Identification of a Genomic Region Containing a Novel Promoter Resistant to Glucose Repression and Over-Expression of β-Glucosidase Gene in Hypocrea orientalis EU7-22

Abstract
:1. Introduction
2. Results and Discussion
2.1. Promoter proA Cloning and Sequence Analysis
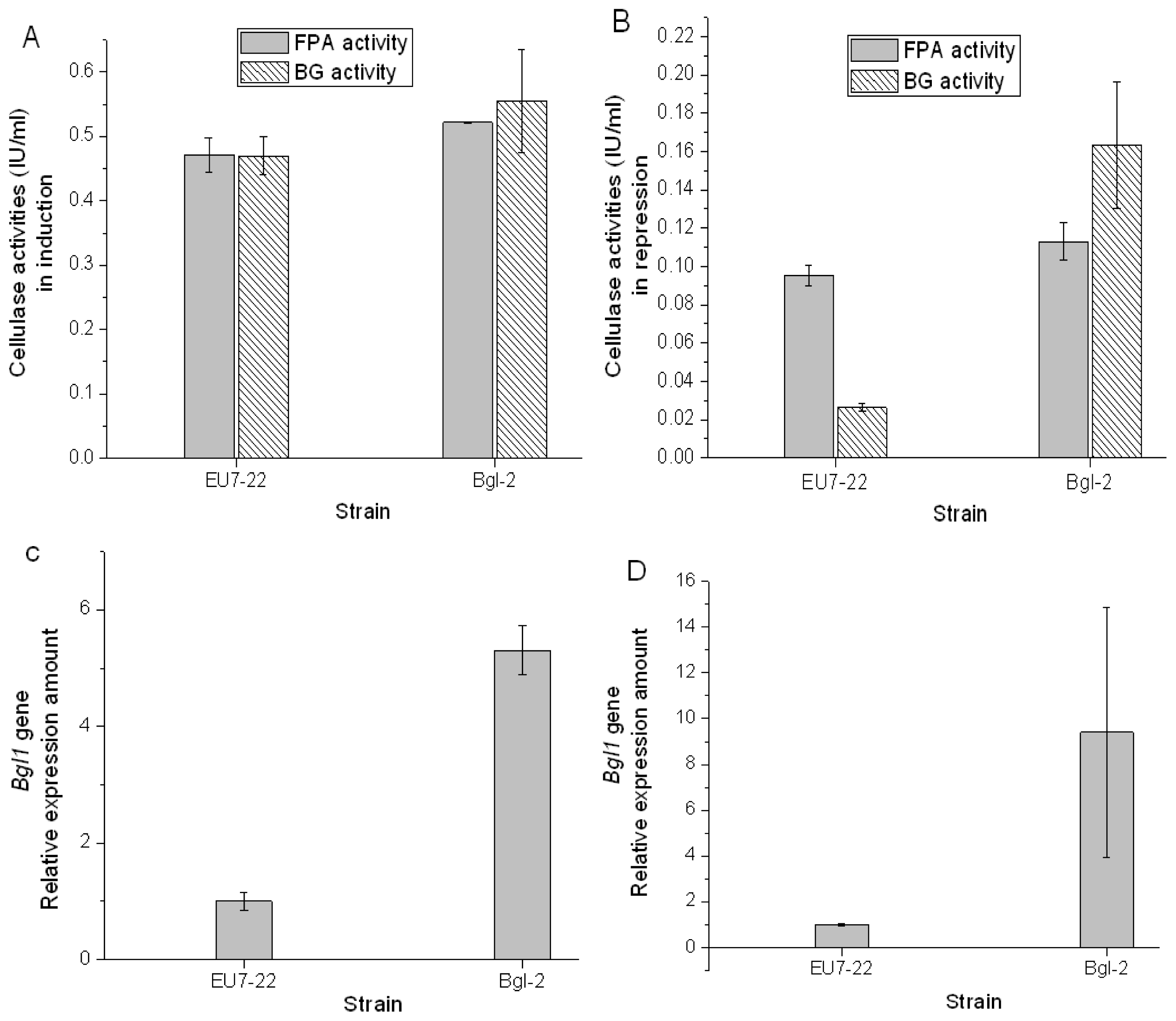
2.2. Over-Expression of β-Glucosidase Gene in H. orientalis
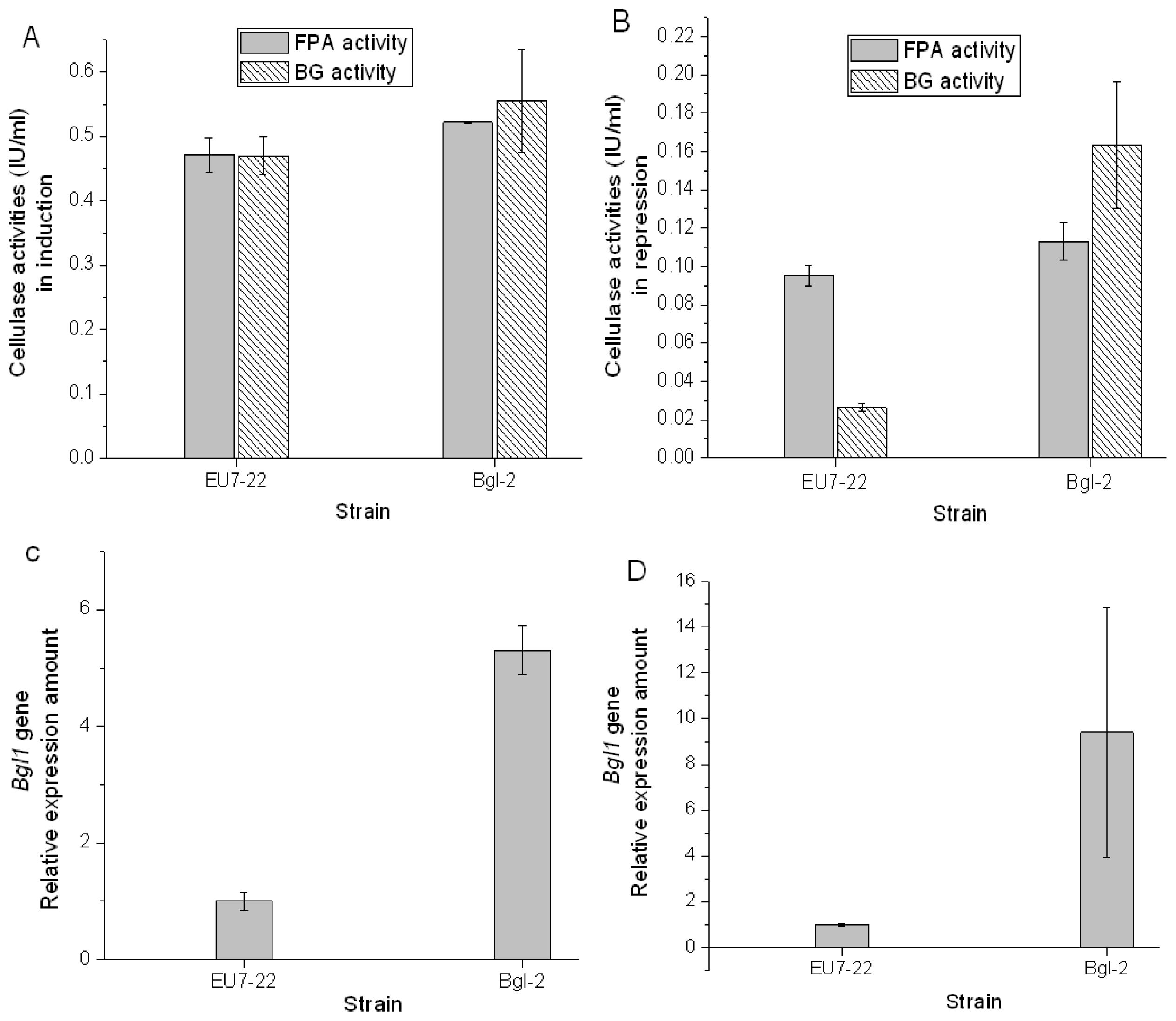
2.3. Comparison Analysis of Cellulase Production of Transformants and Host Strain EU7-22
3. Experimental Section
