Characterization of the Interaction between Eupatorin and Bovine Serum Albumin by Spectroscopic and Molecular Modeling Methods

Abstract
:1. Introduction
2. Results and Discussion
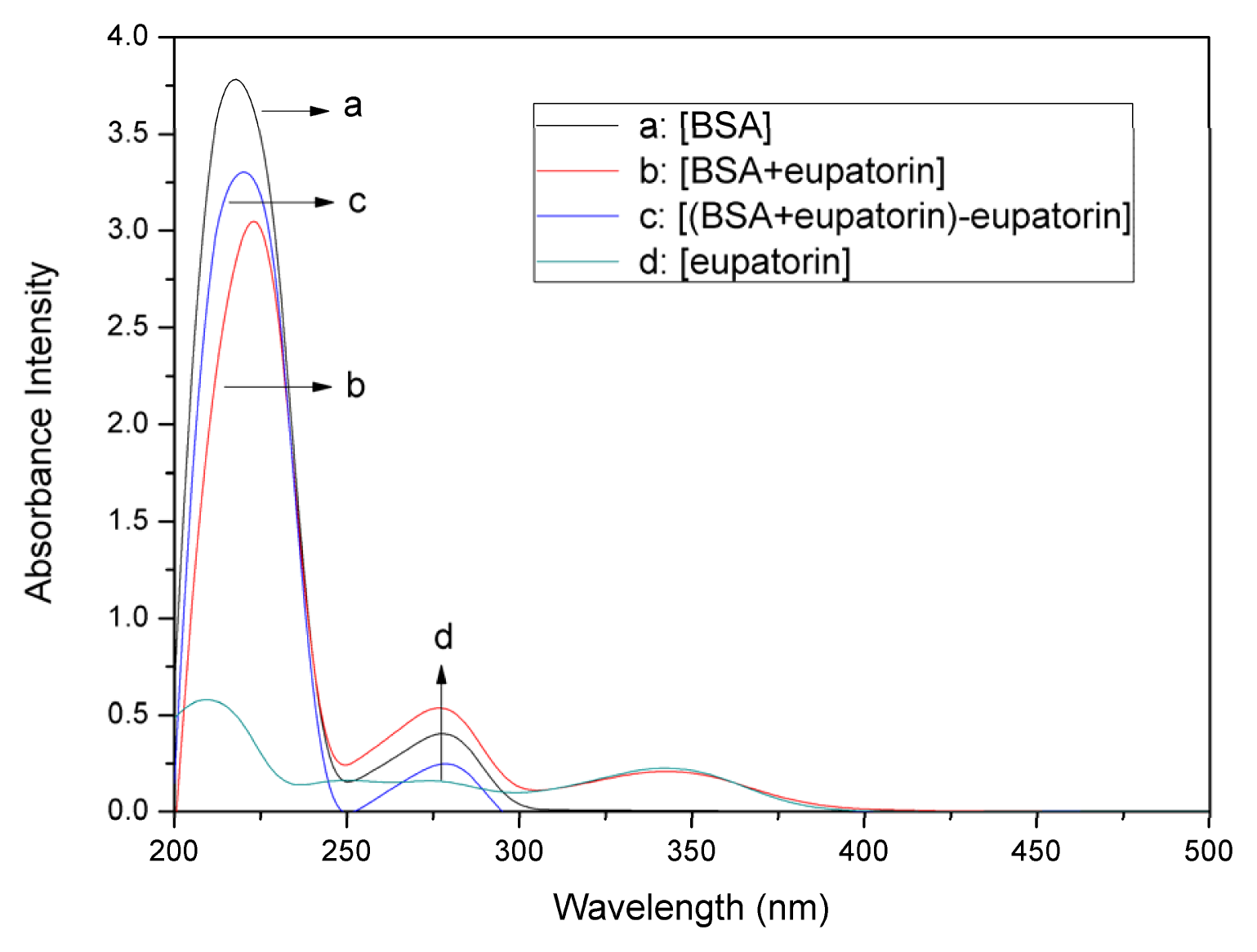
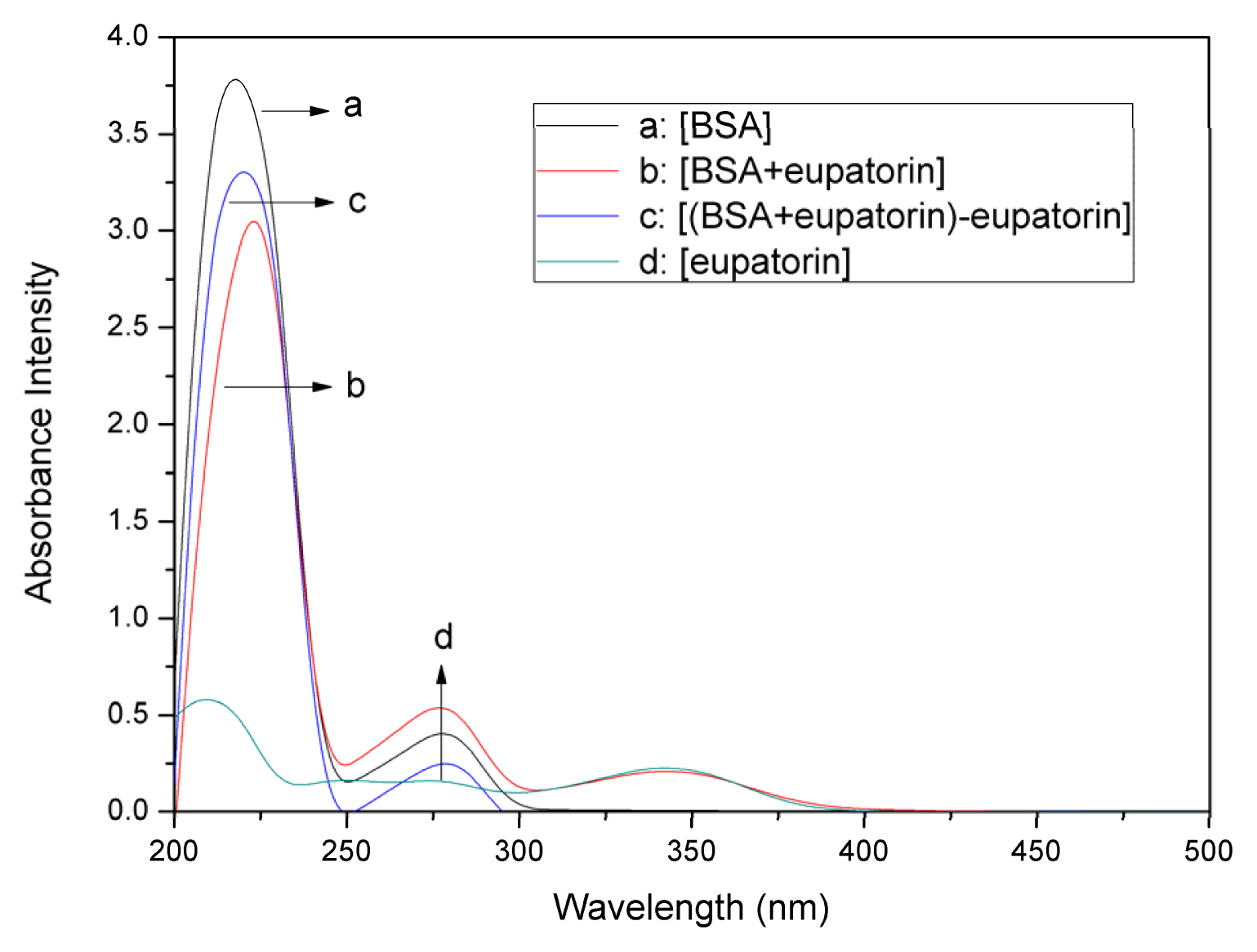
2.1. UV–vis Absorption Spectra Experiments
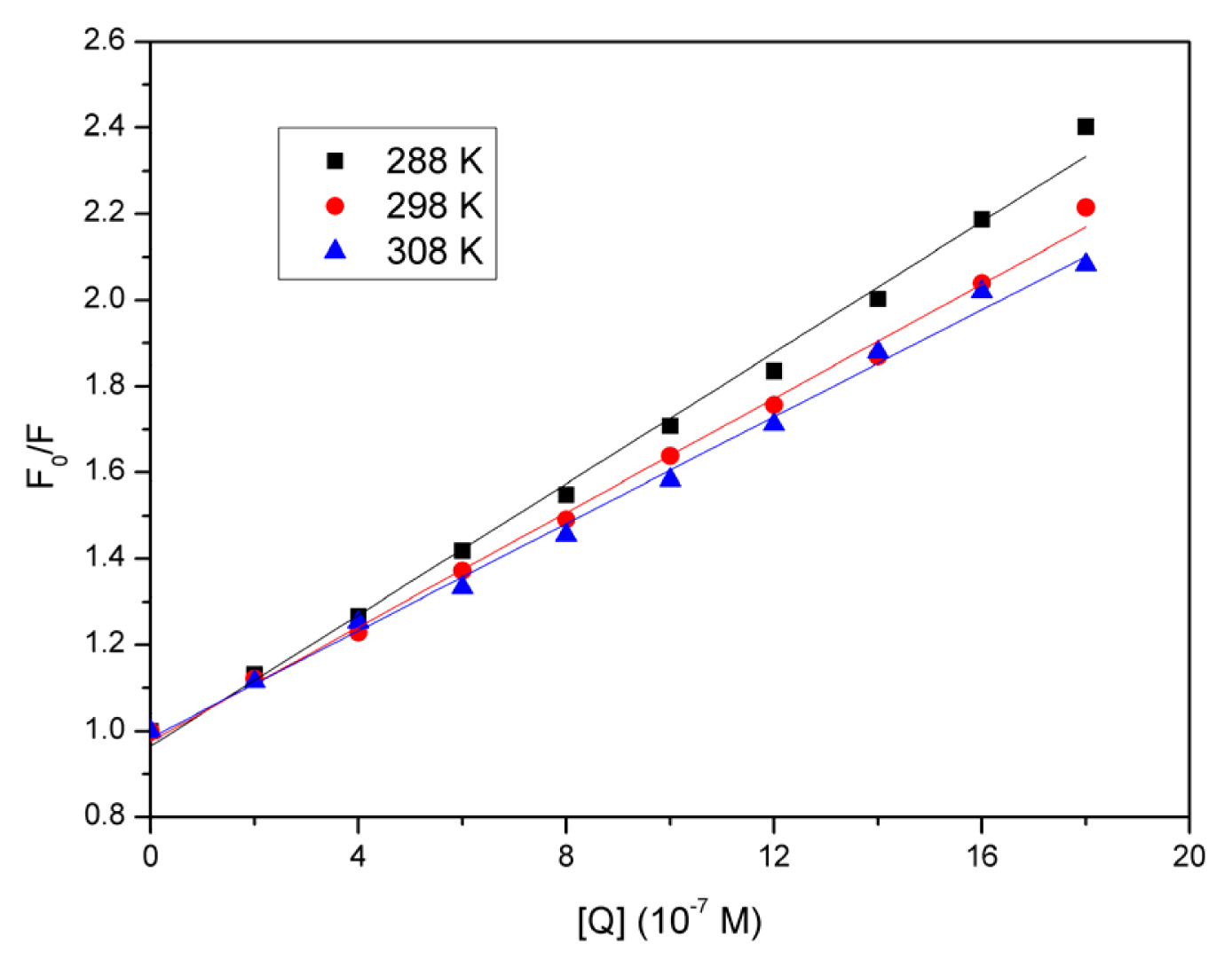
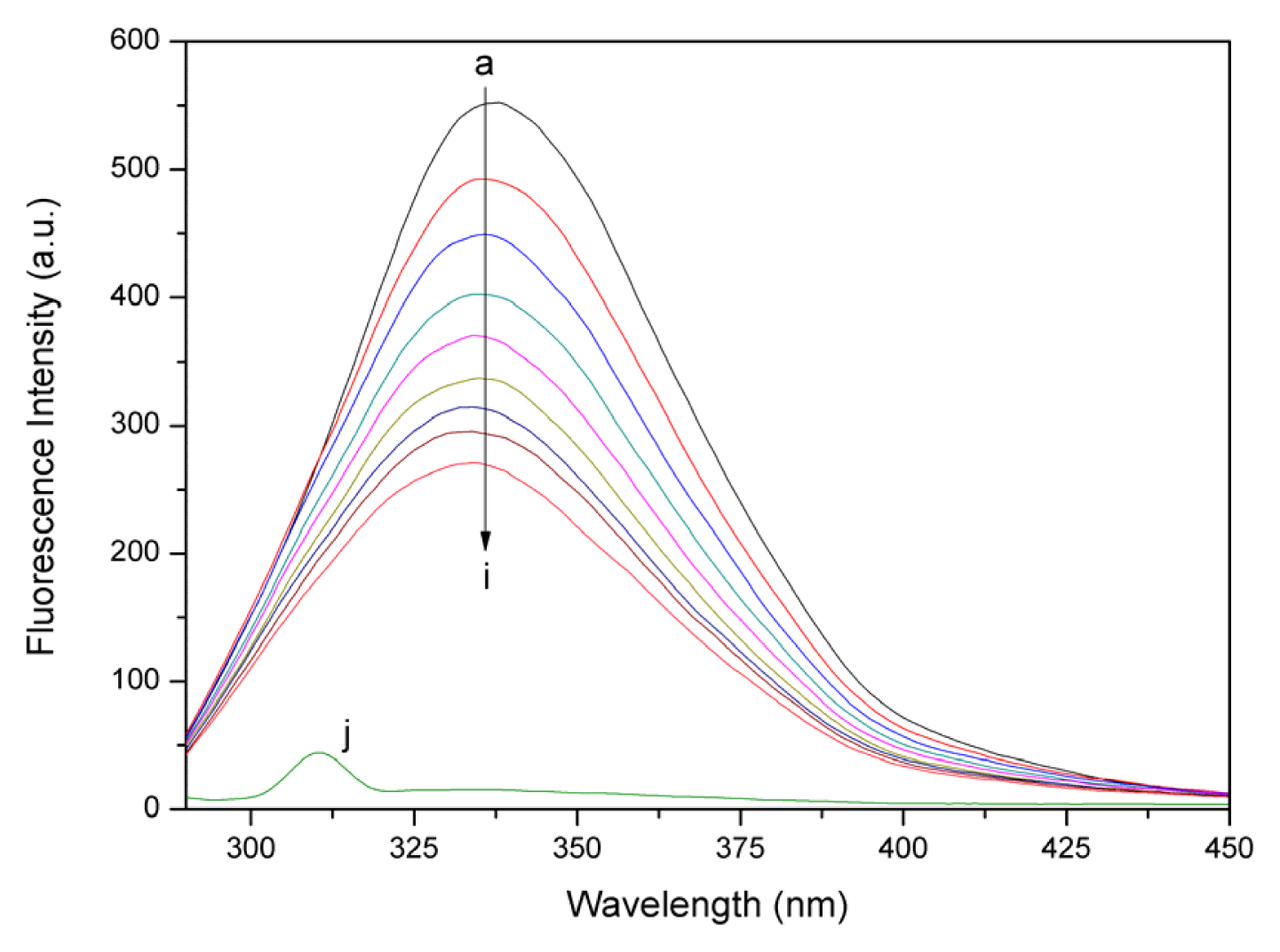
2.2. Fluorescence Quenching Studies of BSA
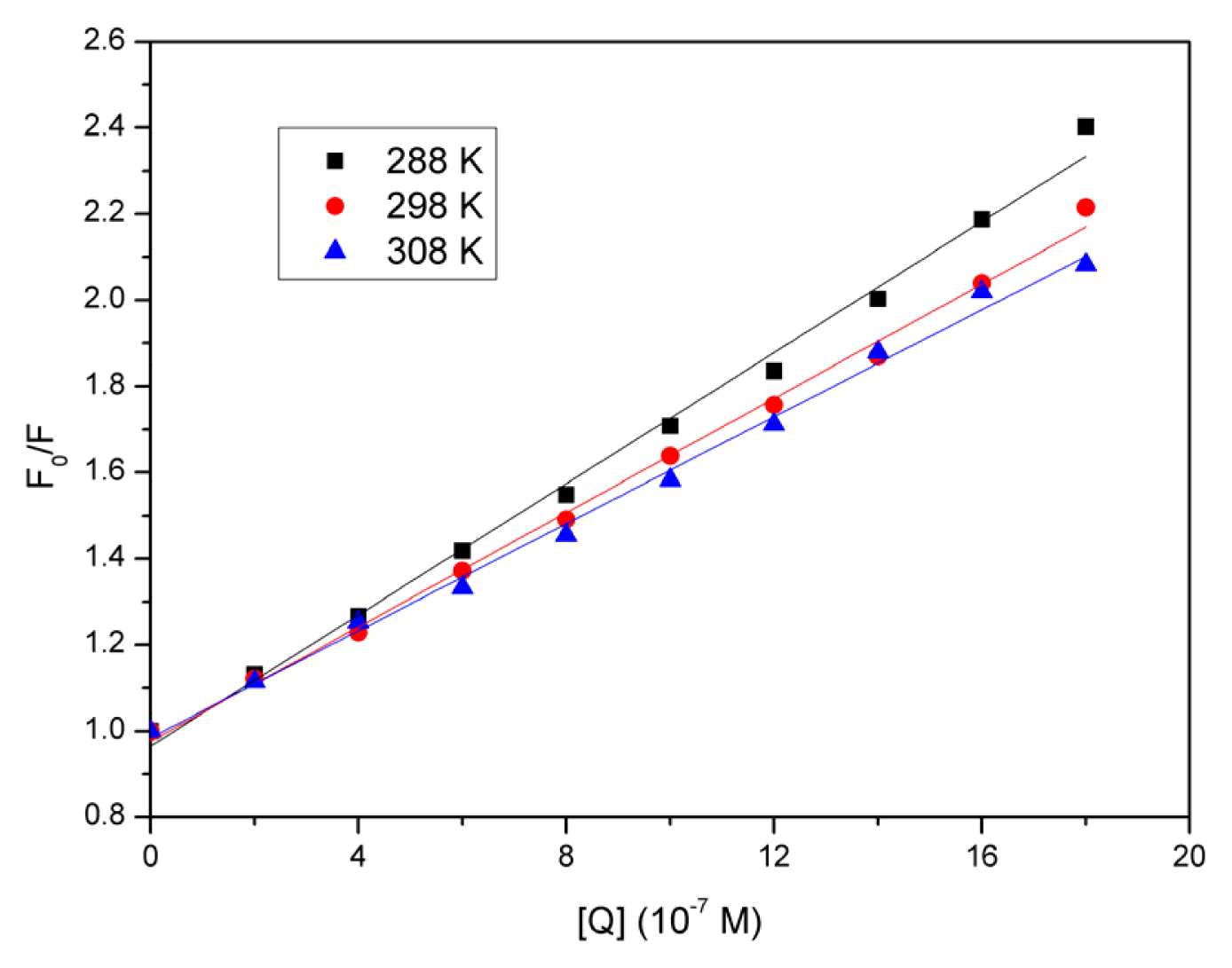
2.3. Quenching Mechanism Analysis
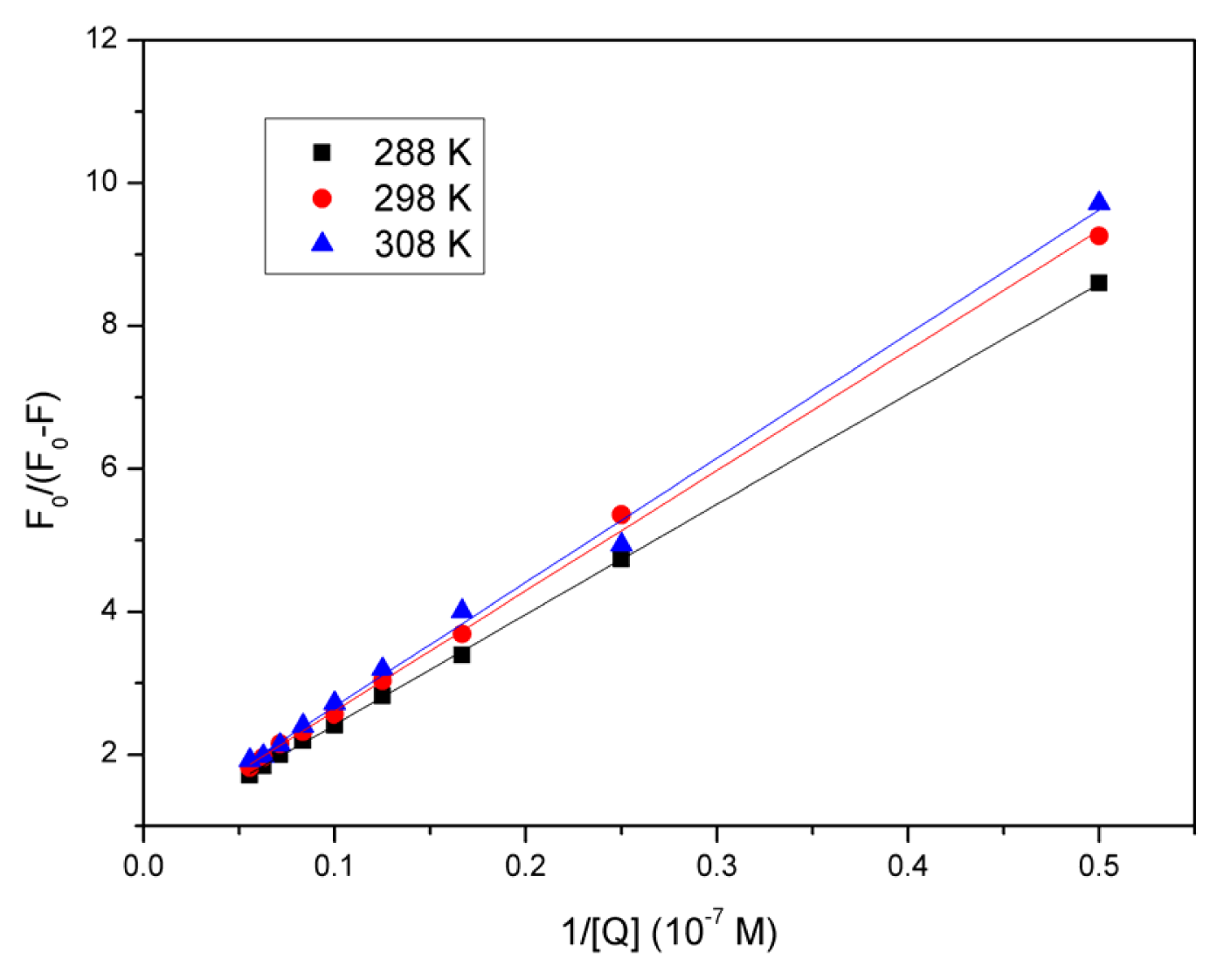
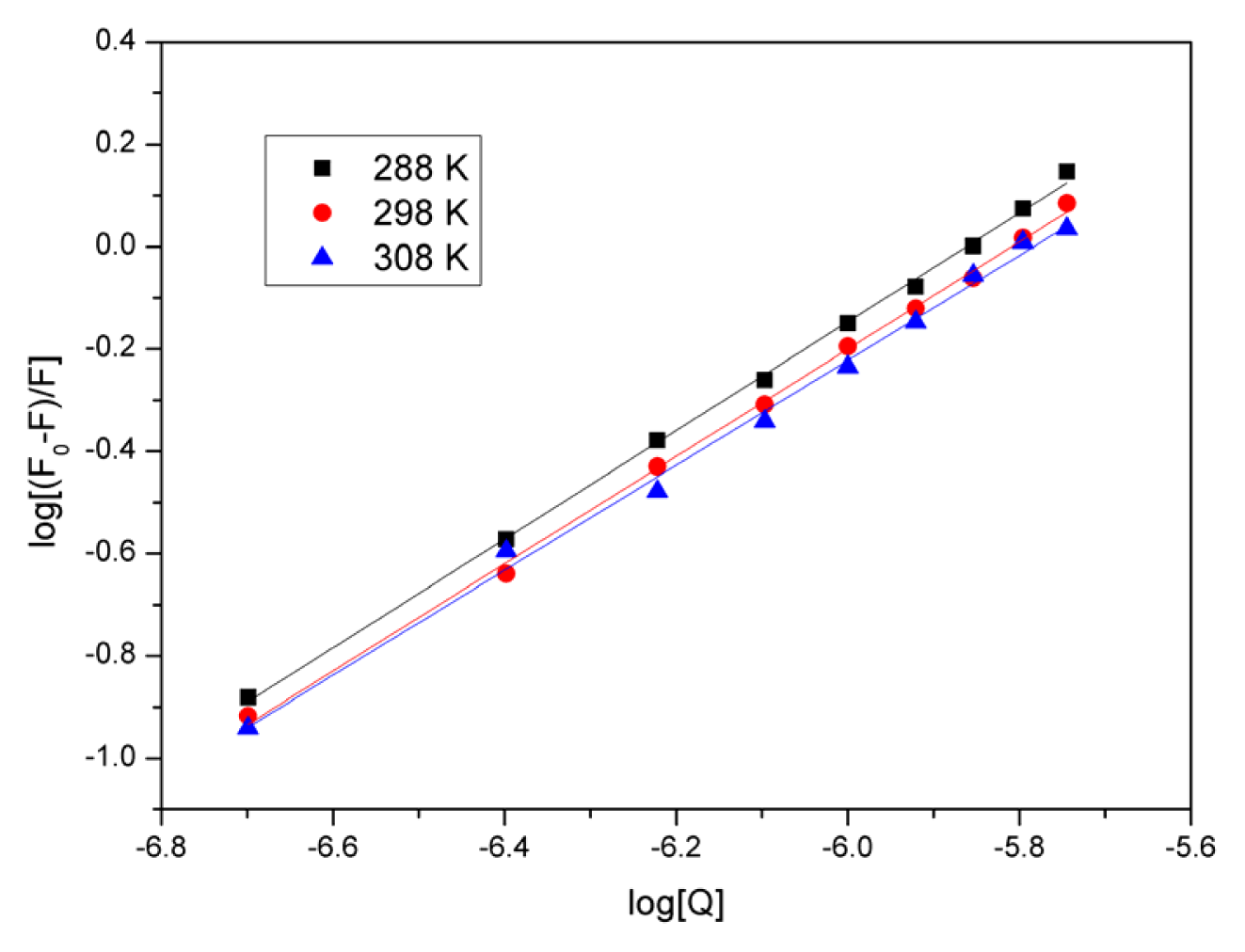
2.4. Evaluation of the Binding Constant and the Number of Binding Sites
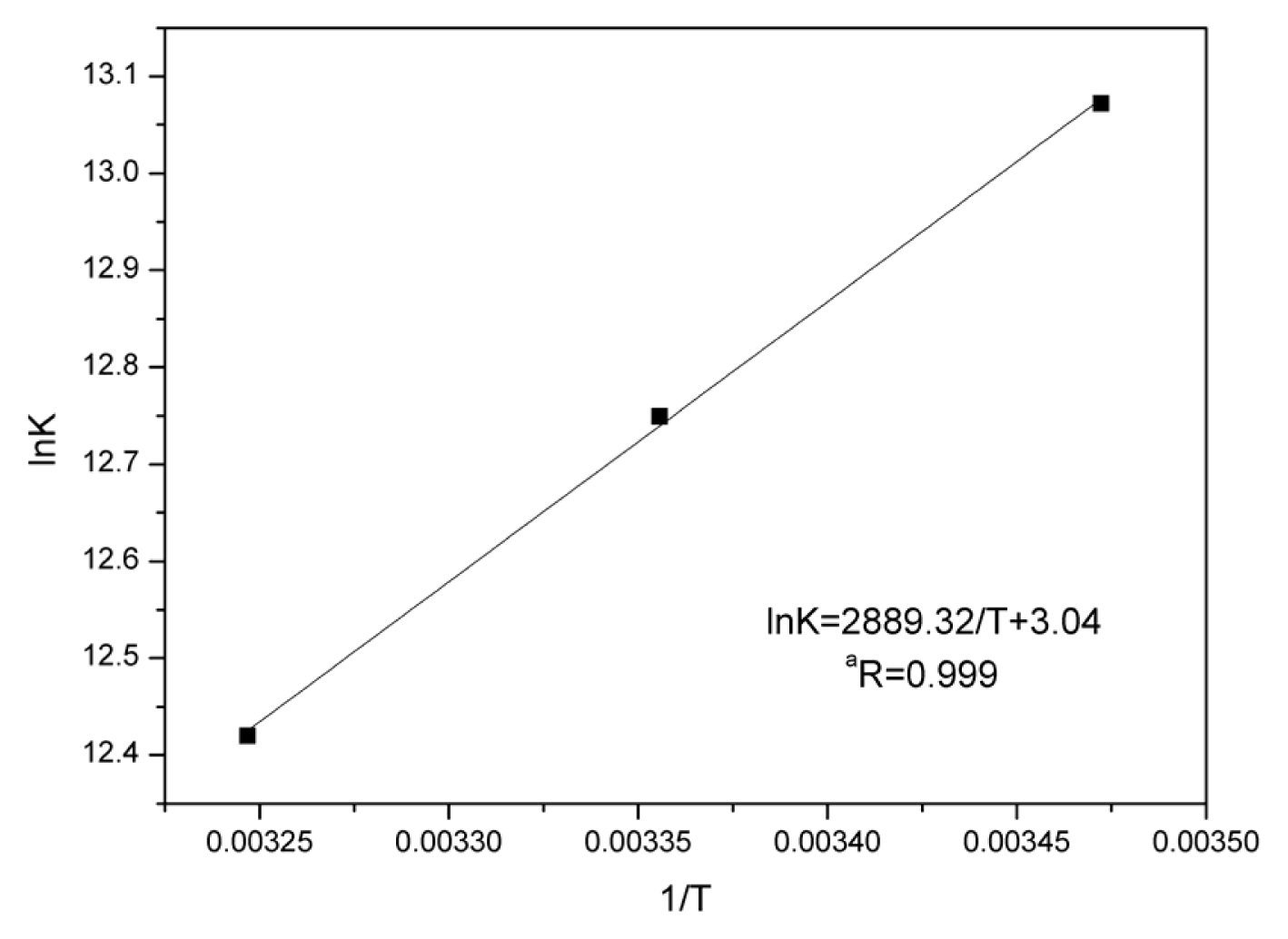
2.5. Thermodynamic Parameters and Nature of the Binding Forces
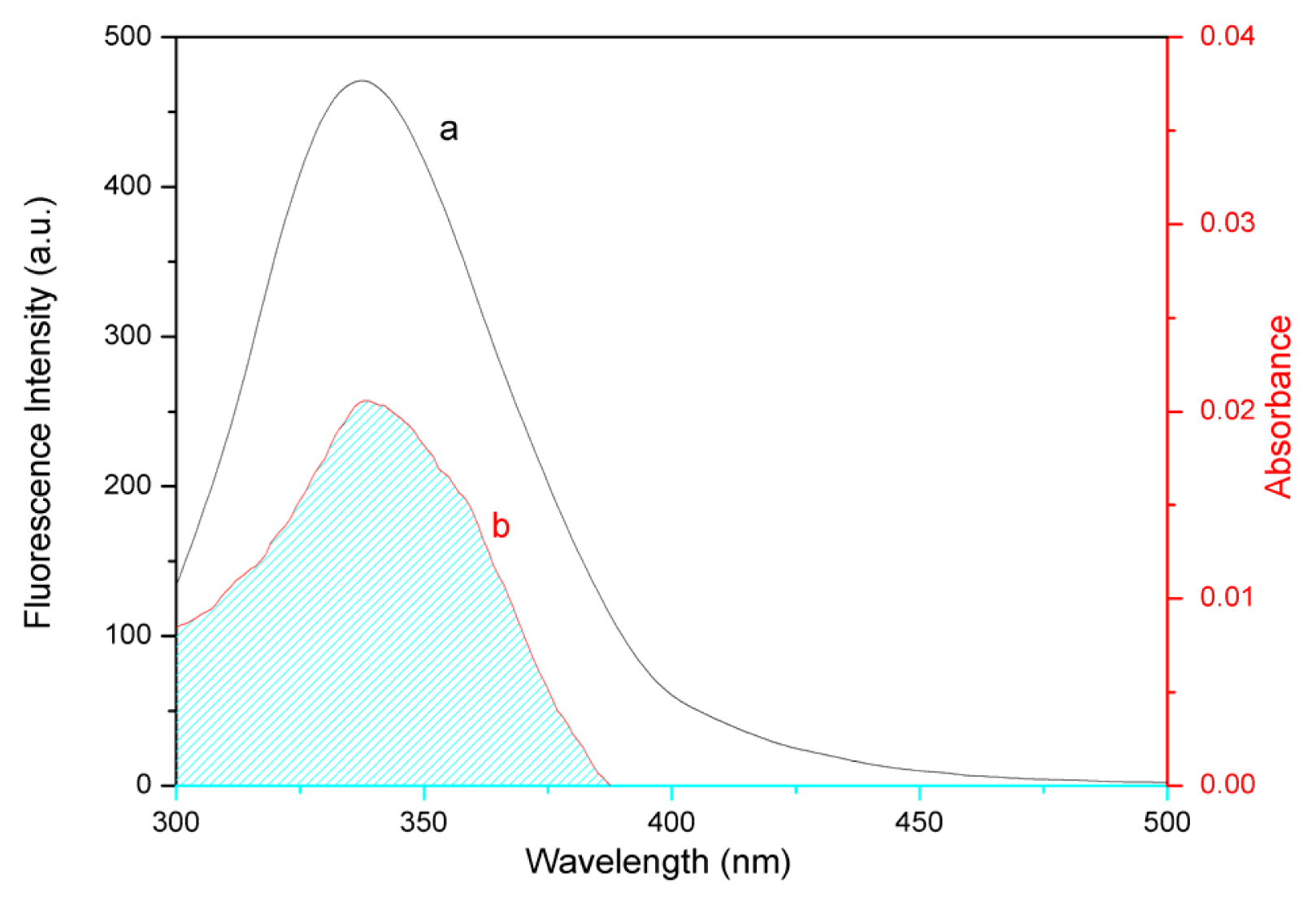
2.6. Energy Transfer from BSA to Eupatorin
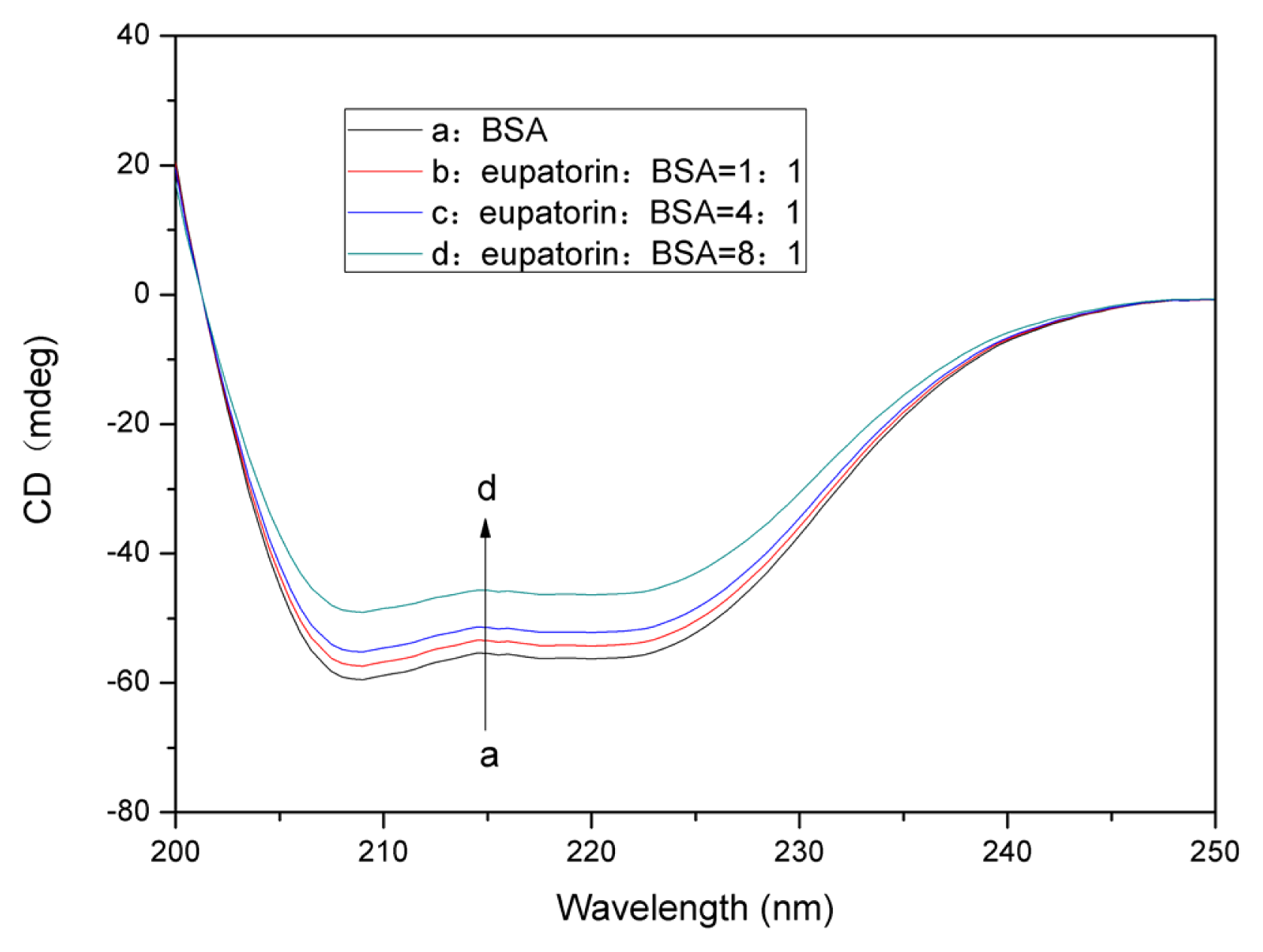
2.7. Analysis of BSA Conformational Changes
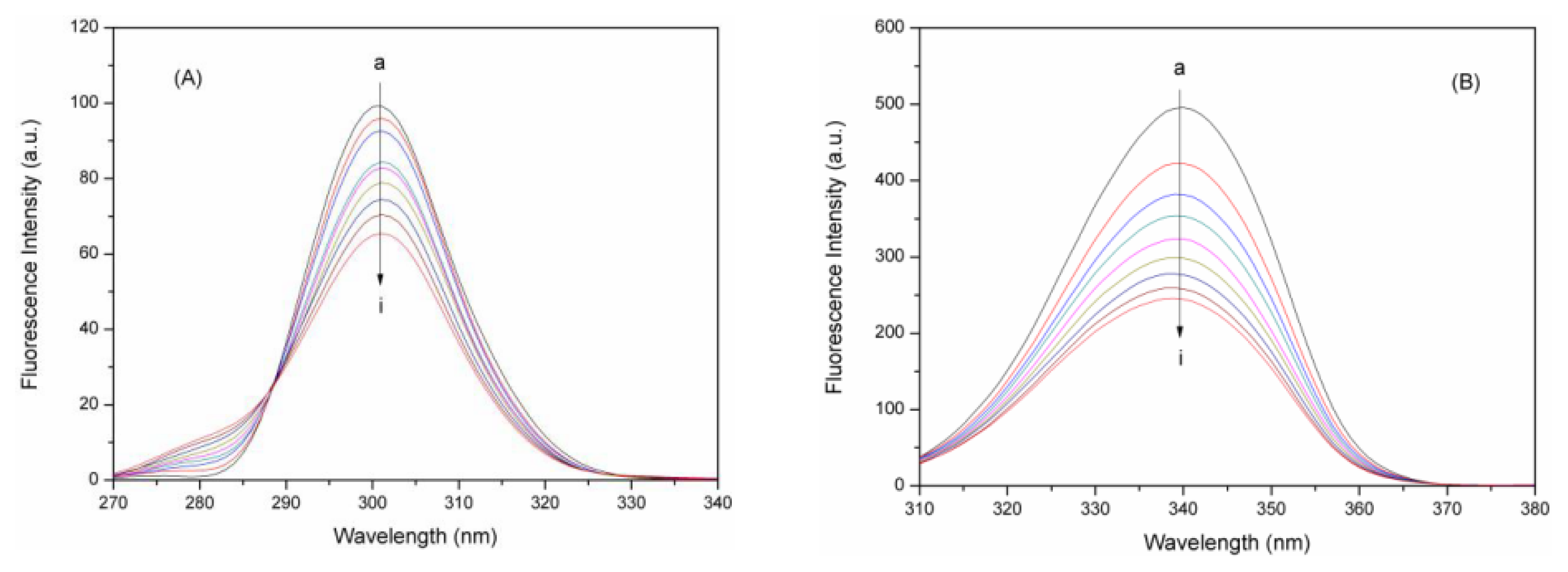
2.7.1. Synchronous Fluorescence Spectroscopic Studies
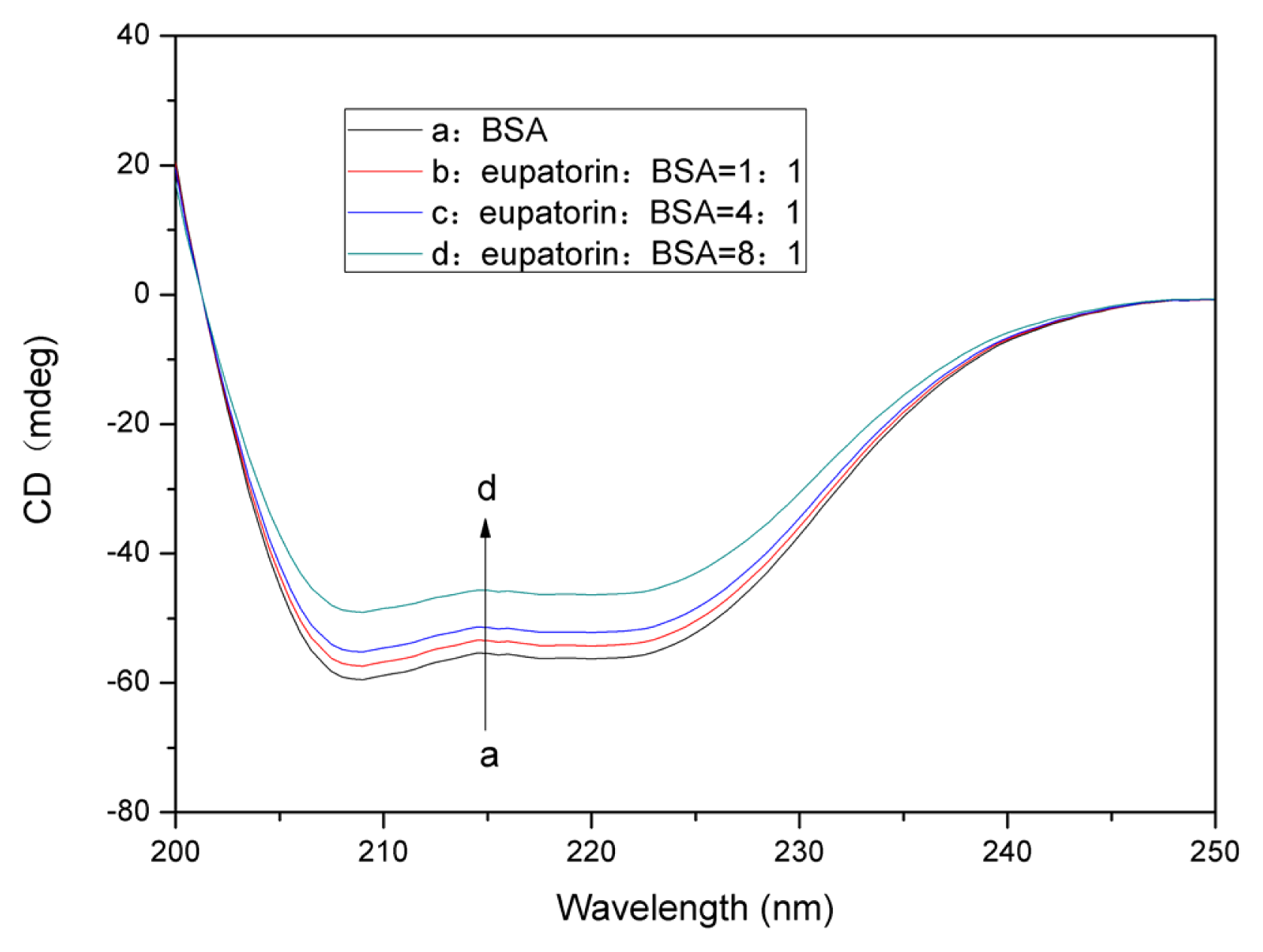
2.7.2. Changes in the BSA Secondary Structure Induced by Eupatorin Binding
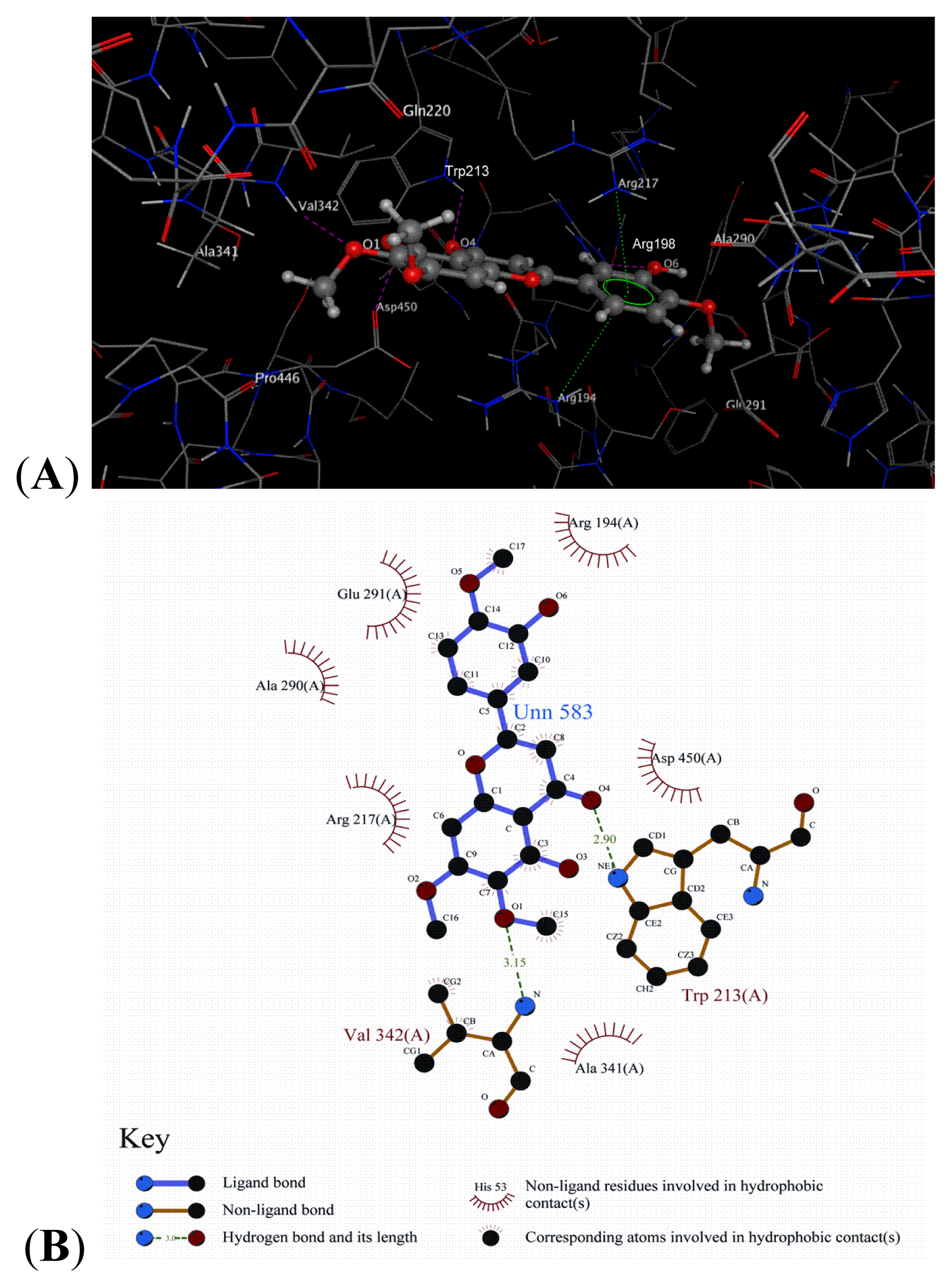
2.8. Docking Analysis
3. Experimental Section
3.1. Materials
3.2. Apparatus
3.3. Procedures
3.3.1. Measurements of UV–vis Absorption Spectra
3.3.2. Measurements of Fluorescence Spectra
3.3.3. Measurements of Synchronous Fluorescence Spectra
3.3.4. Measurements of CD Spectra
3.4. Docking study
