Int6/eIF3e Is Essential for Proliferation and Survival of Human Glioblastoma Cells

Abstract
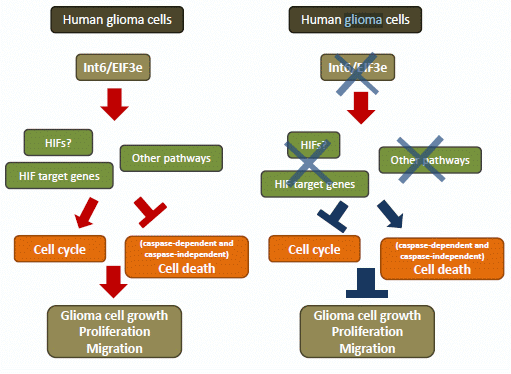
:
1. Introduction
2. Results and Discussion
2.1. Results
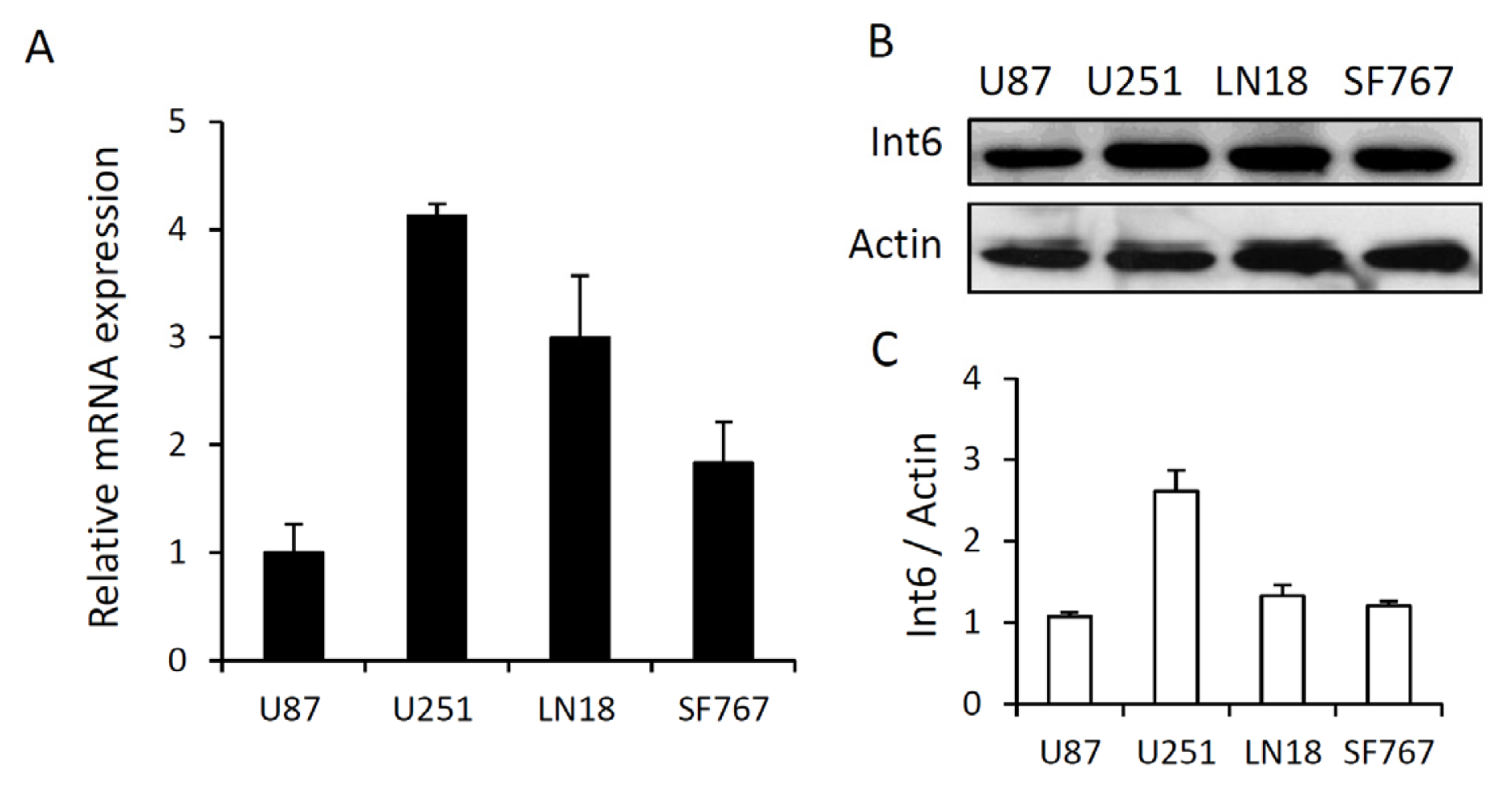
2.1.1. Int6/eIF3e Expression in Human Glioblastoma Cells
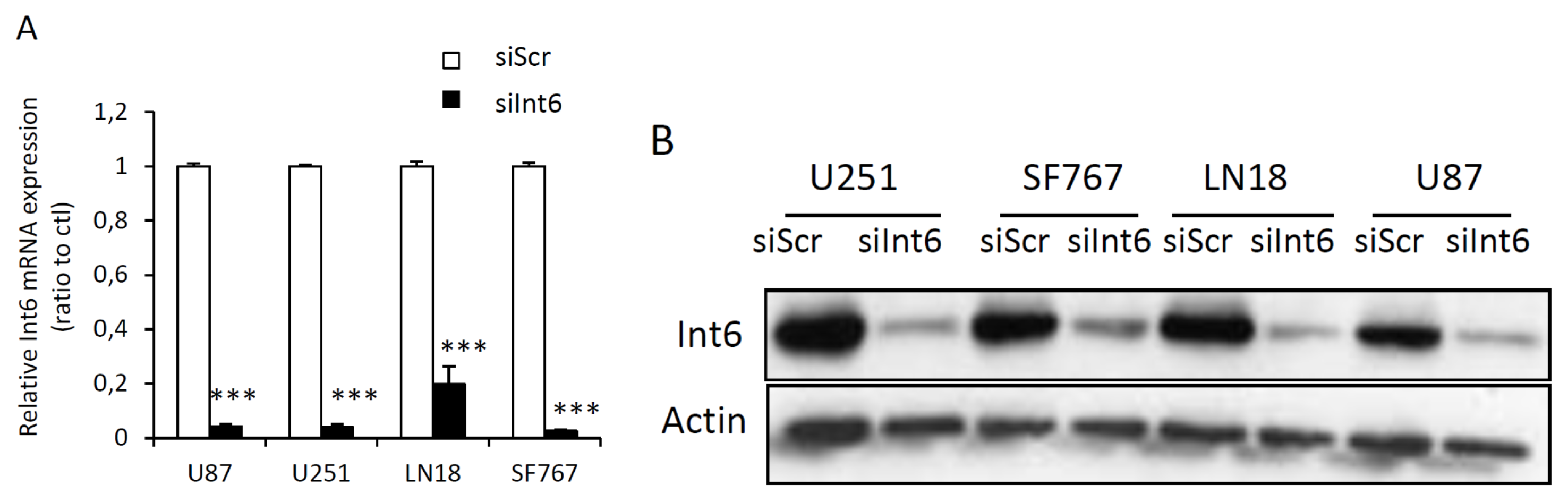
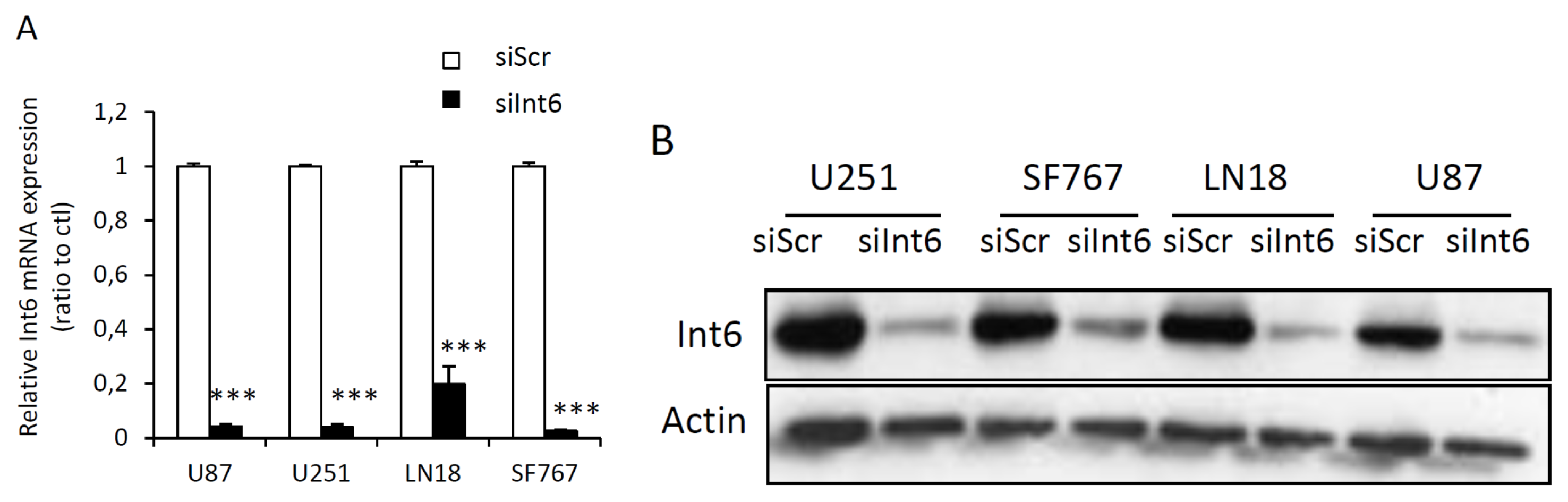
2.1.2. RNA Interference Mediated Int6/EIF3E Silencing in Glioblastoma Cells
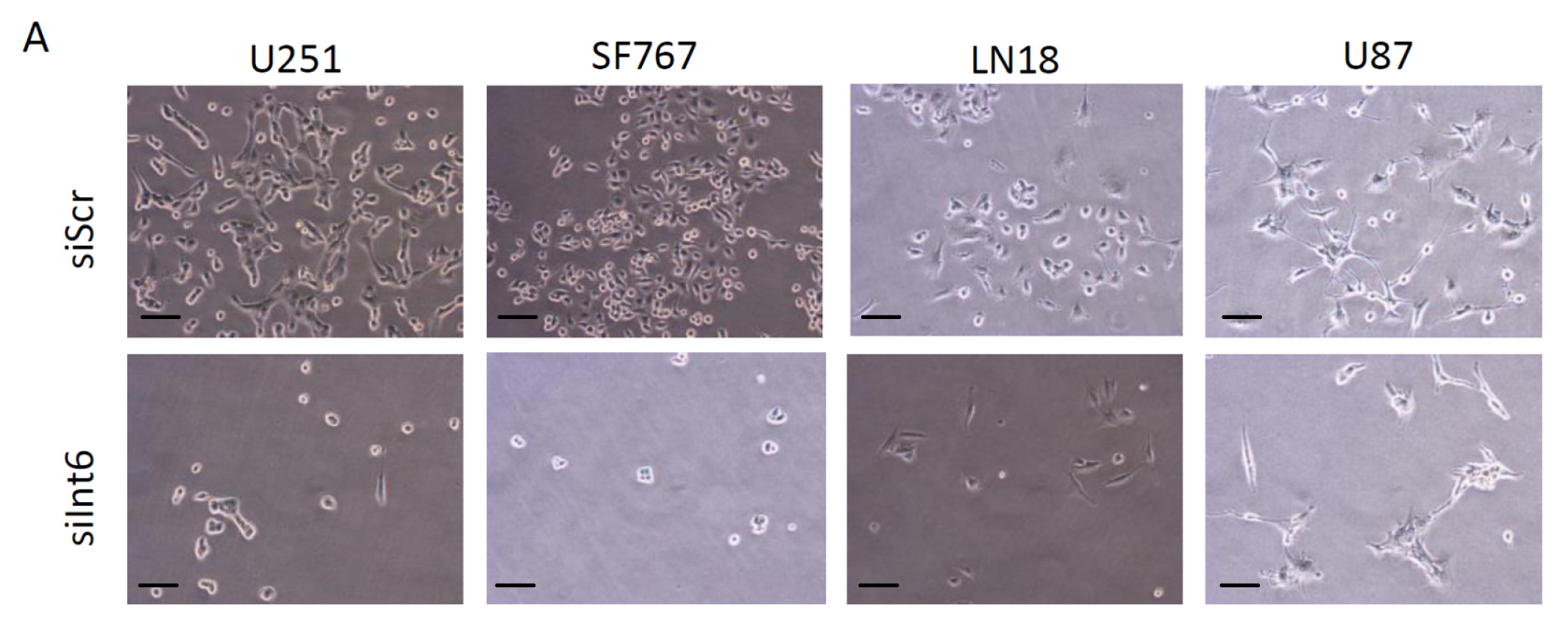
2.1.3. Int6 Inhibition Decreases Glioblastoma Cell Proliferation
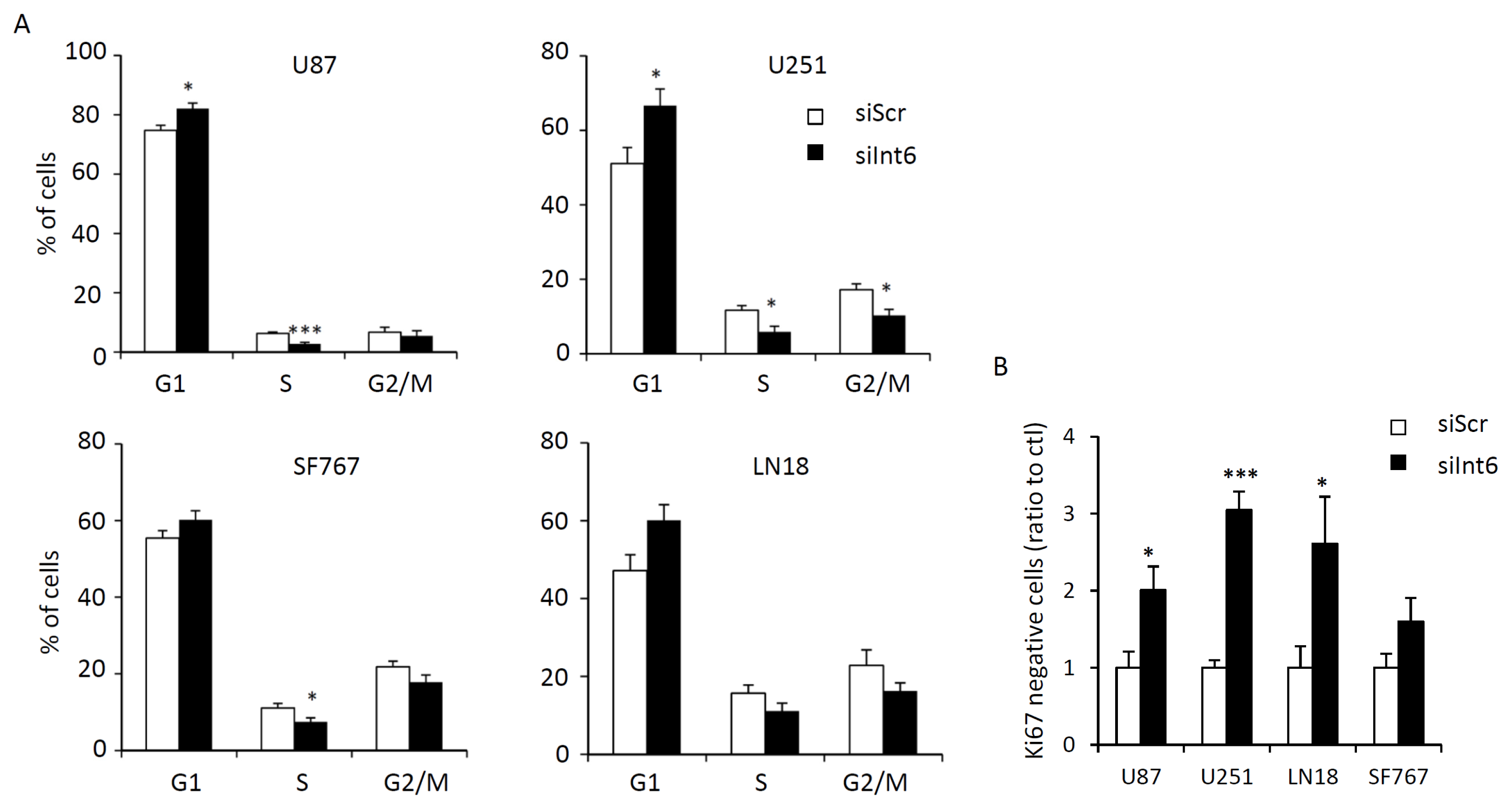
2.1.4. Int6 Inhibition Induces Cell Cycle Arrest and G0 Transition
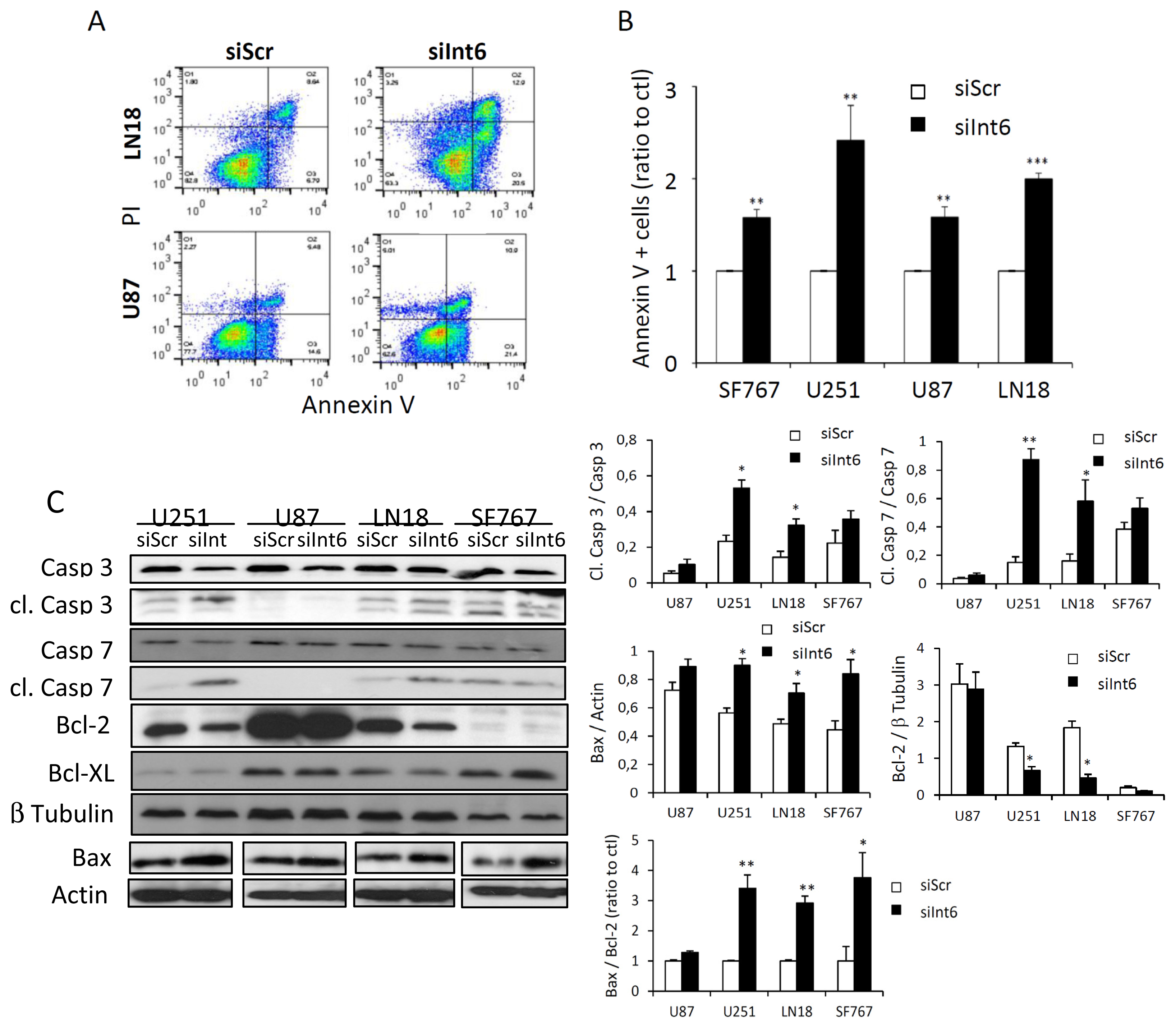
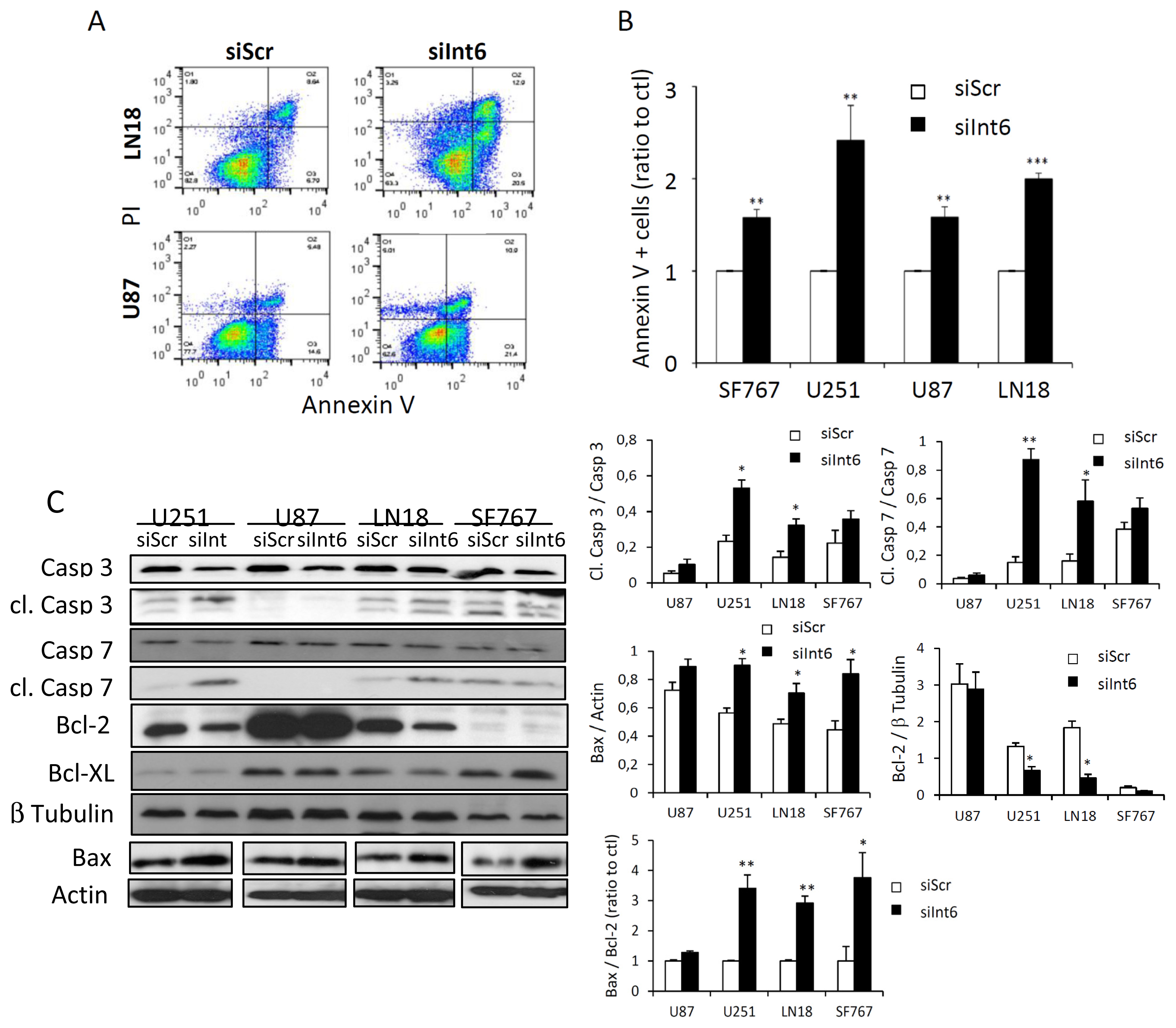
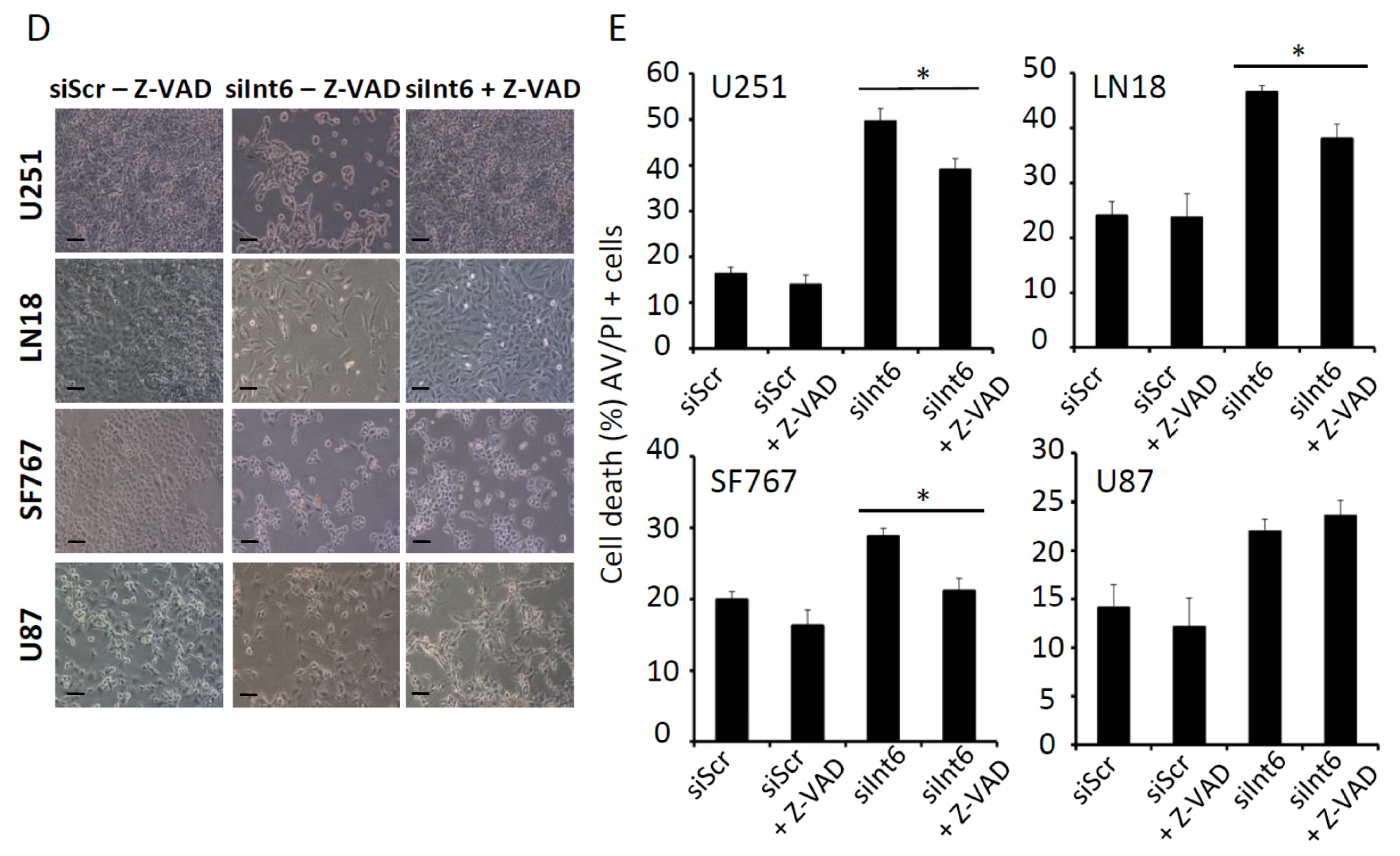
2.1.5. Int6 Inhibition Increases Glioblastoma Cell Apoptosis
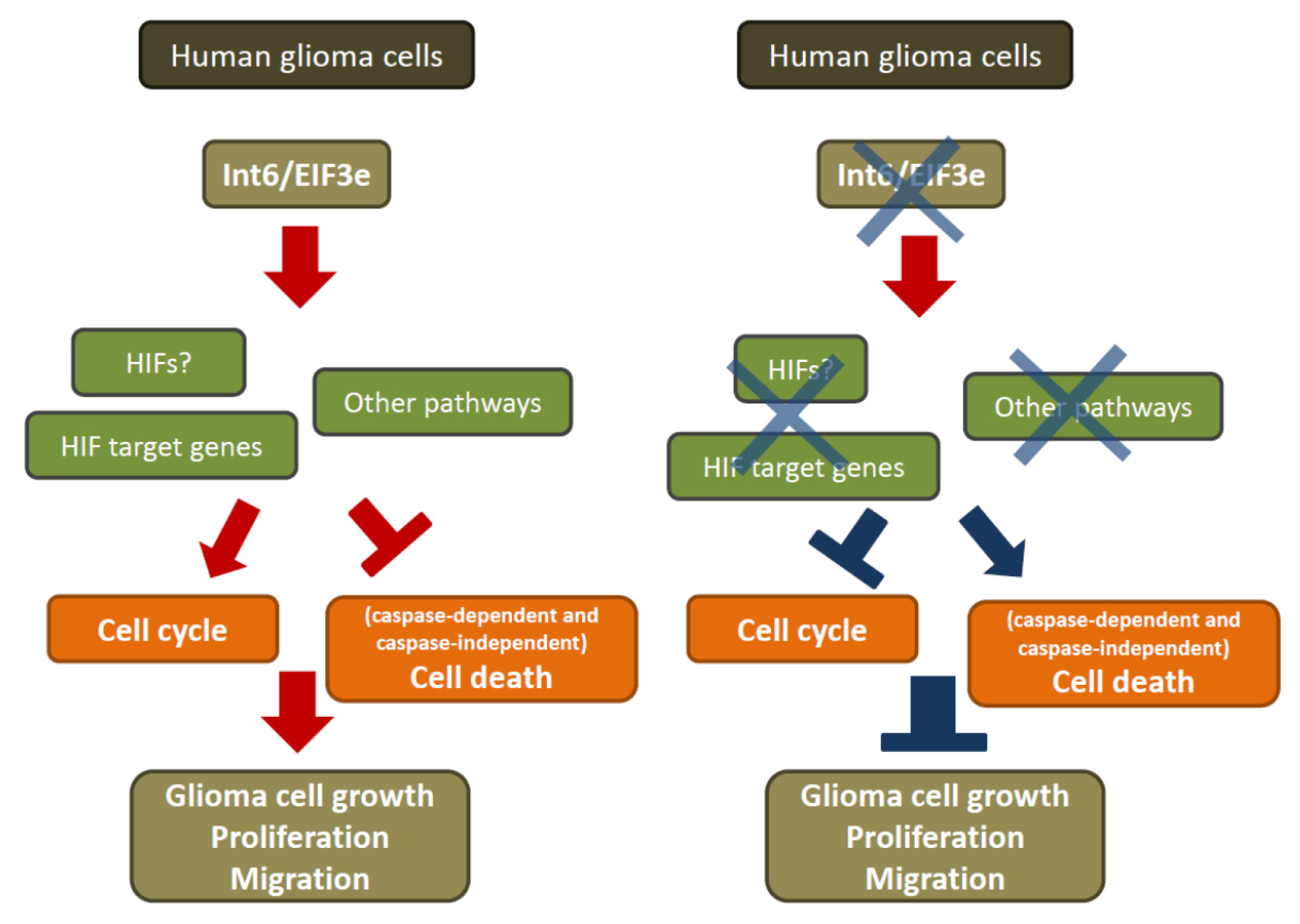
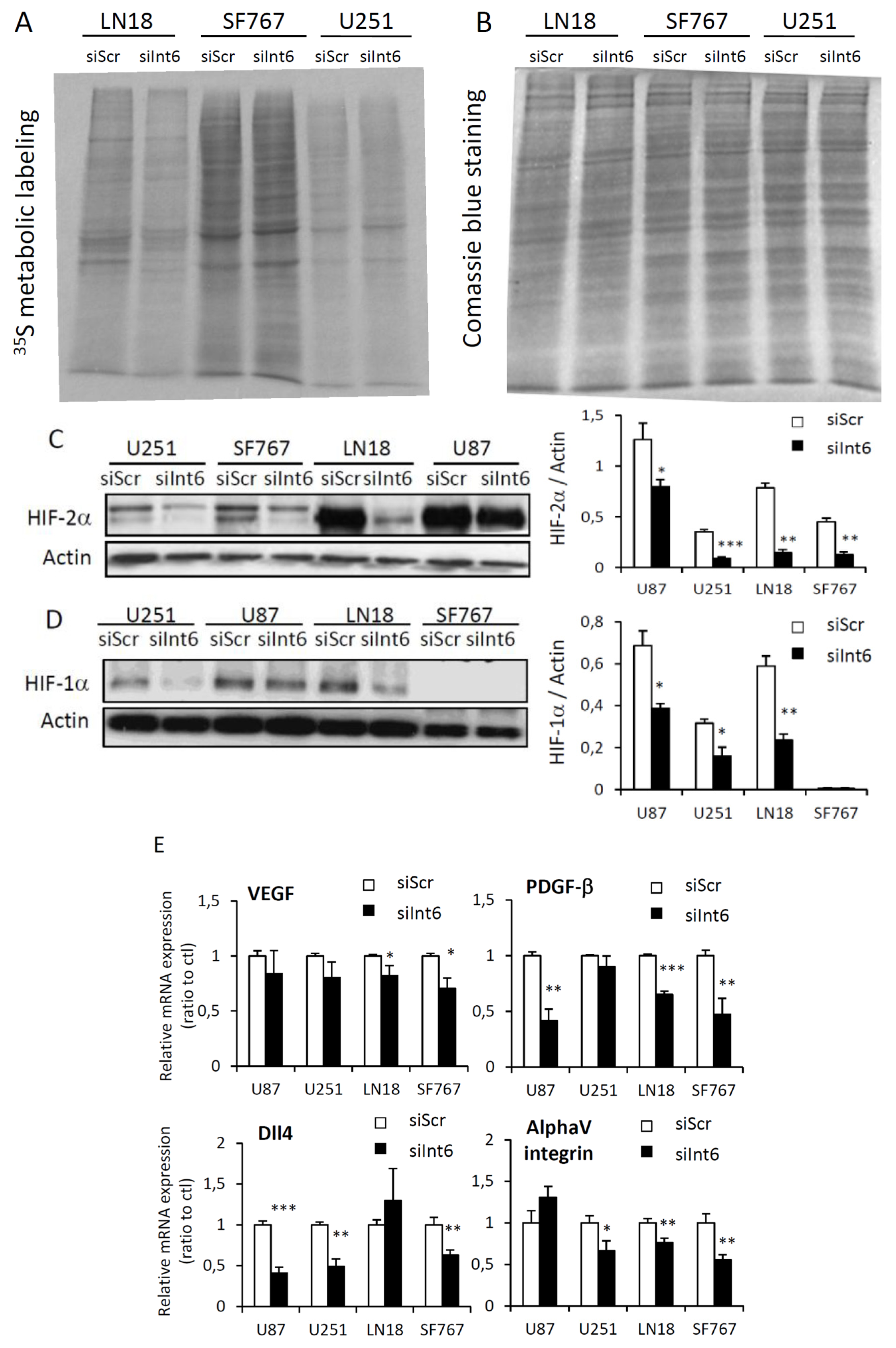
2.1.6. RNA Interference-Mediated Int6/EIF3E Silencing, Translation and HIFs
2.2. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. siRNA Transfection
3.3. Proliferation Assay
3.4. Western Blot Analysis
3.5. RT-qPCR
3.6. Flow Cytometry
3.7. De Novo Protein Synthesis
3.8. Migration/in Vitro Wound-Closure Assay
3.9. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-15-02172-s001.pdfAcknowledgments
Conflicts of Interest
References
- Tanaka, S.; Louis, D.N.; Curry, W.T.; Batchelor, T.T.; Dietrich, J. Diagnostic and therapeutic avenues for glioblastoma: No longer a dead end? Nat. Rev. Clin. Oncol 2012, 10, 14–26. [Google Scholar]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol 2013. [Google Scholar] [CrossRef]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res 2012, 318, 2417–2426. [Google Scholar]
