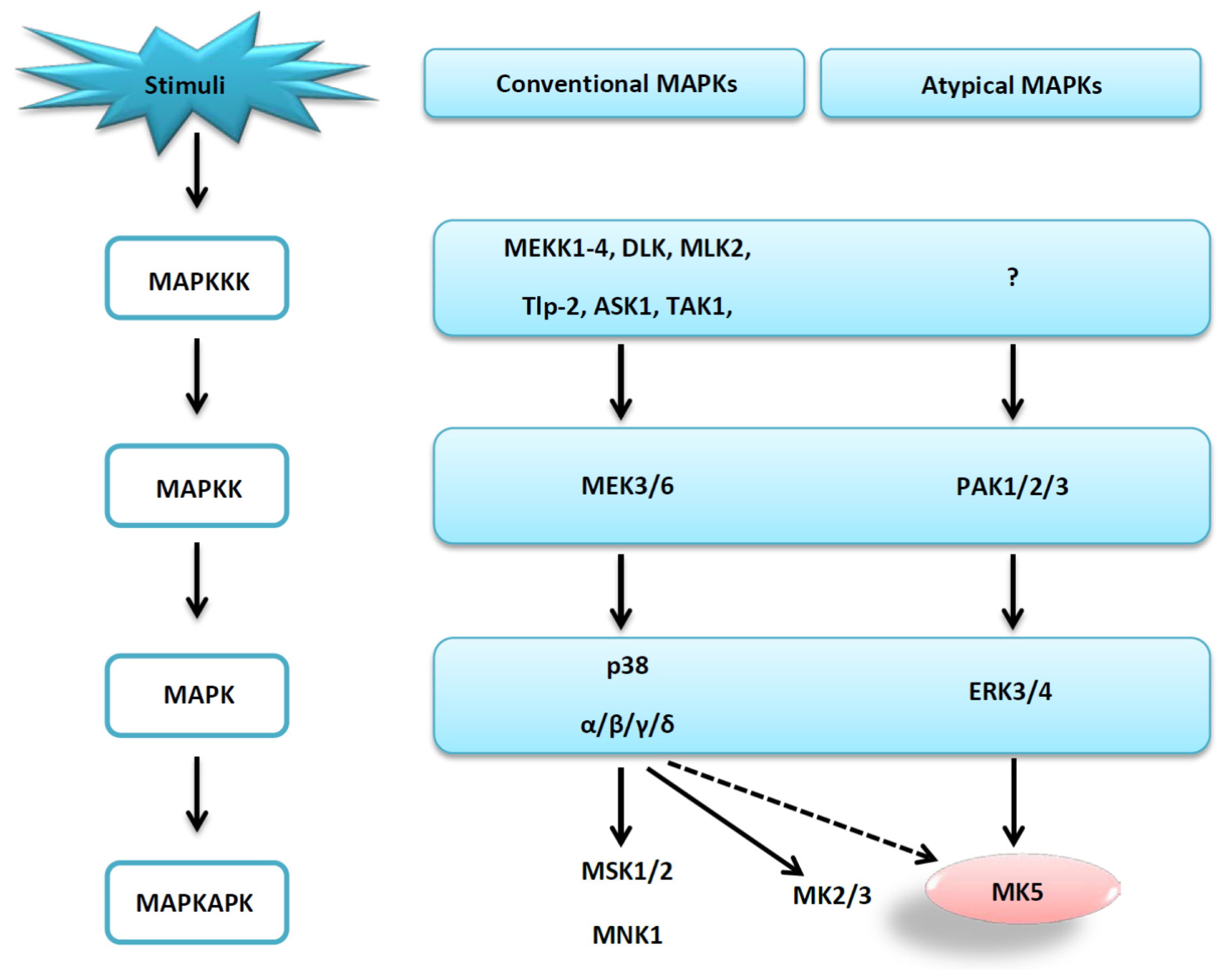
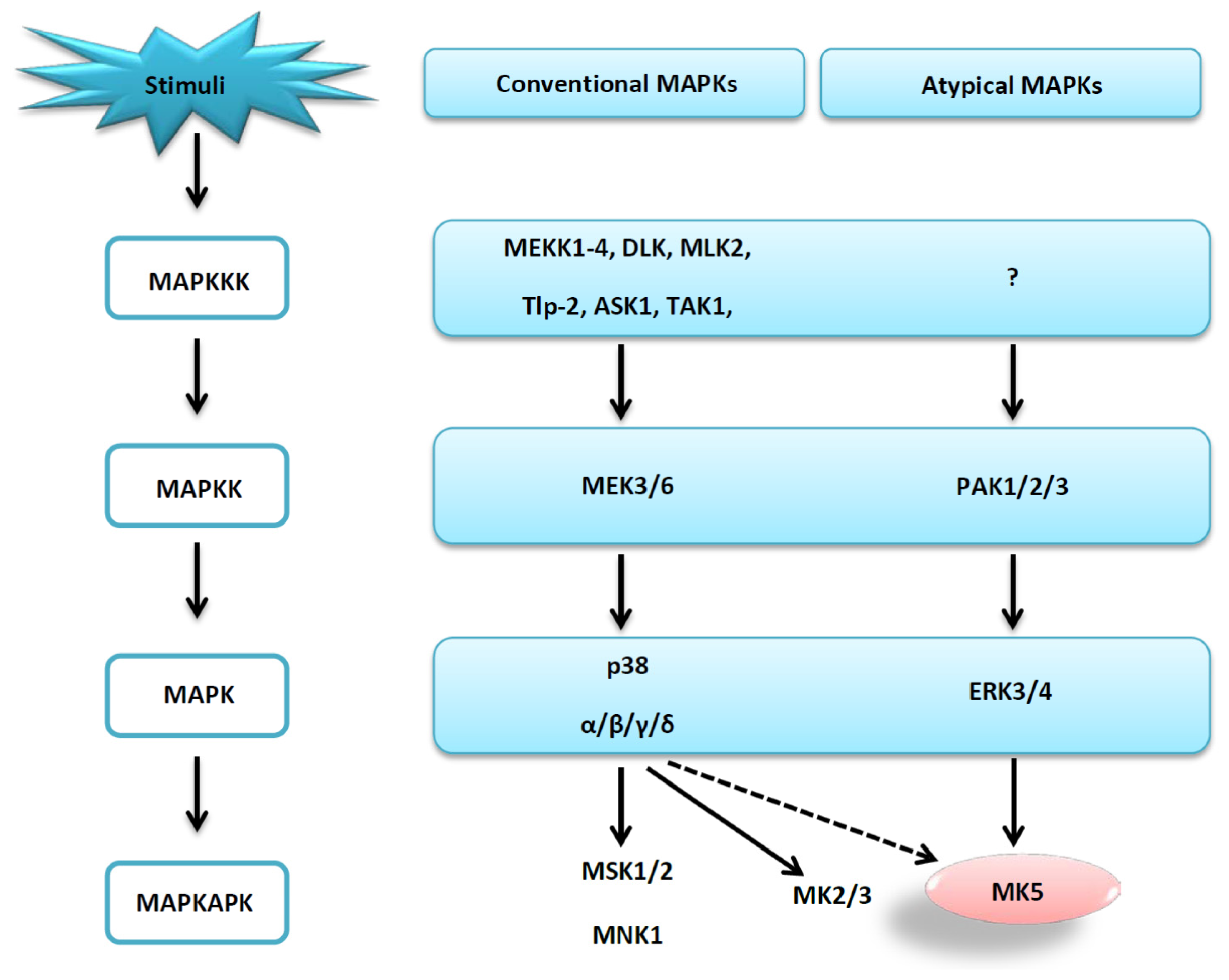
1. Introduction
2. Results and Discussion
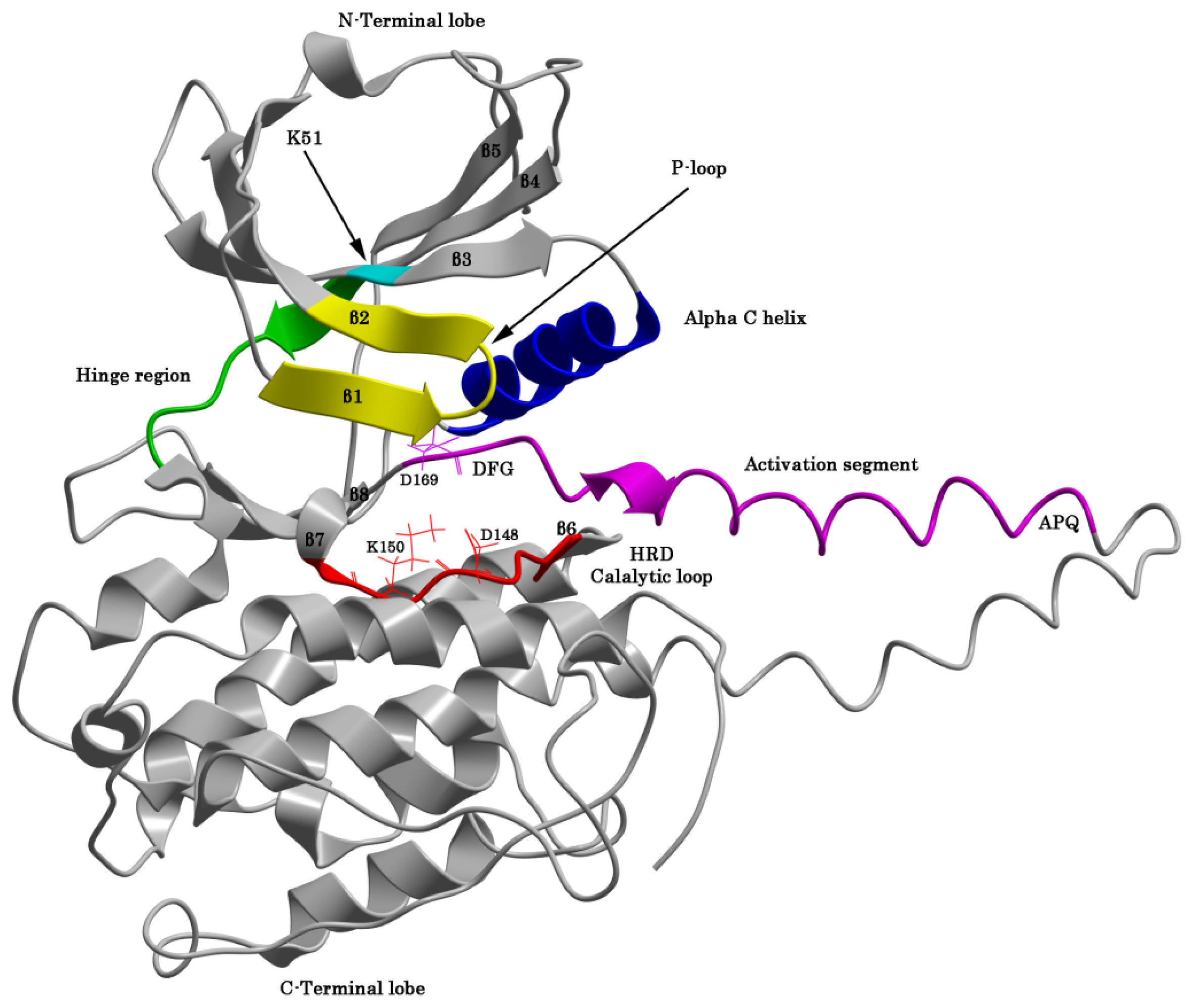
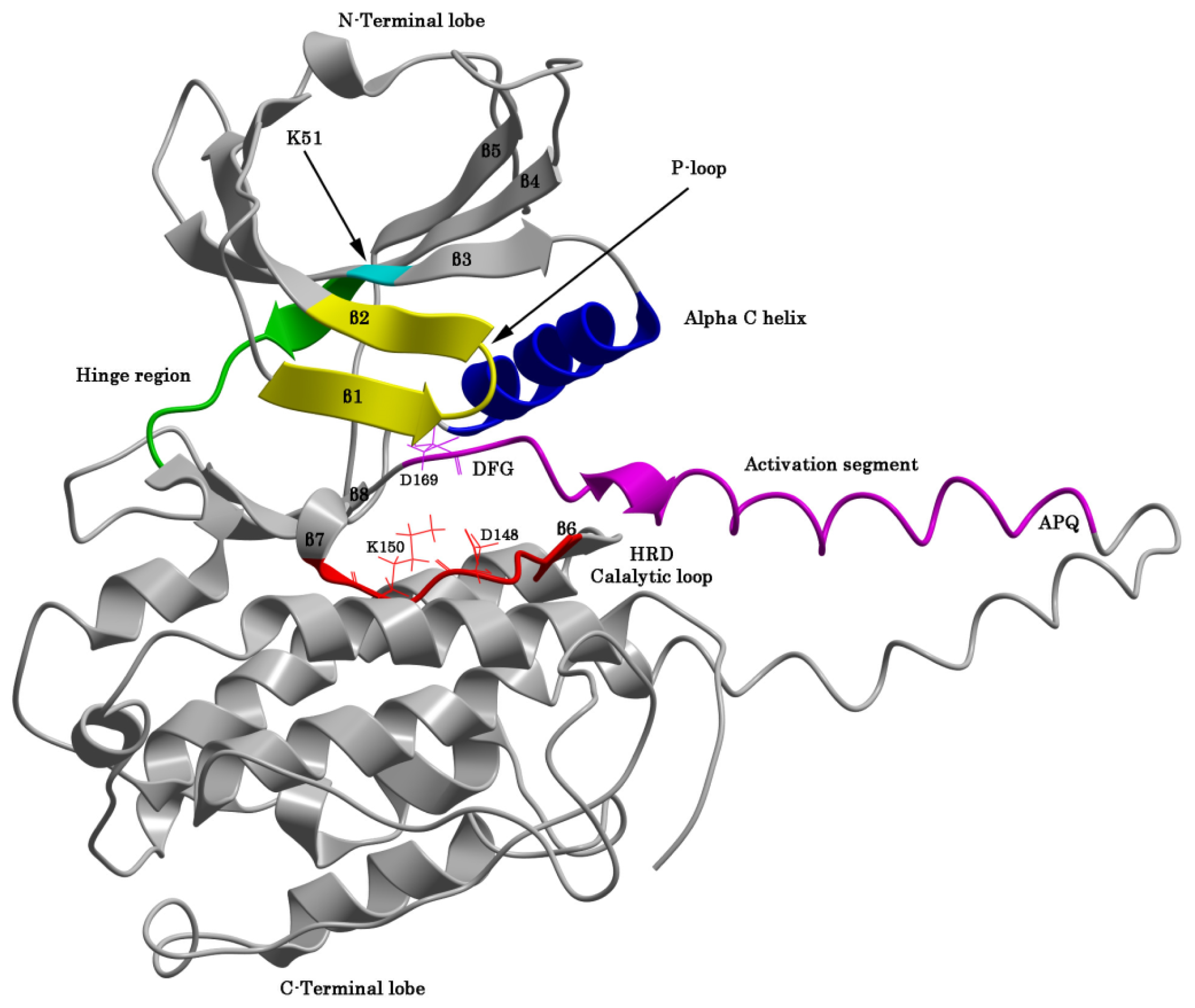
2.1. The Protein Kinase Fold and Homology Models of MK5
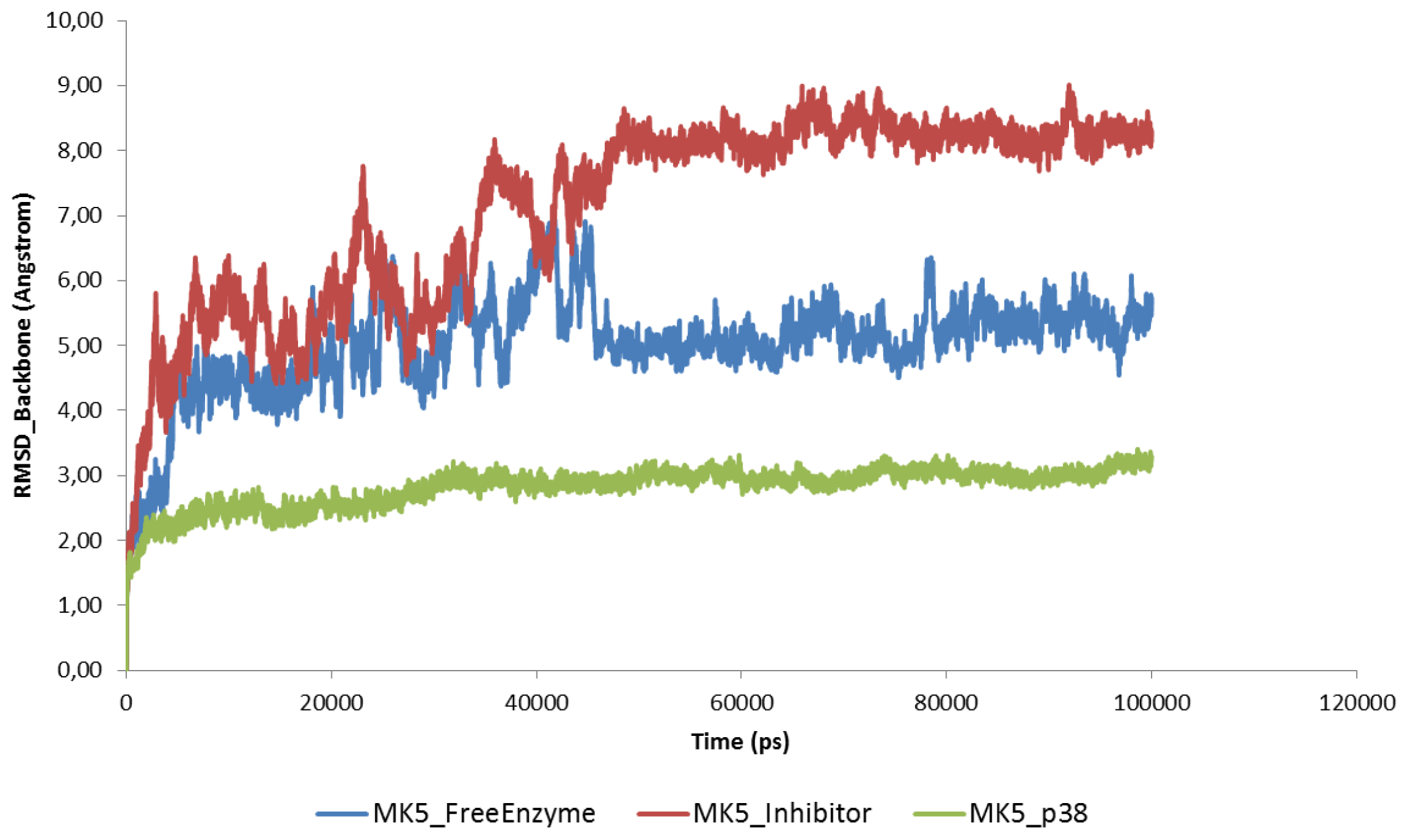
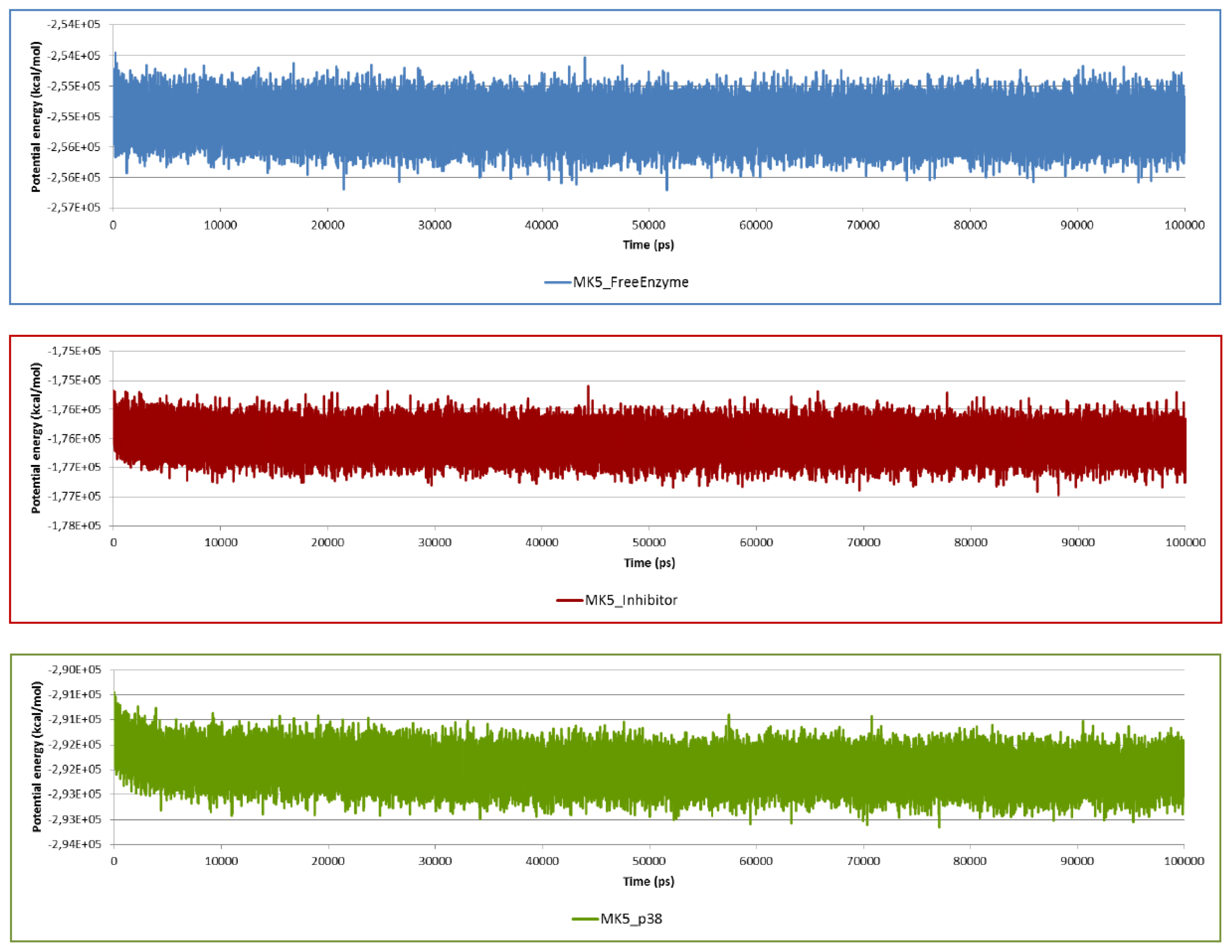
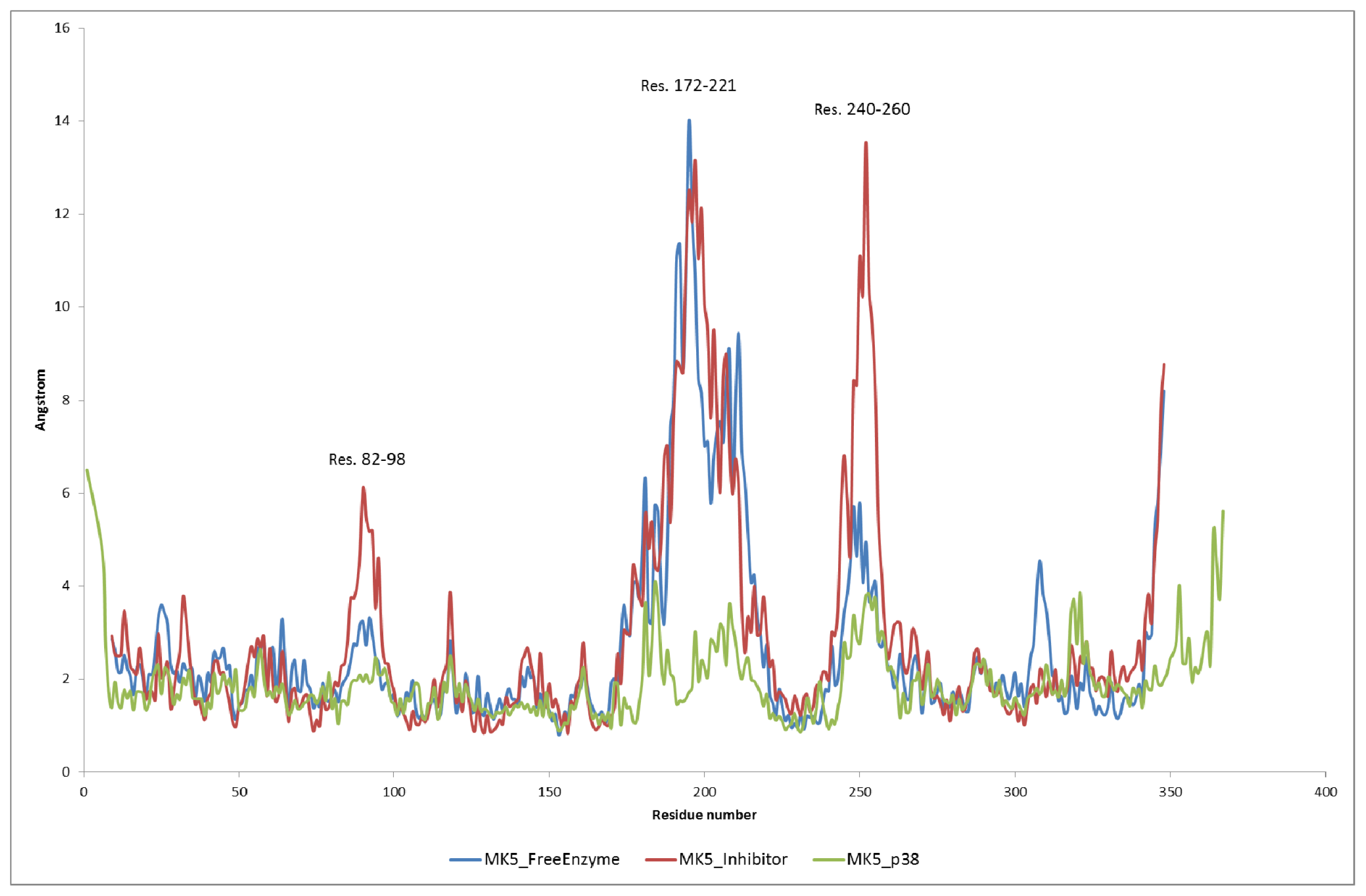
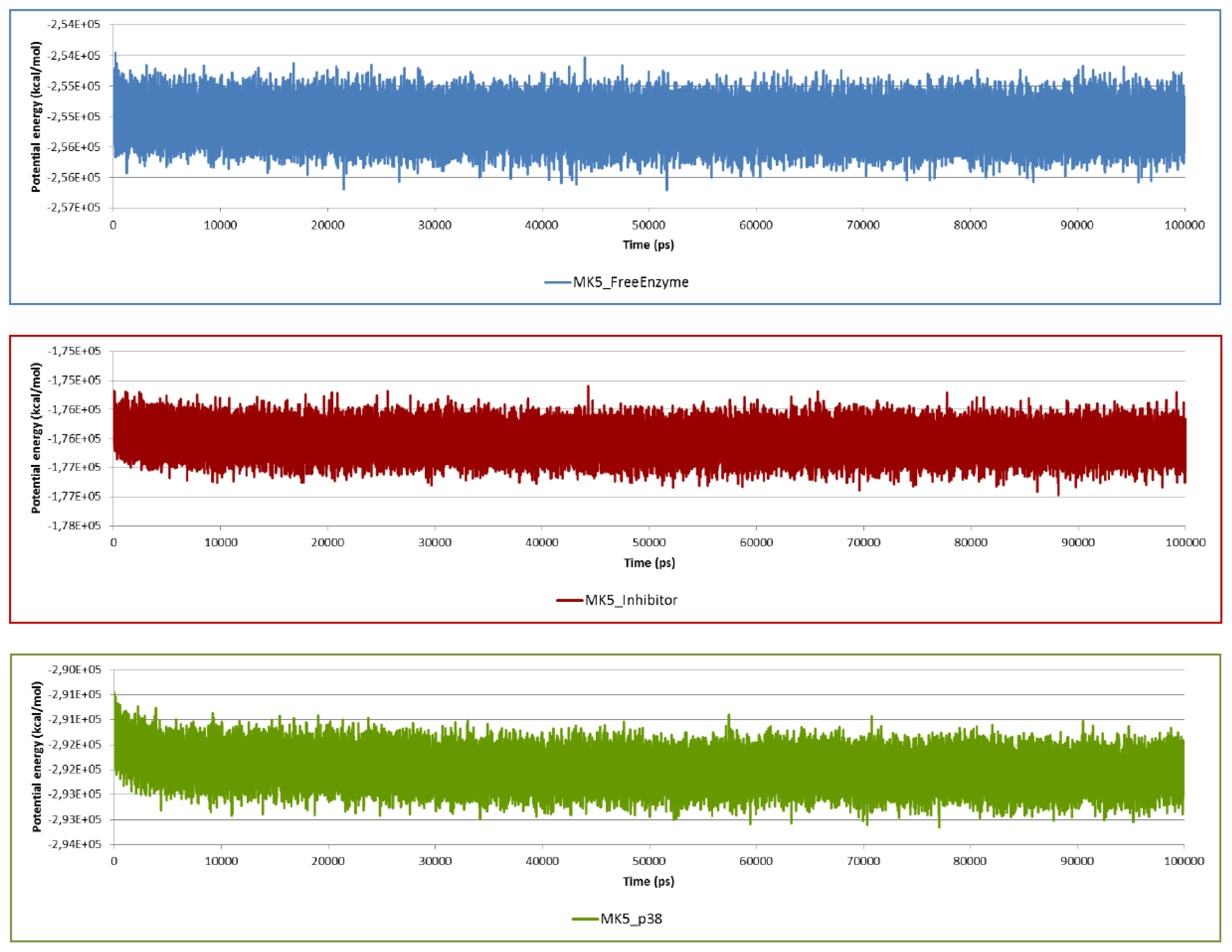
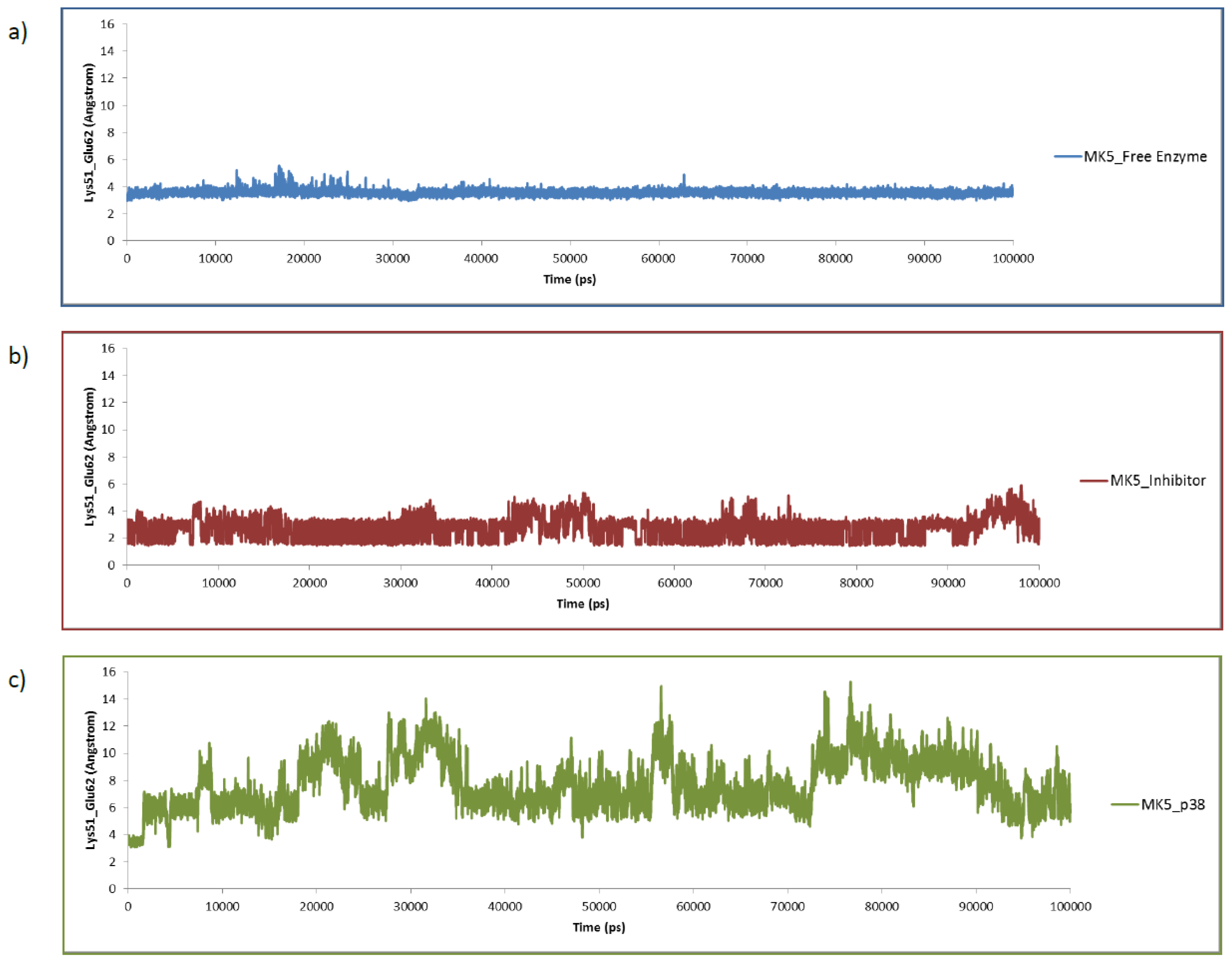
2.2. Stability of the Molecular Systems
2.3. MD of the Free MK5 Model
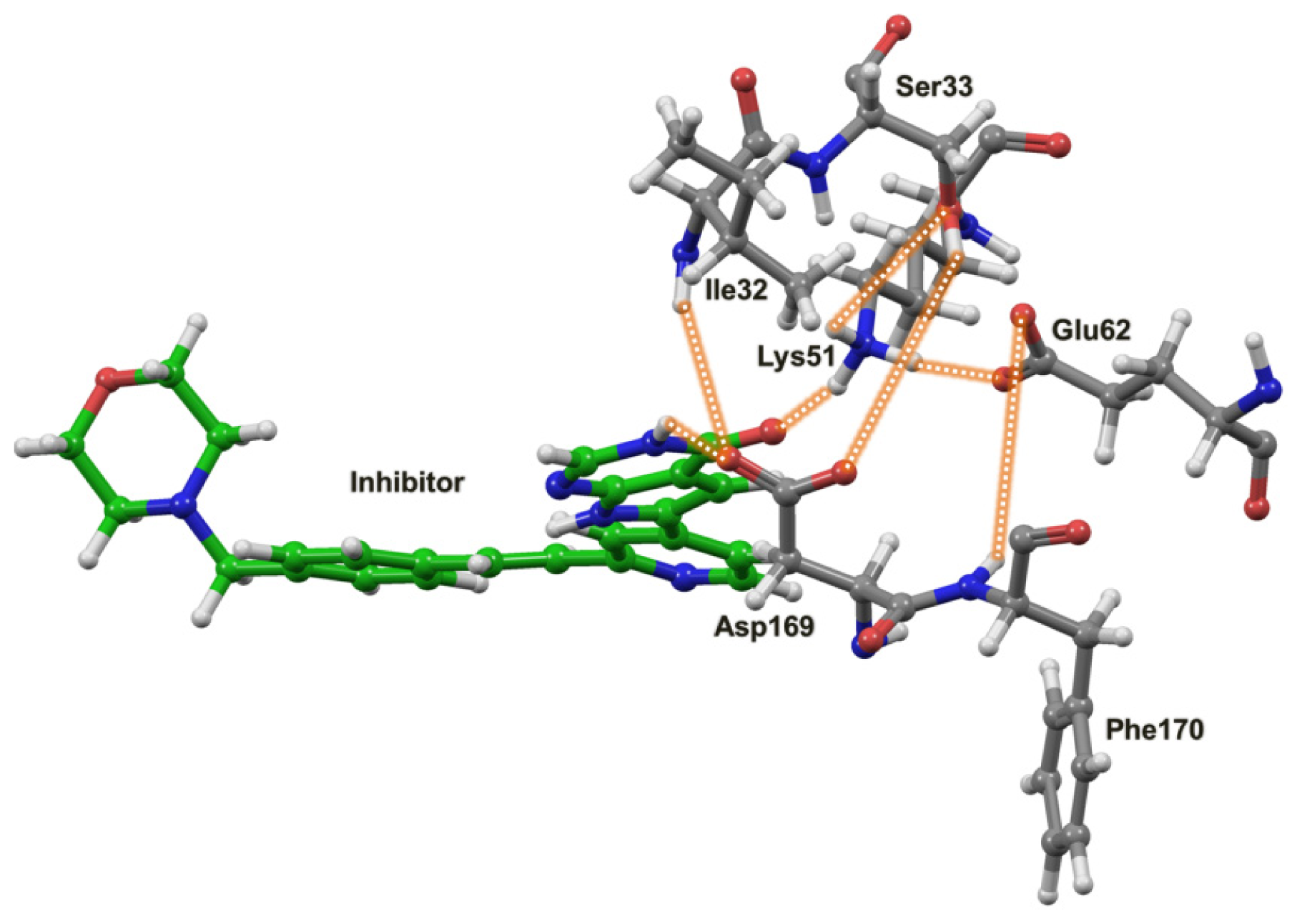
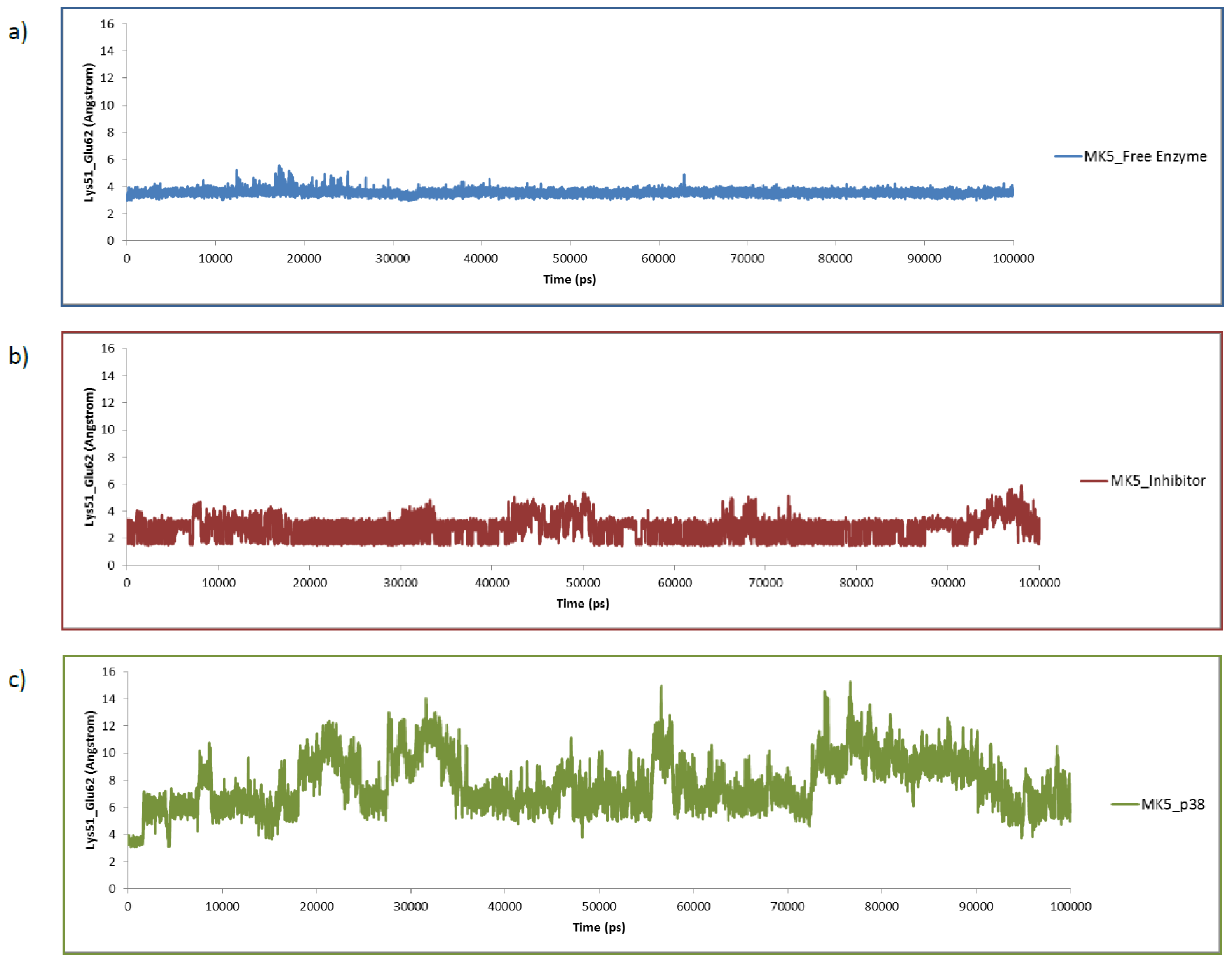
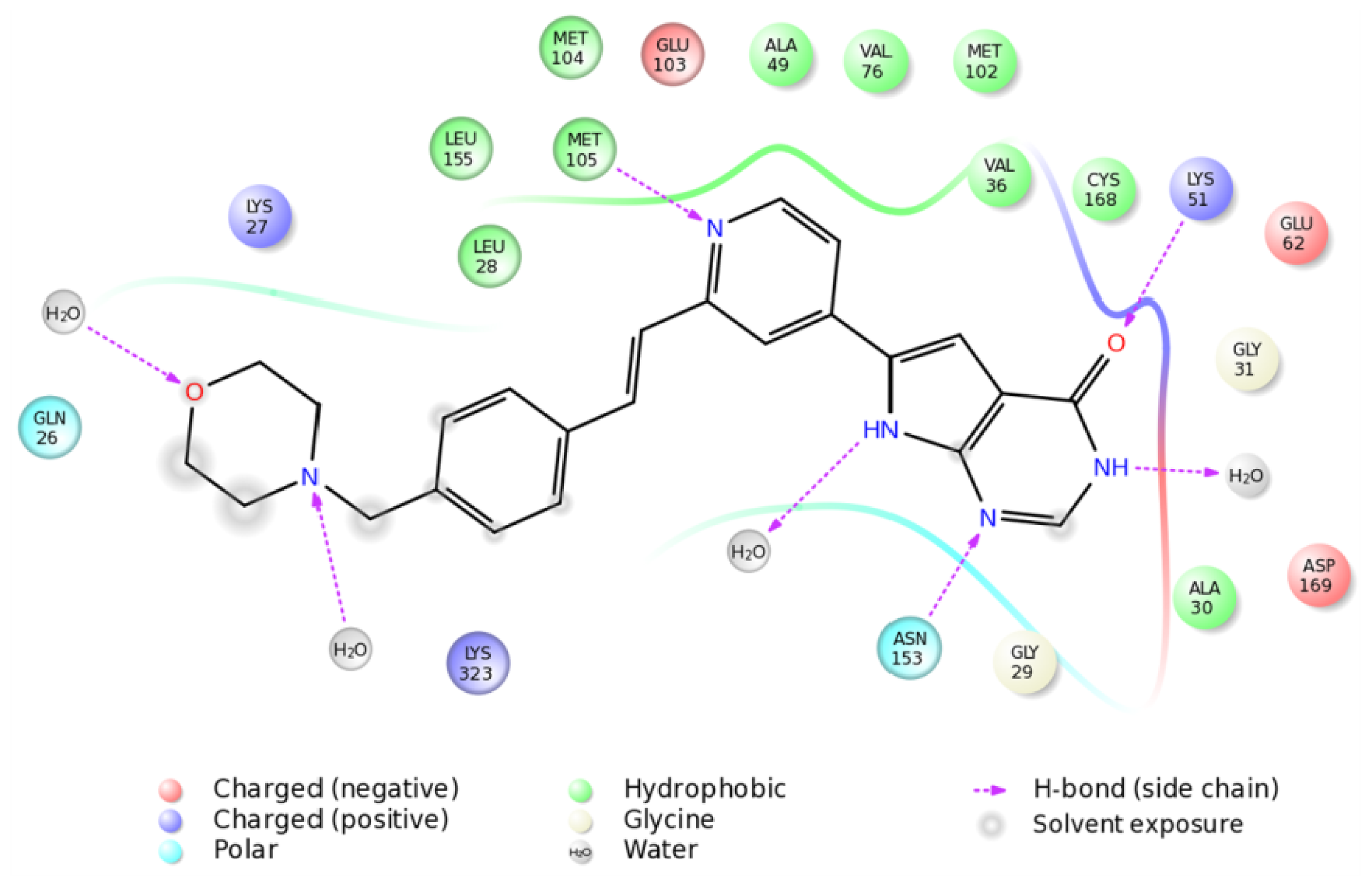
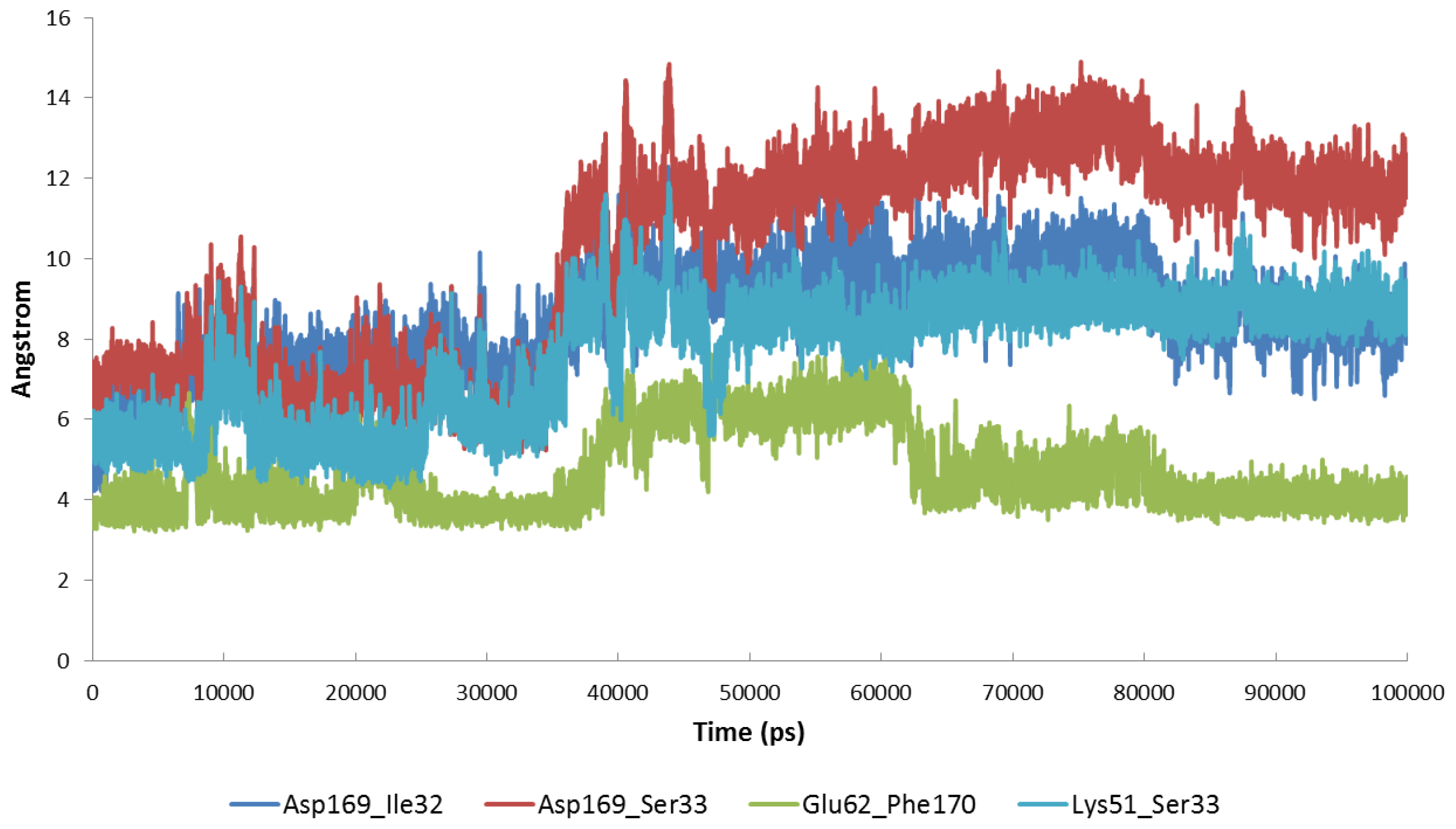
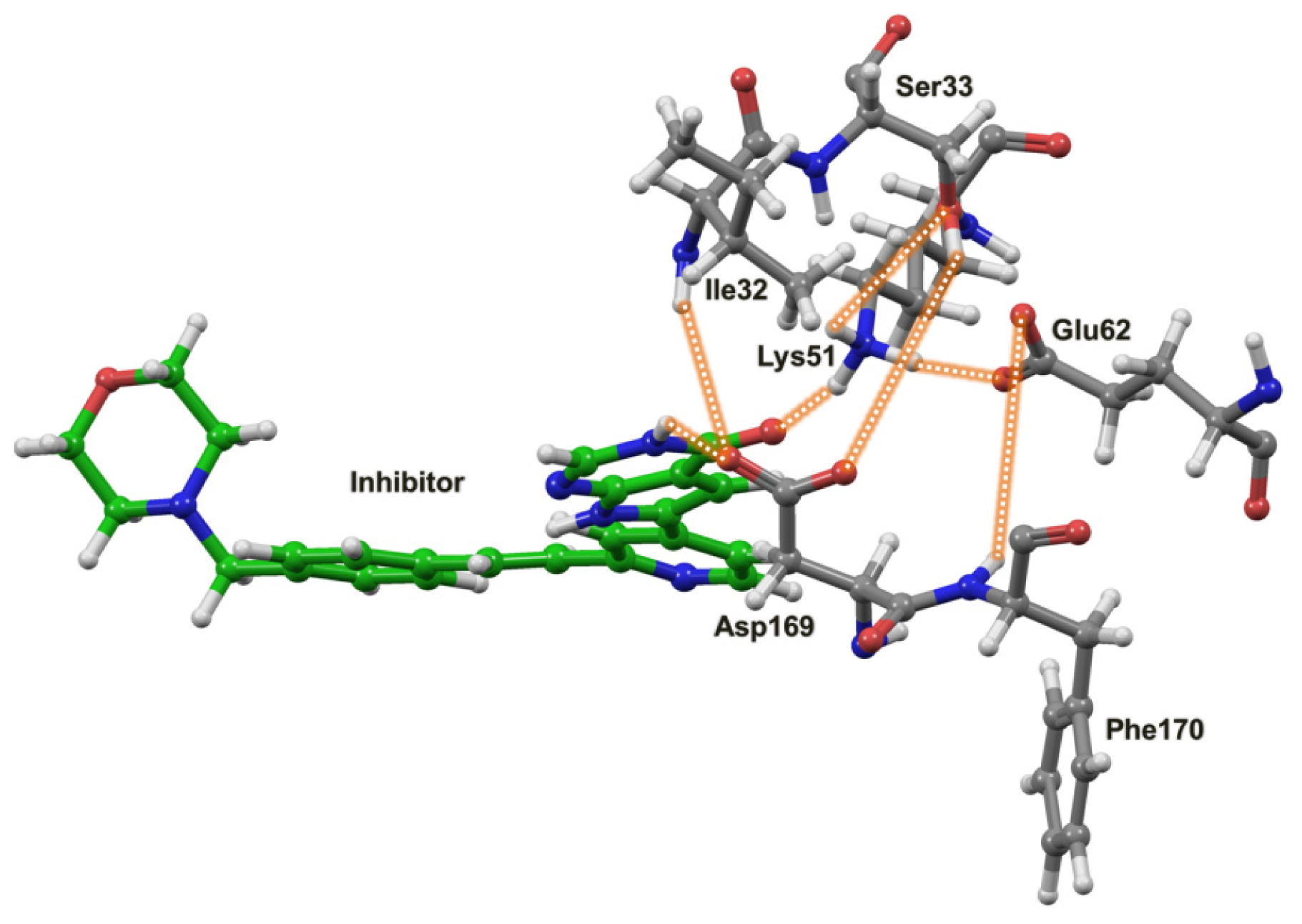
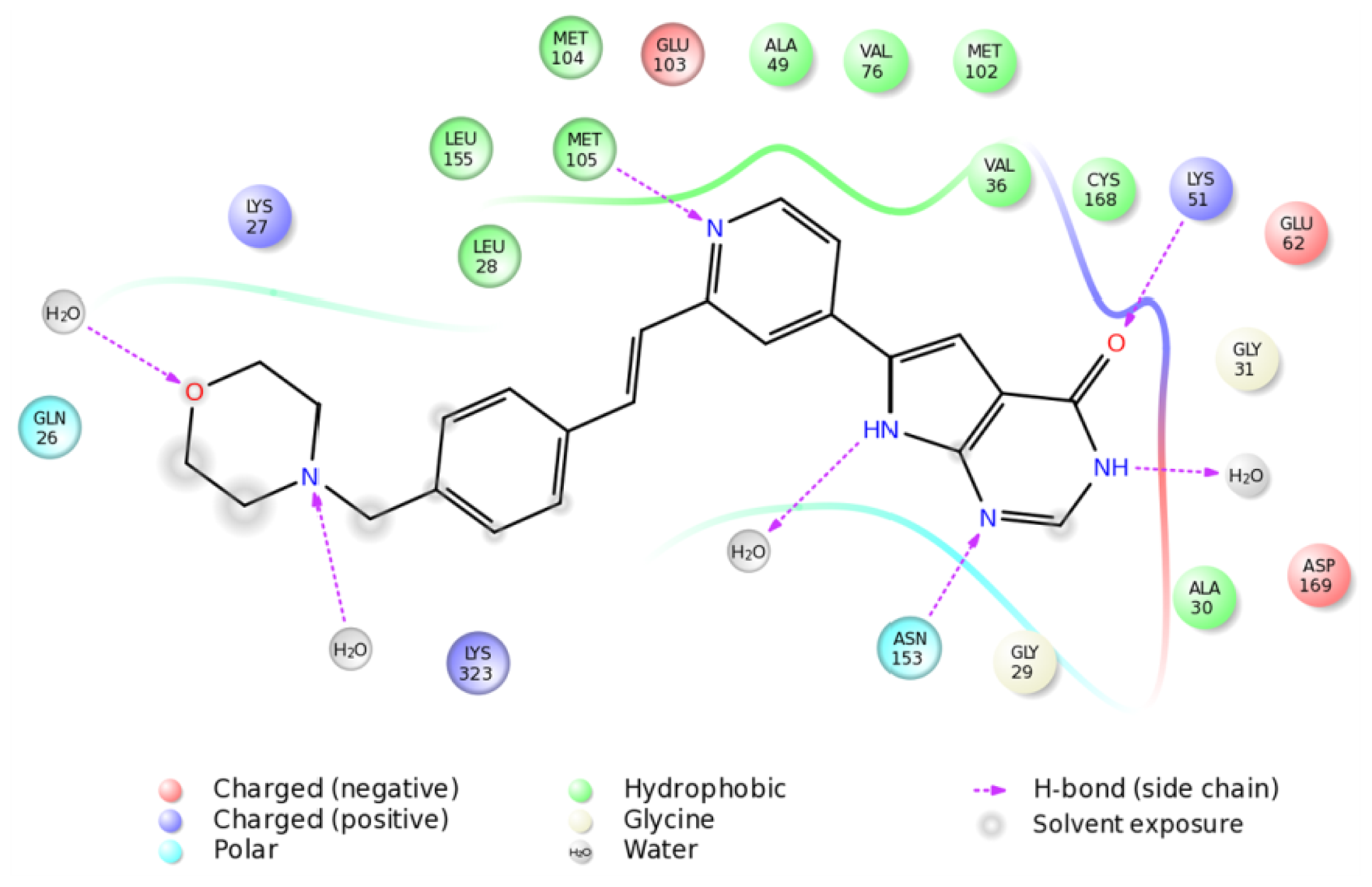
2.4. The MK5-Inhibitor Complex
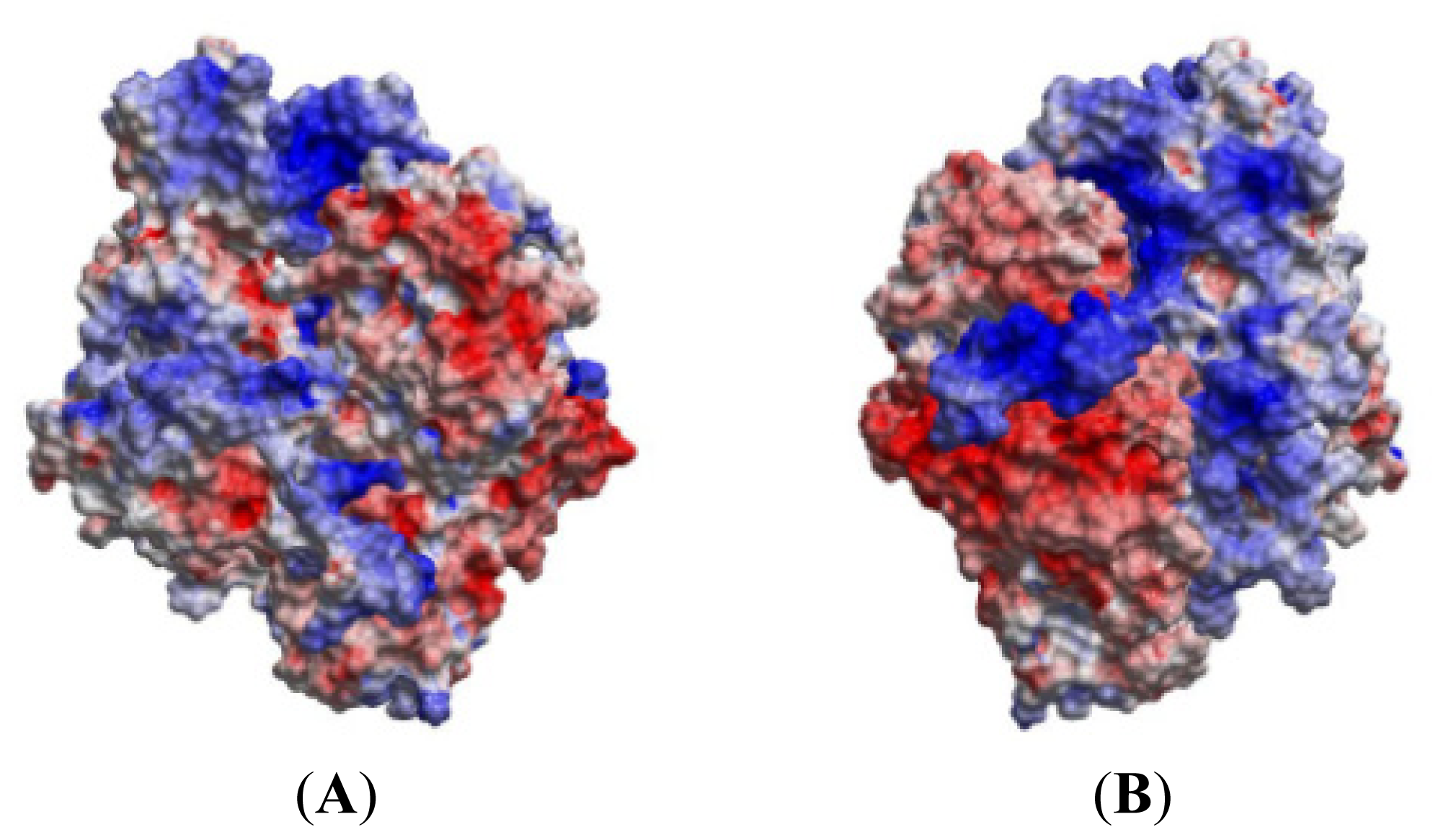
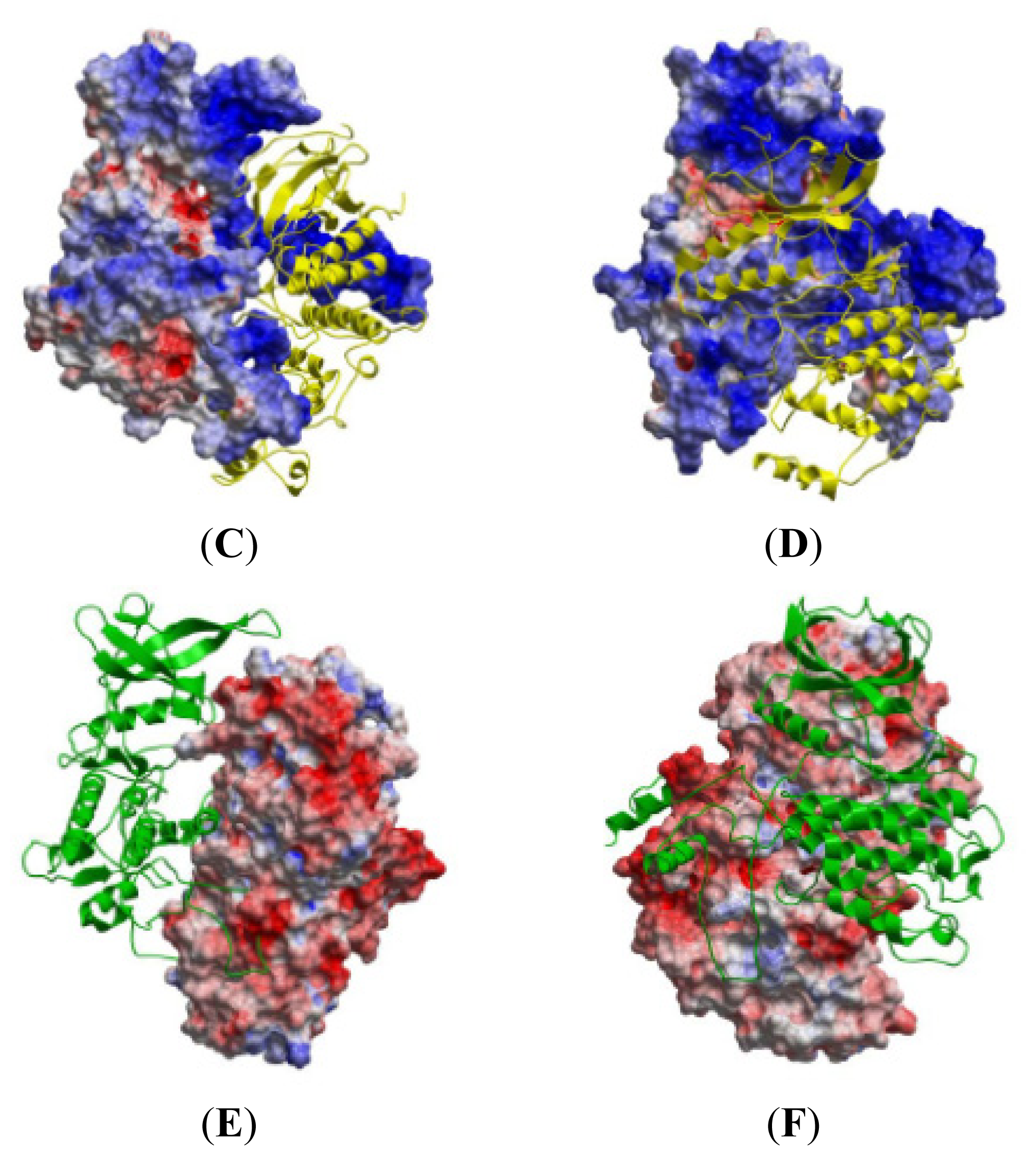
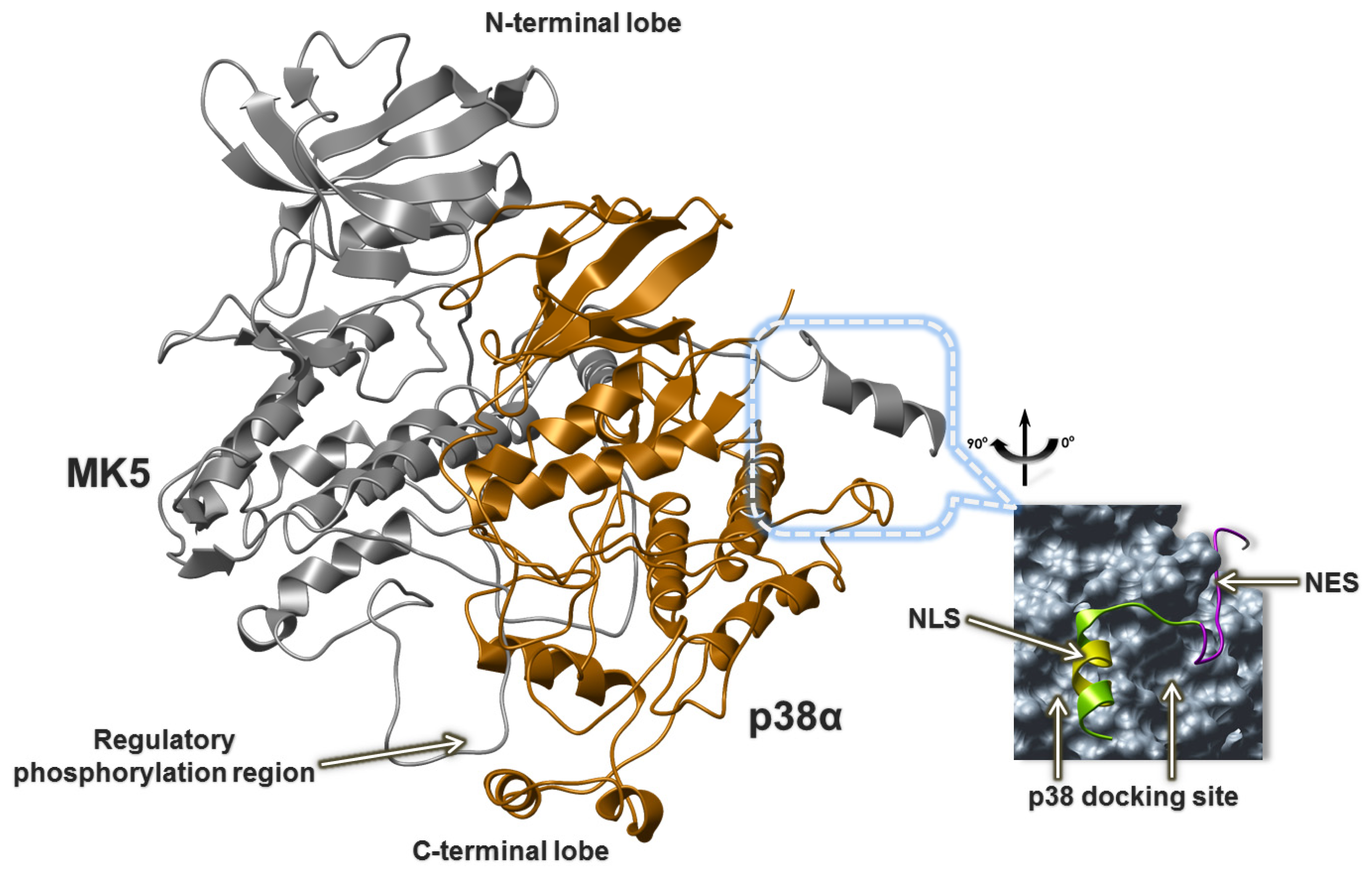
2.5. The MK5-p38 Complex
3. Experimental Section
- 100 ns MD of MK5 alone.
- 100 ns MD of MK5 in complex with a pyrrolopyrimidone-based MK5 inhibitor (Chembel compound id: CHEMBL461139). The inhibitor was in a neutral form.
- 200 ns MD of MK5 in complex with the same inhibitor. The inhibitor was in a neutral form.
- 100 ns of MK5 in complex with p38α.
3.1. Construction of Initial Complexes
3.2. Calculation of Electrostatic Potentials
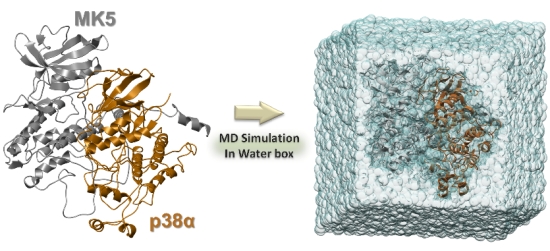
3.3. Solvation and Refinements
3.4. The MD Simulations
3.5. Analysing the Simulations
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar]
- Moens, U.; Kostenko, S. Structure and function of MK5/PRAK: The loner among the mitogen-activated protein kinase-activated protein kinases. Biol. Chem. 2013, 394, 1115–1132. [Google Scholar]
- Kostenko, S.; Shiryaev, A.; Dumitriu, G.; Gerits, N.; Moens, U. Cross-talk between protein kinase A and the MAPK-activated protein kinases RSK1 and MK5. J. Recept. Signal Transduct. Res. 2011, 31, 1–9. [Google Scholar]