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Middleton, E., Jr; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Androutsopoulos, V.; Arroo, R.R.; Hall, J.F.; Surichan, S.; Potter, G.A. Antiproliferative and cytostatic effects of the natural product eupatorin on MDA-MB-468 human breast cancer cells due to CYP1-mediated metabolism. Breast Cancer Res 2008, 10, R39. [Google Scholar]
- Androutsopoulos, V.P.; Li, N.; Arroo, R.R. The methoxylated flavones eupatorin and cirsiliol induce CYP1 enzyme expression in MCF7 cells. J. Nat. Prod 2009, 72, 1390–1394. [Google Scholar]
- Csapi, B.; Hajdú, Z.; Zupkó, I.; Berényi, Á.; Forgo, P.; Szabó, P.; Hohmann, J. Bioactivity-guided isolation of antiproliferative compounds from Centaurea arenaria. Phytother. Res. 2010, 24, 1664–1669. [Google Scholar]
- Salmela, A.L.; Pouwels, J.; Kukkonen-Macchi, A.; Waris, S.; Toivonen, P.; Jaakkola, K.; Jouppilaa, M.; Kallioa, L.; Kallioa, M.J. The flavonoid eupatorin inactivates the mitotic checkpoint leading to polyploidy and apoptosis. Exp. Cell Res 2012, 318, 578–592. [Google Scholar]
- Kratochwil, N.A.; Huber, W.; Muller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol 2002, 64, 1355–1374. [Google Scholar]
- Sharma, R.; Choudhary, S.; Kishore, N. Insights into the binding of the drugs diclofenac sodium and cefotaxime sodium to serum albumin: Calorimetry and spectroscopy. Eur. J. Pharm. Sci 2012, 46, 435–445. [Google Scholar]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209–215. [Google Scholar]
- Peters, T. Serum albumin. Adv. Protein Chem 1985, 37, 161–245. [Google Scholar]