- Krock, B.L.; Skuli, N.; Simon, M.C. Hypoxia-induced angiogenesis: Good and evil. Genes Cancer 2011, 2, 1117–1133. [Google Scholar]
- Keith, B.; Johnson, R.S.; Simon, M.C. Hif1α and hif2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar]
- Chen, L.; Endler, A.; Shibasaki, F. Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Exp. Mol. Med 2009, 41, 849–857. [Google Scholar]
- Pietras, A.; Johnsson, A.S.; Pahlman, S. The hif-2α-driven pseudo-hypoxic phenotype in tumor aggressiveness, differentiation, and vascularization. Curr. Top. Microbiol. Immunol 2010, 345, 1–20. [Google Scholar]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 2009, 15, 501–513. [Google Scholar]
- Chen, L.; Uchida, K.; Endler, A.; Shibasaki, F. Mammalian tumor suppressor int6 specifically targets hypoxia inducible factor 2α for degradation by hypoxia- and pvhl-independent regulation. J. Biol. Chem 2007, 282, 12707–12716. [Google Scholar]
- Miyashita, R.; Chen, L.; Oshiro, H.; Uchino, H.; Shibasaki, F. Int6 silencing causes induction of angiogenic factors in neuronal cells via accumulation of hypoxia-inducible factor 2α and decreases brain damage in rats. Neurosci. Lett 2006, 528, 83–88. [Google Scholar]
- Chen, L.; Endler, A.; Uchida, K.; Horiguchi, S.; Morizane, Y.; Iijima, O.; Toi, M.; Shibasaki, F. Int6/eif3e silencing promotes functional blood vessel outgrowth and enhances wound healing by upregulating hypoxia-induced factor 2α expression. Circulation 2010, 122, 910–919. [Google Scholar]