- Gaestel, M. MAPKAP kinases-MKs—Two’s company three’s a crowd. Nat. Rev. Mol. Cell Biol. 2006, 7, 120–130. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar]
- Schumacher, S.; Laass, K.; Kant, S.; Shi, Y.; Visel, A.; Gruber, A.D.; Kotlyarov, A.; Gaestel, M. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 2004, 23, 4770–4779. [Google Scholar]
- Ronkina, N.; Kotlyarov, A.; Gaestel, M. MK2 and MK3—A pair of isoenzymes? Front. Biosci. 2008, 13, 5511–5521. [Google Scholar]
- Shiryaev, A.; Moens, U. Mitogen-activated protein kinase p38 and MK2 MK3 and MK5: Menage a trois or menage a quatre? Cell. Signal. 2010, 22, 1185–1192. [Google Scholar]
- Ni, H.; Wang, X.S.; Diener, K.; Yao, Z. MAPKAPK5 a novel mitogen-activated protein kinase (MAPK)-activated protein kinase is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem. Biophys. Res. Commun. 1998, 243, 492–496. [Google Scholar]
- New, L.; Jiang, Y.; Zhao, M.; Liu, K.; Zhu, W.; Flood, L.J.; Kato, Y.; Parry, G.C.; Han, J. PRAK a novel protein kinase regulated by the p38 MAP kinase. EMBO J. 1998, 17, 3372–3384. [Google Scholar]
- Gerits, N.; Mikalsen, T.; Kostenko, S.; Shiryaev, A.; Johannessen, M.; Moens, U. Modulation of F-actin rearrangement by the cyclic AMP/cAMP-dependent protein kinase (PKA) pathway is mediated by MAPK-activated protein kinase 5 and requires PKA-induced nuclear export of MK5. J. Biol. Chem. 2007, 282, 37232–37243. [Google Scholar]
- Anderson, D.R.; Meyers, M.J.; Vernier, W.F.; Mahoney, M.W.; Kurumbail, R.G.; Caspers, N.; Poda, G.I.; Schindler, J.F.; Reitz, D.B.; Mourey, R.J. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2). J. Med. Chem. 2007, 50, 2647–2654. [Google Scholar]
- Seternes, O.M.; Johansen, B.; Hegge, B.; Johannessen, M.; Keyse, S.M.; Moens, U. Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress. Mol. Cell. Biol. 2002, 22, 6931–6945. [Google Scholar]
- New, L.; Jiang, Y.; Han, J. Regulation of PRAK subcellular location by p38 MAP kinases. Mol. Biol. Cell 2003, 14, 2603–2616. [Google Scholar]