- Curry, S.; Brick, P.; Franks, N.P. Fatty acid binding to human serum albumin: New insights from crystallographic studies. Biochim. Biophys. Acta 1999, 1441, 131–140. [Google Scholar]
- Majorek, K.A.; Porebski, P.J.; Dayal, A.; Zimmerman, M.D.; Jablonska, K.; Stewart, A.J.; Chruszcz, M.; Minor, W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol 2012, 52, 174–182. [Google Scholar]
- Tayeh, N.; Rungassamy, T.; Albani, J.R. Fluorescence spectral resolution of tryptophan residues in bovine and human serum albumins. J. Pharm. Biomed. Anal 2009, 50, 107–116. [Google Scholar]
- Samari, F.; Hemmateenejad, B.; Shamsipur, M.; Rashidi, M.; Samouei, H. Affinity of two novel five-coordinated anticancer Pt(II) complexes to human and bovine serum albumins: A spectroscopic approach. Inorg. Chem 2012, 51, 3454–3464. [Google Scholar]
- Sułkowska, A. Interaction of drugs with bovine and human serumalbumin. J. Mol. Struct 2002, 614, 227–232. [Google Scholar]
- Chen, T.T.; Zhu, S.J.; Cao, H.; Shang, Y.F.; Wang, M.; Jiang, G.Q.; Shia, Y.J.; Lu, T.H. Studies on the interaction of salvianolic acid B with human hemoglobin by multi-spectroscopic techniques. Spectrochim. Acta A 2011, 78, 1295–1301. [Google Scholar]
- Valeur, B.; Brochon, J.C. New Trends in Fluorescence Spectroscopy, 6th ed.; Springer Press: Berlin, Germany, 1999; pp. 25–28. [Google Scholar]
- Sahoo, B.K.; Ghosh, K.S.; Dasgupta, S. Molecular interactions of isoxazolcurcumin with human serum albumin: Spectroscopic and molecular modeling studies. Biopolymers 2009, 91, 108–119. [Google Scholar]