- Endler, A.; Chen, L.; Li, Q.; Uchida, K.; Hashimoto, T.; Lu, L.; Xu, G.T.; Shibasaki, F. Int6/eif3e silenced hif2α stabilization enhances migration and tube formation of huvecs via IL-6 and IL-8 signaling. Cytokine 2013, 62, 115–122. [Google Scholar]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar]
- Hershey, J.W. Regulation of protein synthesis and the role of eif3 in cancer. Br. J. Med. Biol. Res 2010, 43, 920–930. [Google Scholar]
- Damoc, E.; Fraser, C.S.; Zhou, M.; Videler, H.; Mayeur, G.L.; Hershey, J.W.; Doudna, J.A.; Robinson, C.V.; Leary, J.A. Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Mol. Cell. Proteomics 2007, 6, 1135–1146. [Google Scholar]
- Marchione, R.; Leibovitch, S.A.; Lenormand, J.L. The translational factor eif3f: The ambivalent eif3 subunit. Cell. Mol. Life Sci 2013, 70, 3603–3616. [Google Scholar]
- Morris, C.; Tomimatsu, N.; Richard, D.J.; Cluet, D.; Burma, S.; Khanna, K.K.; Jalinot, P. Int6/eif3e interacts with atm and is required for proper execution of the DNA damage response in human cells. Cancer Res 2012, 72, 2006–2016. [Google Scholar]
- Grzmil, M.; Rzymski, T.; Milani, M.; Harris, A.L.; Capper, R.G.; Saunders, N.J.; Salhan, A.; Ragoussis, J.; Norbury, C.J. An oncogenic role of eif3e/int6 in human breast cancer. Oncogene 2010, 29, 4080–4089. [Google Scholar]
- Gillis, L.D.; Lewis, S.M. Decreased eif3e/int6 expression causes epithelial-to-mesenchymal transition in breast epithelial cells. Oncogene 2012, 32, 3598–3605. [Google Scholar]