- Kostenko, S.; Shiryaev, A.; Gerits, N.; Dumitriu, G.; Klenow, H.; Johannessen, M.; Moens, U. Serine residue 115 of MAPK-activated protein kinase MK5 is crucial for its PKA-regulated nuclear export and biological function. Cell. Mol. Life Sci. 2011, 68, 847–862. [Google Scholar]
- Kant, S.; Schumacher, S.; Singh, M.K.; Kispert, A.; Kotlyarov, A.; Gaestel, M. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J. Biol. Chem. 2006, 281, 35511–35519. [Google Scholar]
- Hansen, C.A.; Bartek, J.; Jensen, S. A functional link between the human cell cycle-regulatory phosphatase Cdc14A and the atypical mitogen-activated kinase Erk3. Cell Cycle 2008, 7, 325–334. [Google Scholar]
- Deleris, P.; Rousseau, J.; Coulombe, P.; Rodier, G.; Tanguay, P.L.; Meloche, S. Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding activation and cytoplasmic relocalization of MK5. J. Cell. Physiol. 2008, 217, 778–788. [Google Scholar]
- Sohal, D.; Yeatts, A.; Ye, K.; Pellagatti, A.; Zhou, L.; Pahanish, P.; Mo, Y.; Bhagat, T.; Mariadason, J.; Boultwood, J.; et al. Meta-analysis of microarray studies reveals a novel hematopoietic progenitor cell signature and demonstrates feasibility of inter-platform data integration. PLoS One 2008, 3, e2965. [Google Scholar]
- Perander, M.; Keyse, S.M.; Seternes, O.M. Does MK5 reconcile classical and atypical MAP kinases? Front. Biosci. 2008, 13, 4617–4624. [Google Scholar]
- Gerits, N.; Shiryaev, A.; Kostenko, S.; Klenow, H.; Shiryaeva, O.; Johannessen, M.; Moens, U. The transcriptional regulation and cell-specific expression of the MAPK-activated protein kinase MK5. Cell. Mol. Biol. Lett. 2009, 14, 548–574. [Google Scholar]
- Tak, H.; Jang, E.; Kim, S.B.; Park, J.; Suk, J.; Yoon, Y.S.; Ahn, J.K.; Lee, J.H.; Joe, C.O. 14-3-3epsilon inhibits MK5-mediated cell migration by disrupting F-actin polymerization. Cell. Signal. 2007, 19, 2379–2387. [Google Scholar]
- Kostenko, S.; Johannessen, M.; Moens, U. PKA-induced F-actin rearrangement requires phosphorylation of Hsp27 by the MAPKAP kinase MK5. Cell. Signal. 2009, 21, 712–718. [Google Scholar]