- Ju, P.; Fan, H.; Liu, T.; Cui, L.; Ai, S.Y. Probing the interaction of flower-like CdSe nanostructure particles targeted to bovine serum albumin using spectroscopic techniques. J. Lumin 2011, 131, 1724–1730. [Google Scholar]
- Liu, X.H.; Xi, P.X.; Chen, F.J.; Xu, Z.H.; Zeng, Z.Z. Spectroscopic studies on binding of 1-phenyl-3-(coumarin-6-yl)sulfonylurea to bovine serum albumin. J. Photochem. Photobiol. B 2008, 92, 98–102. [Google Scholar]
- Wang, Y.Q.; Tang, B.P.; Zhang, H.M.; Zhou, Q.H.; Zhang, G.C. Studies on the interaction between imidacloprid and human serum albumin: Spectroscopic approach. J. Photochem. Photobiol. B 2009, 94, 183–190. [Google Scholar]
- Hu, Y.J.; Liu, Y.; Wang, J.B.; Xiao, X.H.; Qu, S.S. Study of the interaction between monoammonium glycyrrhizinate and bovine serum albumin. J. Pharm. Biomed. Anal 2004, 36, 915–919. [Google Scholar]
- Li, D.J.; Zhu, M.; Xu, C.; Chen, J.J.; Ji, B.M. The effect of Cu2+ or Fe3+ on the noncovalent binding of rutin with bovine serum albumin by spectroscopic analysis. Spectrochim. Acta A 2011, 78, 74–79. [Google Scholar]
- Ware, W.R. Oxygen quenching of fluorescence in solution: An experimental study of the diffusion process. J. Phys. Chem 1962, 66, 455–458. [Google Scholar]
- Li, D.J.; Zhu, M.; Xu, C.; Chen, J.J.; Ji, B.M. Characterization of the baicalein-bovine serum albumin complex without or with Cu2+or Fe3+ by spectroscopic approaches. Eur. J. Med. Chem 2011, 46, 588–599. [Google Scholar]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar]
- Samari, F.; Shamsipur, M.; Hemmateenejad, B.; Samari, T.; Gharaghani, S. Investigation of the interaction between amodiaquine and human serum albumin by fluorescence spectroscopy and molecular modeling. Eur. J. Med. Chem 2012, 54, 255–263. [Google Scholar]