- Cappuzzo, F.; Varella-Garcia, M.; Rossi, E.; Gajapathy, S.; Valente, M.; Drabkin, H.; Gemmill, R. Myc and eif3h coamplification significantly improve response and survival of non-small cell lung cancer patients (nsclc) treated with gefitinib. J. Thorac. Oncol 2009, 4, 472–478. [Google Scholar]
- Nupponen, N.N.; Porkka, K.; Kakkola, L.; Tanner, M.; Persson, K.; Borg, A.; Isola, J.; Visakorpi, T. Amplification and overexpression of p40 subunit of eukaryotic translation initiation factor 3 in breast and prostate cancer. Am. J. Pathol 1999, 154, 1777–1783. [Google Scholar]
- Scoles, D.R.; Yong, W.H.; Qin, Y.; Wawrowsky, K.; Pulst, S.M. Schwannomin inhibits tumorigenesis through direct interaction with the eukaryotic initiation factor subunit c (eif3c). Hum. Mol. Genet 2006, 15, 1059–1070. [Google Scholar]
- Liang, H.; Ding, X.; Zhou, C.; Zhang, Y.; Xu, M.; Zhang, C.; Xu, L. Knockdown of eukaryotic translation initiation factors 3b (eif3b) inhibits proliferation and promotes apoptosis in glioblastoma cells. Neurol. Sci 2012, 33, 1057–1062. [Google Scholar]
- Miyazaki, S.; Kozak, C.A.; Marchetti, A.; Buttitta, F.; Gallahan, D.; Callahan, R. The chromosomal location of the mouse mammary tumor gene int6 and related pseudogenes in the mouse genome. Genomics 1995, 27, 420–424. [Google Scholar]
- Miyazaki, S.; Imatani, A.; Ballard, L.; Marchetti, A.; Buttitta, F.; Albertsen, H.; Nevanlinna, H.A.; Gallahan, D.; Callahan, R. The chromosome location of the human homolog of the mouse mammary tumor-associated gene int6 and its status in human breast carcinomas. Genomics 1997, 46, 155–158. [Google Scholar]