- Kostenko, S.; Dumitriu, G.; Laegreid, K.J.; Moens, U. Physiological roles of mitogen-activated-protein- kinase-activated p38-regulated/activated protein kinase. World J. Biol. Chem. 2011, 2, 73–89. [Google Scholar]
- Stohr, N.; Kohn, M.; Lederer, M.; Glass, M.; Reinke, C.; Singer, R.H.; Huttelmaier, S. IGF2BP1 promotes cell migration by regulating MK5 and PTEN signaling. Genes Dev. 2012, 26, 176–189. [Google Scholar]
- Yoshizuka, N.; Chen, R.M.; Xu, Z.; Liao, R.; Hong, L.; Hu, W.Y.; Yu, G.; Han, J.; Chen, L.; Sun, P. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cell. Biol. 2012, 32, 606–618. [Google Scholar]
- Yoshizuka, N.; Lai, M.; Liao, R.; Cook, R.; Xiao, C.; Han, J.; Sun, P. PRAK suppresses oncogenic ras-induced hematopoietic cancer development by antagonizing the JNK pathway. Mol. Cancer Res. 2012, 10, 810–820. [Google Scholar]
- Kress, T.R.; Cannell, I.G.; Brenkman, A.B.; Samans, B.; Gaestel, M.; Roepman, P.; Burgering, B.M.; Bushell, M.; Rosenwald, A.; Eilers, M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol. Cell 2011, 41, 445–457. [Google Scholar]
- Tanguay, P.L.; Rodier, G.; Meloche, S. C-terminal domain phosphorylation of ERK3 controlled by Cdk1 and Cdc14 regulates its stability in mitosis. Biochem. J. 2010, 428, 103–111. [Google Scholar]
- Coulombe, P.; Rodier, G.; Pelletier, S.; Pellerin, J.; Meloche, S. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell. Biol. 2003, 23, 4542–4558. [Google Scholar]
- Chow, K.T.; Timblin, G.A.; McWhirter, S.M.; Schlissel, M.S. MK5 activates Rag transcription via Foxo1 in developing B cells. J. Exp. Med. 2013, 210, 1621–1634. [Google Scholar]
- Koh, G.C.; Schreiber, M.F.; Bautista, R.; Maude, R.R.; Dunachie, S.; Limmathurotsakul, D.; Day, N.P.; Dougan, G.; Peacock, S.J. Host responses to melioidosis and tuberculosis are both dominated by interferon-mediated signaling. PLoS One 2013, 8, e54961. [Google Scholar]