- Wang, Z.; Li, D.J.; Jin, J. Study on the interaction of puerarin with lysozyme by spectroscopic methods. Spectrochim. Acta A 2008, 70, 866–870. [Google Scholar]
- Li, D.J.; Zhu, J.F.; Jin, J.; Yao, X.J. Studies on the binding of nevadensin to human serum albumin by molecular spectroscopy and modeling. J. Mol. Struct 2007, 846, 34–41. [Google Scholar]
- Tian, J.N.; Liu, J.; Hu, Z.D.; Chen, X.G. Interaction of wogonin with bovine serum albumin. Bioorg. Med. Chem 2005, 13, 4124–4129. [Google Scholar]
- Rahman, M.H.; Maruyama, T.; Okada, T.; Yamasaki, K.; Otagiri, M. Study of interaction of carprofen and its enantiomers with human serum albumin—I: Mechanism of binding studied by dialysis and spectroscopic methods. Biochem. Pharmacol 1993, 46, 1721–1731. [Google Scholar]
- Ross, P.D.; Subramanian, S. Thermodynamics of protein association reactions: Forces contributing to stability. Biochemistry 1981, 20, 3096–3102. [Google Scholar]
- Förster, T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann. Phys 1948, 2, 55–75. [Google Scholar]
- Sklar, L.A.; Hudson, B.S.; Simoni, R.D. Conjugated polyene fatty acids as fluorescent probes: Binding to bovine serum albumin. Biochemistry 1977, 16, 5100–5108. [Google Scholar]
- Wu, P.; Brand, L. Resonance energy transfer: Methods and applications. Anal. Biochem 1994, 218, 1–13. [Google Scholar]
- Hossain, M.; Khan, A.Y.; Kumar, G.S. Interaction of the anticancer plant alkaloid sanguinarine with bovine serum albumin. PLoS One 2011, 6, e18333. [Google Scholar]