- Buttitta, F.; Martella, C.; Barassi, F.; Felicioni, L.; Salvatore, S.; Rosini, S.; D’Antuono, T.; Chella, A.; Mucilli, F.; Sacco, R.; et al. Int6 expression can predict survival in early-stage non-small cell lung cancer patients. Clin. Cancer Res 2005, 11, 3198–3204. [Google Scholar]
- Umar, A.; Kang, H.; Timmermans, A.M.; Look, M.P.; Meijer-van Gelder, M.E.; den Bakker, M.A.; Jaitly, N.; Martens, J.W.; Luider, T.M.; Foekens, J.A.; et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol. Cell. Proteomics 2009, 8, 1278–1294. [Google Scholar]
- Suo, J.; Snider, S.J.; Mills, G.B.; Creighton, C.J.; Chen, A.C.; Schiff, R.; Lloyd, R.E.; Chang, E.C. Int6 regulates both proteasomal degradation and translation initiation and is critical for proper formation of acini by human mammary epithelium. Oncogene 2010, 30, 724–736. [Google Scholar]
- Morris, C.; Jalinot, P. Silencing of human int-6 impairs mitosis progression and inhibits cyclin b-cdk1 activation. Oncogene 2005, 24, 1203–1211. [Google Scholar]
- Grzmil, M.; Whiting, D.; Maule, J.; Anastasaki, C.; Amatruda, J.F.; Kelsh, R.N.; Norbury, C.J.; Patton, E.E. The int6 cancer gene and mek signaling pathways converge during zebrafish development. PLoS One 2007, 2, e959. [Google Scholar]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. Hif-1α inhibition by sirna or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via ca9 expression. BMC Cancer 2010, 10, 605. [Google Scholar]