- Gerits, N.; van Belle, W.; Moens, U. Transgenic mice expressing constitutive active MAPKAPK5 display gender-dependent differences in exploration and activity. Behav. Brain Funct. 2007, 3, 58. [Google Scholar]
- Brand, F.; Schumacher, S.; Kant, S.; Menon, M.B.; Simon, R.; Turgeon, B.; Britsch, S.; Meloche, S.; Gaestel, M.; Kotlyarov, A. The extracellular signal-regulated kinase 3 (mitogen-activated protein kinase 6 [MAPK6])-MAPK-activated protein kinase 5 signaling complex regulates septin function and dendrite morphology. Mol. Cell. Biol. 2012, 32, 2467–2478. [Google Scholar]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar]
- Shi, Y.; Kotlyarov, A.; Laabeta, K.; Gruber, A.D.; Butt, E.; Marcus, K.; Meyer, H.E.; Friedrich, A.; Volk, H.D.; Gaestel, M. Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol. Cell. Biol. 2003, 23, 7732–7741. [Google Scholar]
- White, A.; Pargellis, C.A.; Studts, J.M.; Werneburg, B.G.; Farmer, B.T., 2nd. Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 6353–6358. [Google Scholar]
- Underwood, K.W.; Parris, K.D.; Federico, E.; Mosyak, L.; Czerwinski, R.M.; Shane, T.; Taylor, M.; Svenson, K.; Liu, Y.; Hsiao, C.L.; et al. Catalytically active MAP KAP kinase 2 structures in complex with staurosporine and ADP reveal differences with the autoinhibited enzyme. Structure 2003, 11, 627–636. [Google Scholar]
- Revesz, L.; Schlapbach, A.; Aichholz, R.; Feifel, R.; Hawtin, S.; Heng, R.; Hiestand, P.; Jahnke, W.; Koch, G.; Kroemer, M.; et al. In vivo and in vitro SAR of tetracyclic MAPKAP-K2 (MK2) inhibitors. Part I. Bioorg. Med. Chem. Lett. 2010, 20, 4715–4718. [Google Scholar]
- Revesz, L.; Schlapbach, A.; Aichholz, R.; Dawson, J.; Feifel, R.; Hawtin, S.; Littlewood-Evans, A.; Koch, G.; Kroemer, M.; Mobitz, H.; et al. In vivo and in vitro SAR of tetracyclic MAPKAP-K2 (MK2) inhibitors. Part II. Bioorg. Med. Chem. Lett. 2010, 20, 4719–4723. [Google Scholar]