- Abert, W.C.; Gregory, W.M.; Allan, G.S. The binding interaction of Coomassie blue with proteins. Anal. Biochem 1993, 213, 407–413. [Google Scholar]
- Ashoka, S.; Seetharamappa, J.; Kandagal, P.B.; Shaikh, S.M.T. Investigation of the interaction between trazodone hydrochloride and bovine serum albumin. J. Lumin 2006, 121, 179–186. [Google Scholar]
- Vekshin, N.L. Separation of the tyrosine and tryptophan components of fluorescence using synchronous scanning method. Biofizika 1996, 41, 1176–1179. [Google Scholar]
- Miller, J.N. Recent advances in molecular luminescence analysis. Proc. Anal. Div. Chem. Soc 1979, 16, 203–208. [Google Scholar]
- Yang, P.; Gao, F. The Principle of Bioinorganic Chemistry; Science Press: Beijing, China, 2002; p. 349. [Google Scholar]
- Greenfield, N.; Fasman, G.D. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry 1969, 8, 4108–4116. [Google Scholar]
- Staprans, I.; Watanabe, S. Optical properties of troponin, tropomyosin, and relaxing protein of rabbit skeletal muscle. J. Biol. Chem 1970, 245, 5962–5966. [Google Scholar]
- Chen, Y.H.; Yang, J.T.; Martinez, H.M. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 1972, 11, 4120–4131. [Google Scholar]
- Gao, H.; Lei, L.D.; Liu, J.Q.; Kong, Q.; Chen, X.G.; Hu, Z.D. The study on the interaction between human serum albumin and a new reagent with antitumour activity by spectrophotometric methods. J. Photochem. Photobiol. A 2004, 167, 213–221. [Google Scholar]
- Petitpas, I.; Petersen, C.E.; Ha, C.E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar]