- Jensen, R.L.; Ragel, B.T.; Whang, K.; Gillespie, D. Inhibition of hypoxia inducible factor-1α (hif-1α) decreases vascular endothelial growth factor (vegf) secretion and tumor growth in malignant gliomas. J. Neurooncol 2006, 78, 233–247. [Google Scholar]
- Gillespie, D.L.; Whang, K.; Ragel, B.T.; Flynn, J.R.; Kelly, D.A.; Jensen, R.L. Silencing of hypoxia inducible factor-1α by rna interference attenuates human glioma cell growth in vivo. Clin. Cancer Res. 2007, 13, 2441–2448. [Google Scholar]
- Franovic, A.; Holterman, C.E.; Payette, J.; Lee, S. Human cancers converge at the hif-2α oncogenic axis. Proc. Natl. Acad. Sci. USA 2009, 106, 21306–21311. [Google Scholar]
- Morris, C.; Wittmann, J.; Jack, H.M.; Jalinot, P. Human int6/eif3e is required for nonsense-mediated mrna decay. EMBO Rep 2007, 8, 596–602. [Google Scholar]
- Zhang, X.; Chen, T.; Zhang, J.; Mao, Q.; Li, S.; Xiong, W.; Qiu, Y.; Xie, Q.; Ge, J. Notch1 promotes glioma cell migration and invasion by stimulating β-catenin and NF-κB signaling via akt activation. Cancer Sci 2012, 103, 181–190. [Google Scholar]
- Kil, W.J.; Tofilon, P.J.; Camphausen, K. Post-radiation increase in vegf enhances glioma cell motility in vitro. Radiat. Oncol. 2012, 7, 25. [Google Scholar]
- Xu, C.; Wu, X.; Zhu, J. Vegf promotes proliferation of human glioblastoma multiforme stem-like cells through vegf receptor 2. Sci. World J 2013, 2013, 417413. [Google Scholar]
- Nazarenko, I.; Hede, S.M.; He, X.; Hedren, A.; Thompson, J.; Lindstrom, M.S.; Nister, M. Pdgf and pdgf receptors in glioma. Ups. J. Med. Sci 2012, 117, 99–112. [Google Scholar]