- Fujino, A.; Fukushima, K.; Kubota, T.; Kosugi, T.; Takimoto-Kamimura, M. Crystal structure of human cyclin-dependent kinase-2 complex with MK2 inhibitor TEI-I01800: Insight into the selectivity. J. Synchrotron Radiat. 2013, 20(Pt. 6), 905–909. [Google Scholar]
- Barf, T.; Kaptein, A.; de Wilde, S.; van der Heijden, R.; van Someren, R.; Demont, D.; Schultz-Fademrecht, C.; Versteegh, J.; van Zeeland, M.; Seegers, N.; et al. Structure-based lead identification of ATP-competitive MK2 inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 3818–3822. [Google Scholar]
- Cheng, R.; Felicetti, B.; Palan, S.; Toogood-Johnson, I.; Scheich, C.; Barker, J.; Whittaker, M.; Hesterkamp, T. High-resolution crystal structure of human Mapkap kinase 3 in complex with a high affinity ligand. Protein Sci. 2010, 19, 168–173. [Google Scholar]
- Lindin, I.; Wuxiuer, Y.; Kufareva, I.; Abagyan, R.; Moens, U.; Sylte, I.; Ravna, A.W. Homology modeling and ligand docking of Mitogen-activated protein kinase-activated protein kinase 5 (MK5). Theor. Biol. Med. Model. 2013, 10, 56. [Google Scholar]
- Emrick, M.A.; Lee, T.; Starkey, P.J.; Mumby, M.C.; Resing, K.A.; Ahn, N.G. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 18101–18106. [Google Scholar]
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure function and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar]
- Nicolau, N., Jr; Giuliatti, S. Modeling and molecular dynamics of the intrinsically disordered e7 proteins from high- and low-risk types of human papillomavirus. J. Mol. Model. 2013, 19, 4025–4037. [Google Scholar]
- Cashman, D.J.; Ortega, D.R.; Zhulin, I.B.; Baudry, J. Homology modeling of the CheW coupling protein of the chemotaxis signaling complex. PLoS One 2013, 8, e70705. [Google Scholar]
- Wang, Z.; Li, S.; Sun, L.; Fan, J.; Liu, Z. Comparative analyses of lipoprotein lipase hepatic lipase and endothelial lipase and their binding properties with known inhibitors. PLoS One 2013, 8, e72146. [Google Scholar]