- Wu, X.H.; Liu, J.J.; Wang, Q.; Xue, W.W.; Yao, X.J.; Zhang, Y.; Jin, J. Spectroscopic and molecular modeling evidence of clozapine binding to human serum albumin at subdomain IIA. Spectrochim. Acta A 2011, 79, 1202–1209. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar]
- Lin, S.Y.; Wei, Y.S.; Li, M.J.; Wang, S.L. Effect of ethanol or/and captopril on the secondary structure of human serum albumin and after protein binding. Eur. J. Pharm. Biopharm 2004, 57, 457–464. [Google Scholar]
- Lerman, L.S. Structural considerations in the interaction of DNA and acridines. J. Mol. Biol 1961, 3, 18–30. [Google Scholar]
- Sun, Y.T.; Zhang, H.T.; Sun, Y.; Zhang, Y.P.; Liu, H.; Cheng, J.H.; Bi, S.Y.; Zhang, H.Q. Study of interaction between protein and mainactive components in Citrus aurantium L. by optical spectroscopy. J. Lumin 2010, 130, 270–279. [Google Scholar]
- Dennis, E.E.; Thomas, J.R.; Valeria, C.; Augusto, C.; Moreno, Z. Determination of the affinity of drugs toward serum albumin by measurement of the quenching of the intrinsic tryptophan fluorescence of the protein. J. Pharm. Pharmacol 1999, 51, 41–48. [Google Scholar]
- Silva, K.P.; Seraphim, T.V.; Borges, J.C. Structural and functional studies of Leishmania braziliensis Hsp90. Biochim. Biophys. Acta 2013, 1834, 351–361. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [Google Scholar]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, USA, 2002. Available online: http://www.pymol.org (on accessed 5 July 2012).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | T (K) | Ksv (105 M−1) | Kq (1013 M−1s−1) | Ra |