- Taga, T.; Suzuki, A.; Gonzalez-Gomez, I.; Gilles, F.H.; Stins, M.; Shimada, H.; Barsky, L.; Weinberg, K.I.; Laug, W.E. αV-integrin antagonist emd 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int. J. Cancer 2002, 98, 690–697. [Google Scholar]
- MacDonald, T.J.; Ladisch, S. Antisense to integrin alpha v inhibits growth and induces apoptosis in medulloblastoma cells. Anticancer Res 2001, 21, 3785–3791. [Google Scholar]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar]
- Skuli, N.; Monferran, S.; Delmas, C.; Lajoie-Mazenc, I.; Favre, G.; Toulas, C.; Cohen-Jonathan-Moyal, E. Activation of rhob by hypoxia controls hypoxia-inducible factor-1α stabilization through glycogen synthase kinase-3 in u87 glioblastoma cells. Cancer Res 2006, 66, 482–489. [Google Scholar]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial hif-2α regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig 2012, 122, 1427–1443. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Sequence |
|---|---|
| Qiagen FlexiTube Gene Solution GS3646 for EIF3E, 1 nmol | SI02662499: 5′-CCCAAAGGUCGCGAUAAUAUU-3′ SI02661981: 5′-AAGCUGGCCUCUGAAAUCUUA-3′ SI02656094: 5′-AAGCUGAAAGGUGGAUUGUAA-3′ SI03153136: 5′-AUGGAAGACCUUACACGGUUA-3′ |
| Qiagen SI02662499 (FlexiTube EIF3E siRNA, 20 nmol) | 5′-CCCAAAGGUCGCGAUAAUAUU-3′ |
| Dharmacon (ON-TARGET plus SMART pool EIF3E siRNA 3646, 10 nmol) | #1: 5′-UGGCUUGUCUUGAGGAUUU-3′ #2: 5′-GGAUCGGCAUCUAGUCUUU-3′ #3: 5′-GGGUAACAAUGCAGUCUCA-3′ #4: 5′-AAAGGUCGCGAUAAUAUUA-3′ |