- Gabrielsen, M.; Ravna, A.W.; Kristiansen, K.; Sylte, I. Substrate binding and translocation of the serotonin transporter studied by docking and molecular dynamics simulations. J. Mol. Model. 2012, 18, 1073–1085. [Google Scholar]
- Rahman, K.S.; Cui, G.; Harvey, S.C.; McCarty, N.A. Modeling the conformational changes underlying channel opening in CFTR. PLoS One 2013, 8, e74574. [Google Scholar]
- Yuan, S.; Wu, R.; Latek, D.; Trzaskowski, B.; Filipek, S. Lipid receptor S1P(1) activation scheme concluded from microsecond all-atom molecular dynamics simulations. PLoS Comput. Biol. 2013, 9, e1003261. [Google Scholar]
- Yuan, S.; Vogel, H.; Filipek, S. The role of water and sodium ions in the activation of the mu-opioid receptor. Angew. Chem. Int. Ed. Engl. 2013, 52, 10112–10115. [Google Scholar]
- Arumugam, K.; Crouzy, S.; Chevigne, A.; Seguin-Devaux, C.; Schmit, J.C. Structure prediction of GPCRs using piecewise homologs and application to the human CCR5 chemokine receptor: Validation through agonist and antagonist docking. J. Biomol. Struct. Dyn. 2013. [Google Scholar] [CrossRef]
- Schlapbach, A.; Feifel, R.; Hawtin, S.; Heng, R.; Koch, G.; Moebitz, H.; Revesz, L.; Scheufler, C.; Velcicky, J.; Waelchli, R.; et al. Pyrrolo-pyrimidones: A novel class of MK2 inhibitors with potent cellular activity. Bioorg. Med. Chem. Lett. 2008, 18, 6142–6146. [Google Scholar]
- Li, Q.; Zhang, N.; Zhang, D.; Wang, Y.; Lin, T.; Zhou, H.; Ye, Z.; Zhang, F.; Lin, S.C.; Han, J. Determinants that control the distinct subcellular localization of p38alpha-PRAK and p38beta-PRAK complexes. J. Biol. Chem. 2008, 283, 11014–11023. [Google Scholar]
- Shan, Y.; Seeliger, M.A.; Eastwood, M.P.; Frank, F.; Xu, H.; Jensen, M.O.; Dror, R.O.; Kuriyan, J.; Shaw, D.E. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 139–144. [Google Scholar]
- Totrov, M.; Abagyan, R. Rapid boundary element solvation electrostatics calculations in folding simulations: Successful folding of a 23-residue peptide. Biopolymers 2001, 60, 124–133. [Google Scholar]
- Maestro9.1; Schrödinger: LLC, New York, NY, USA, 2010.