|---|---|---|---|---|
| 7.4 | 288 | 7.60 | 7.60 | 0.994 |
| 298 | 6.63 | 6.63 | 0.996 | |
| 308 | 6.21 | 6.21 | 0.995 |
| pH | T (K) | Kq (105 M−1) | Ra |
|---|---|---|---|
| 7.4 | 288 | 5.68 | 0.999 |
| 298 | 5.54 | 0.998 | |
| 308 | 5.39 | 0.996 |
| pH | T (K) | Kb (106 M−1) | n | Ra |
|---|---|---|---|---|
| 7.4 | 288 | 1.679 | 1.062 | 0.998 |
| 298 | 1.230 | 1.048 | 0.998 | |
| 308 | 0.857 | 1.026 | 0.995 |
| pH | T (K) | ΔG0 (kJ/mol) | ΔH0 (kJ/mol) | ΔS0 (J/mol·K) | Ra |
|---|---|---|---|---|---|
| 7.4 | 288 | −31.31 | |||
| 298 | −31.56 | −24.02 | 25.31 | 0.999 | |
| 308 | −31.82 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, H.; Yao, N.; Xu, H.; Wang, T.; Li, G.; Li, Z. Characterization of the Interaction between Eupatorin and Bovine Serum Albumin by Spectroscopic and Molecular Modeling Methods. Int. J. Mol. Sci. 2013, 14, 14185-14203. https://doi.org/10.3390/ijms140714185
Xu H, Yao N, Xu H, Wang T, Li G, Li Z. Characterization of the Interaction between Eupatorin and Bovine Serum Albumin by Spectroscopic and Molecular Modeling Methods. International Journal of Molecular Sciences. 2013; 14(7):14185-14203. https://doi.org/10.3390/ijms140714185
Chicago/Turabian StyleXu, Hongliang, Nannan Yao, Haoran Xu, Tianshi Wang, Guiying Li, and Zhengqiang Li. 2013. "Characterization of the Interaction between Eupatorin and Bovine Serum Albumin by Spectroscopic and Molecular Modeling Methods" International Journal of Molecular Sciences 14, no. 7: 14185-14203. https://doi.org/10.3390/ijms140714185