| Targets | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| β2m | ACCCCCACTGAAAAAGATGA | ATCTTCAAACCTCCATGATG |
| 18S | GTCTGTGATGCCCTTAGATG | CGTACAGGATGATGTCCGTATACCT |
| Int6 | TTCTTCAATCACCCCAAAGG | TAGAACCTGCCGACGTTTTC |
| HIF-2α | CCACCAGCTTCACTCTCTCC | TCAGAAAAAGGCCACTGCTT |
| Dll4 | TGGGTCAGAACTGGTTATTGGA | GTCATTGCGCTTCTTGCACAG |
| PDGFβ | GATCCCTCCTTTGATGATCTC | TCCAACTCGGCCCCATCT |
| VEGF | CTACCTCCACCATGCCAAGT | GCAGTAGCTGCGCTGATAGA |
| αV Integrin | GGAGCAATTCGACGAGCACT | TTCATCCCGCAGATACGCTA |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sesen, J.; Cammas, A.; Scotland, S.J.; Elefterion, B.; Lemarié, A.; Millevoi, S.; Mathew, L.K.; Seva, C.; Toulas, C.; Moyal, E.C.-J.; et al. Int6/eIF3e Is Essential for Proliferation and Survival of Human Glioblastoma Cells. Int. J. Mol. Sci. 2014, 15, 2172-2190. https://doi.org/10.3390/ijms15022172
Sesen J, Cammas A, Scotland SJ, Elefterion B, Lemarié A, Millevoi S, Mathew LK, Seva C, Toulas C, Moyal EC-J, et al. Int6/eIF3e Is Essential for Proliferation and Survival of Human Glioblastoma Cells. International Journal of Molecular Sciences. 2014; 15(2):2172-2190. https://doi.org/10.3390/ijms15022172
Chicago/Turabian StyleSesen, Julie, Anne Cammas, Sarah J. Scotland, Bertand Elefterion, Anthony Lemarié, Stefania Millevoi, Lijoy K. Mathew, Cathy Seva, Christine Toulas, Elizabeth Cohen-Jonathan Moyal, and et al. 2014. "Int6/eIF3e Is Essential for Proliferation and Survival of Human Glioblastoma Cells" International Journal of Molecular Sciences 15, no. 2: 2172-2190. https://doi.org/10.3390/ijms15022172