- Jorgensen, J.H.; Swenson, J.M.; Tenover, F.C.; Barry, A.; Ferraro, M.J.; Murray, P.R.; Reller, L.B. Development of interpretive criteria and quality control limits for macrolide and clindamycin susceptibility testing of Streptococcus pneumoniae. J. Clin. Microbiol. 1996, 34, 2679–2684. [Google Scholar]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635. [Google Scholar]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-time-step molecular dynamics algorithm for macromolecules. J. Chem. Phys. 1994, 98, 6885–6892. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MK5 | p38α | Percentages of frames with the atomic distance < 3.5 Å | Minimum atomic distance (Å) during 100 ns MD | |
|---|---|---|---|---|
| Location | Amino acid | Amino acid | ||
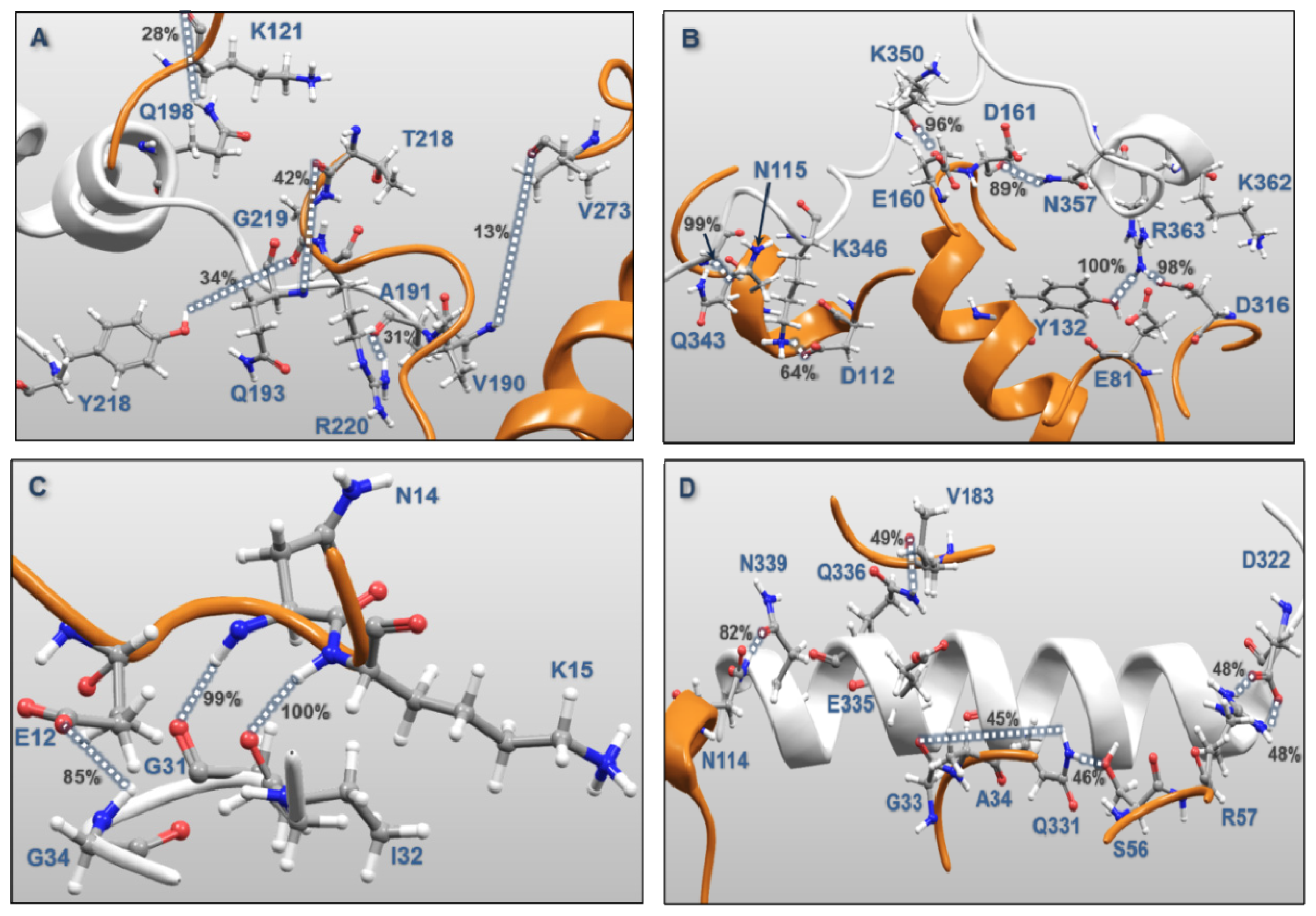
| Activation Segment (Figure 12A) | Val190 | Val273 | 12.8 | 1.57 |
| Gln193 | Arg220 | 1.5 | 1.59 | |
| Gln193 | Thr218 | 41.2 | 1.55 | |
| Tyr218 | Gly219 | 34.1 | 1.49 | |
| Gln198 | Lys121 | 28.5 | 1.56 | |
| Ala191 | Arg220 | 31.1 | 1.47 | |
| C-terminal End (Figure 12B) | Lys362 | Glu81 | 0.7 | 2.75 |
| Lys362 | Asp316 | 15.3 | 1.43 | |
| Arg363 | Asp316 | 0.7 | 1.61 | |
| Arg363 | Asp316 | 98.2 | 1.41 | |
| Arg363 | Tyr132 | 99.9 | 1.48 | |
| Asn357 | Asp161 | 89.3 | 1.51 | |
| Lys350 | Asp161 | 24.0 | 1.42 | |
| Lys350 | Glu160 | 96.1 | 1.50 | |
| Gln343 | Asn115 | 99.4 | 1.48 | |
| Lys346 | Asp112 | 63.8 | 1.38 | |
| P-loop (Figure 12C) | Gly31 | Lys15 | 100.0 | 1.55 |
| Ile32 | Asn114 | 99.1 | 1.53 | |
| Gly34 | Glu12 | 84.7 | 1.50 | |
| αH helix (Figure 12D) | Asp322 | Arg57 | 48.4 | 1.40 |
| Asp322 | Arg57 | 48.6 | 1.40 | |
| Gln331 | Ser56 | 46.0 | 1.57 | |
| Gln331 | Gly33 | 45.3 | 1.51 | |
| Gln335 | Ala34 | 28.4 | 1.52 | |
| Gln336 | Val183 | 49.2 | 1.57 | |
| Asn339 | Asn114 | 81.6 | 1.52 | |
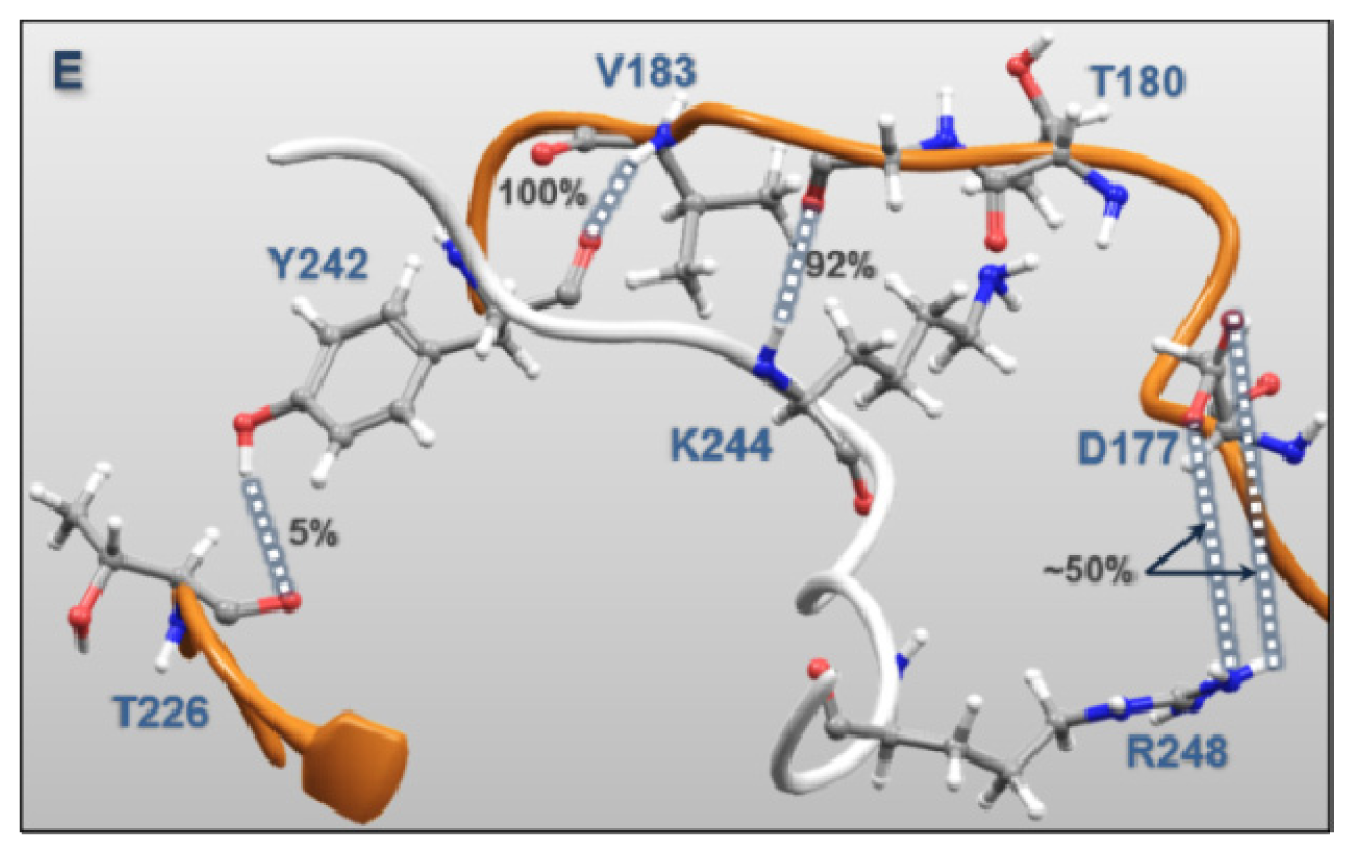
| Regulatory Phosphorylation Region (Figure 12E) | Tyr242 | Val183 | 100.0 | 1.51 |
| Lys244 | Gly181 | 92.0 | 1.47 | |
| Tyr242 | Thr226 | 4.8 | 1.57 | |
| Lys244 | Thr180 | 9.2 | 1.59 | |
| Arg248 | Asp177 | 49.1 | 1.39 | |
| Arg248 | Asp177 | 55.4 | 1.40 | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindin, I.; Wuxiuer, Y.; Ravna, A.W.; Moens, U.; Sylte, I. Comparative Molecular Dynamics Simulations of Mitogen-Activated Protein Kinase-Activated Protein Kinase 5. Int. J. Mol. Sci. 2014, 15, 4878-4902. https://doi.org/10.3390/ijms15034878
Lindin I, Wuxiuer Y, Ravna AW, Moens U, Sylte I. Comparative Molecular Dynamics Simulations of Mitogen-Activated Protein Kinase-Activated Protein Kinase 5. International Journal of Molecular Sciences. 2014; 15(3):4878-4902. https://doi.org/10.3390/ijms15034878
Chicago/Turabian StyleLindin, Inger, Yimingjiang Wuxiuer, Aina Westrheim Ravna, Ugo Moens, and Ingebrigt Sylte. 2014. "Comparative Molecular Dynamics Simulations of Mitogen-Activated Protein Kinase-Activated Protein Kinase 5" International Journal of Molecular Sciences 15, no. 3: 4878-4902. https://doi.org/10.3390/ijms15034878
APA StyleLindin, I., Wuxiuer, Y., Ravna, A. W., Moens, U., & Sylte, I. (2014). Comparative Molecular Dynamics Simulations of Mitogen-Activated Protein Kinase-Activated Protein Kinase 5. International Journal of Molecular Sciences, 15(3), 4878-4902. https://doi.org/10.3390/ijms15034878