3.1. Microorganism Strains and Culture Conditions
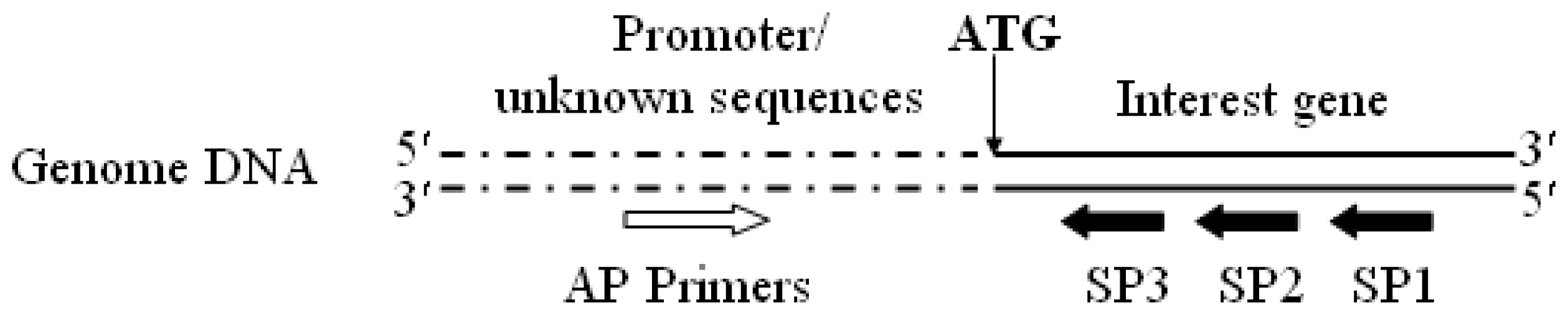
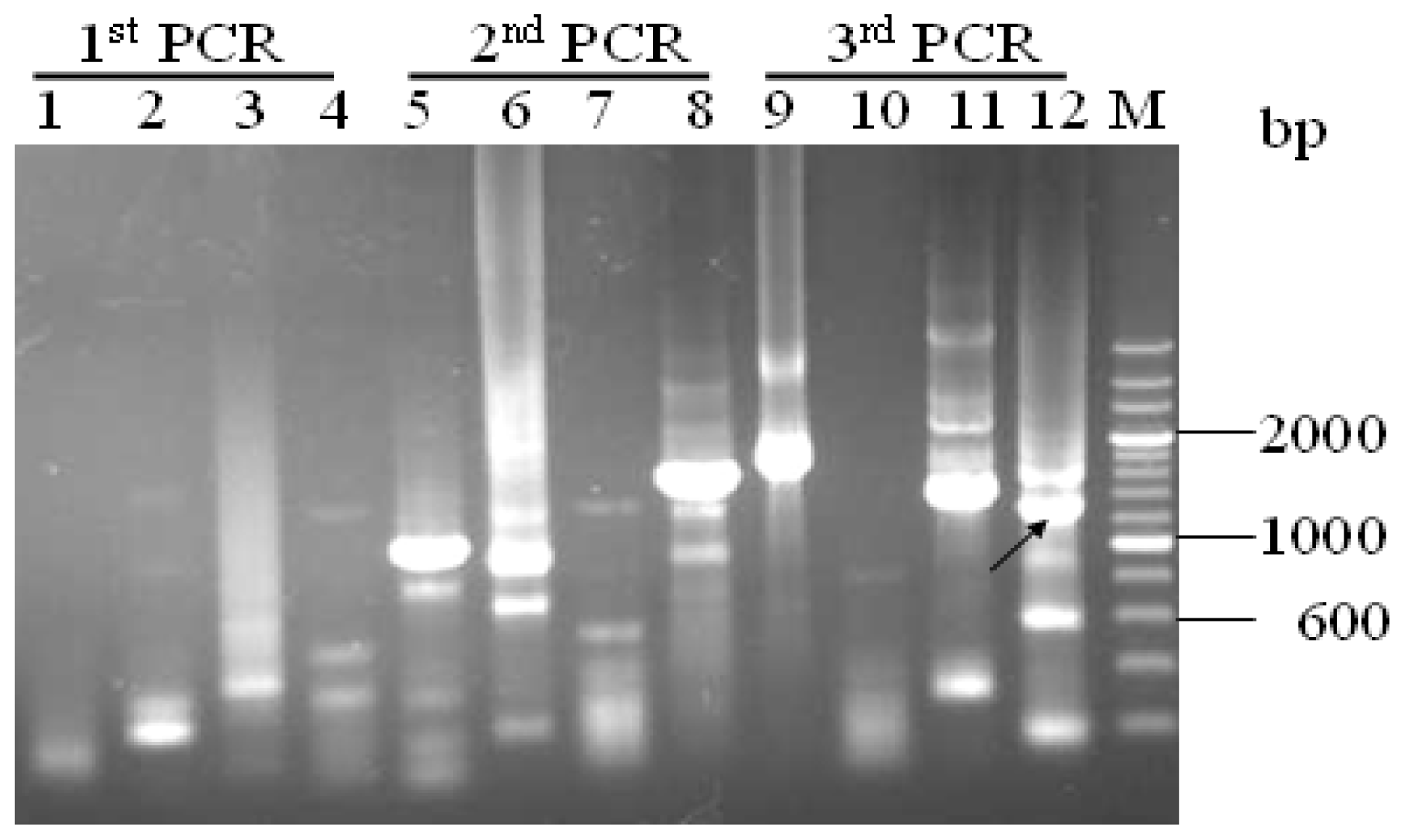
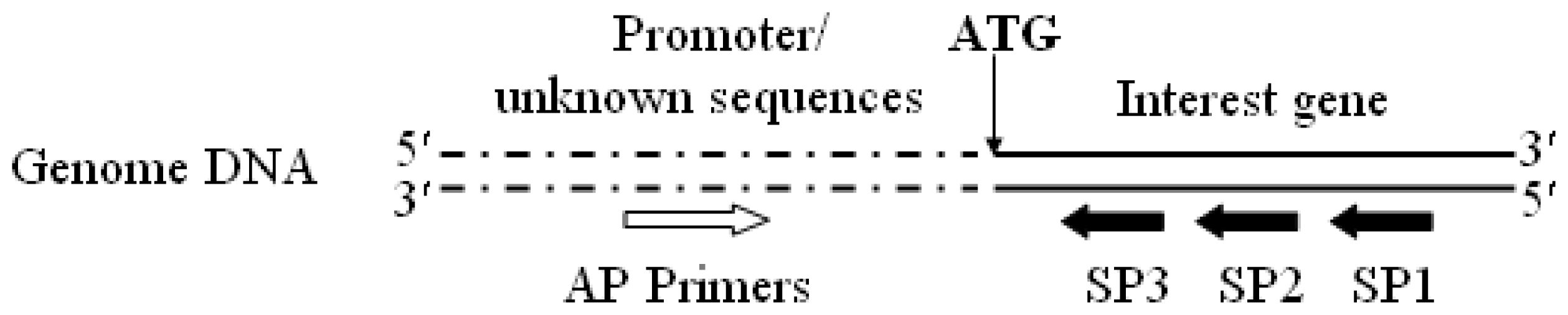
3.2. Promoter proA Cloning and Analysis
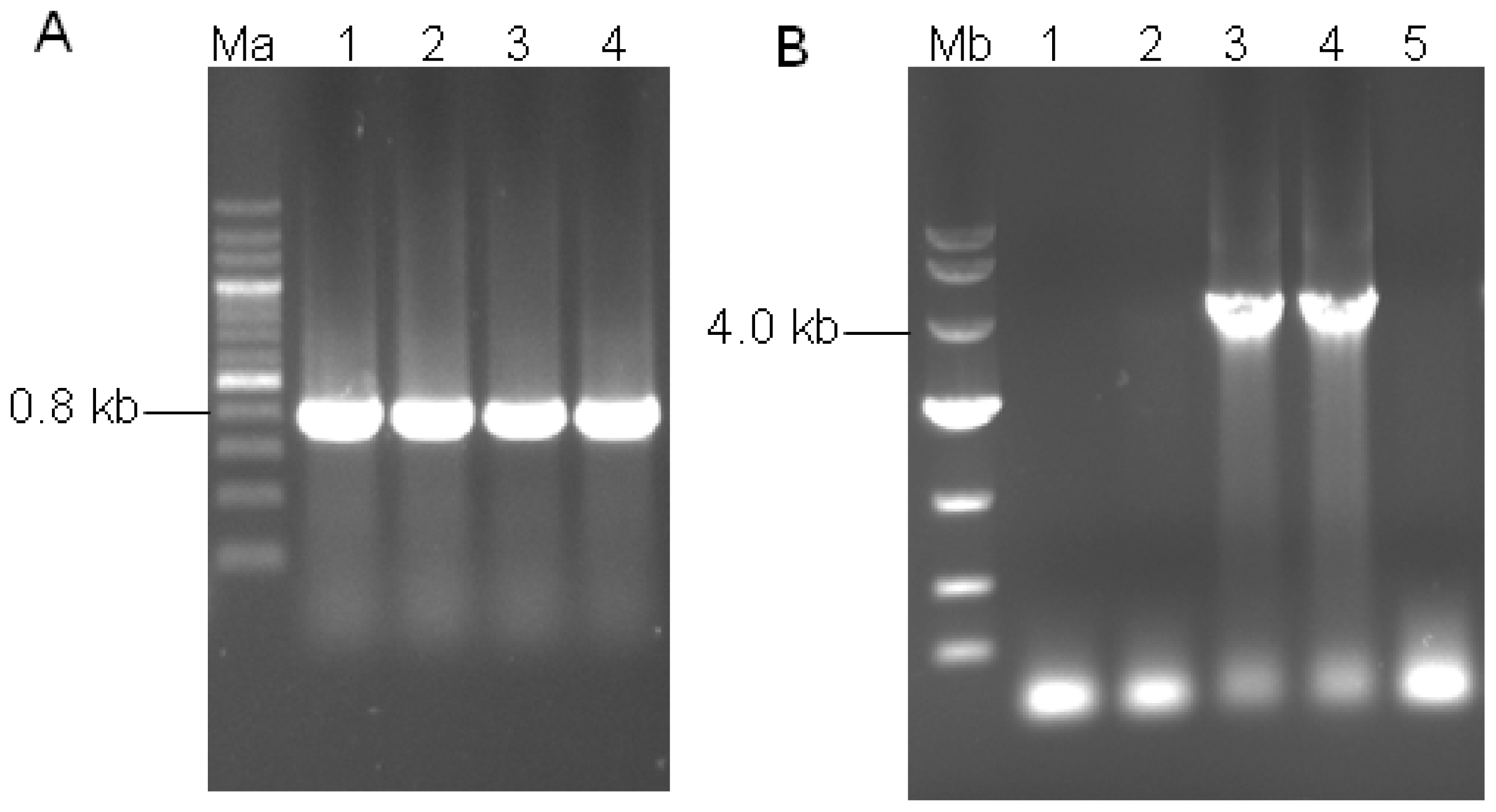
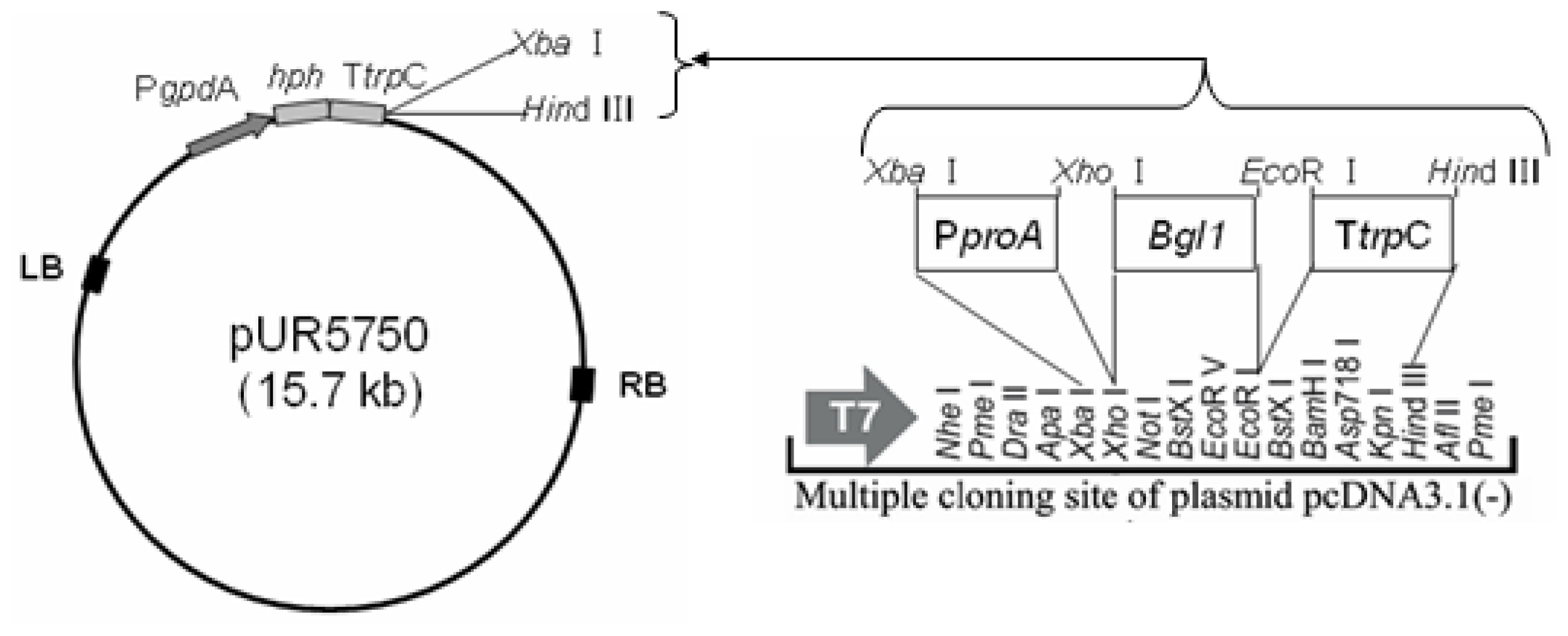
3.3. Construction of Binary Expression Vectors pUR5750-Bgl1
3.4. Agrobacterium-Tumefaciens-Mediated-Transformation (ATMT) in H. orientalis EU7-22
3.5. Transformant Stability and Molecular Analysis of the Transformants
3.6. Enzymatic Analysis
3.7. Transcription Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Kamm, B.; Kamm, M. Biorefineries–Multi product processes. Adv. Biochem. Eng. Biotechnol 2007, 105, 175–204. [Google Scholar]
- Kumar, R.; Singh, S.; Singh, O.V. Bioconversion of lignocellulosic biomass: Biochemical and molecular perspectives. J. Ind. Microbiol. Biotechnol 2008, 35, 377–391. [Google Scholar]
- Kovács, K.; Szakacs, G.; Zacchi, G. Comparative enzymatic hydrolysis of pretreated spruce by supernatants, whole fermentation broths and washed mycelia of Trichoderma reesei and Trichoderma atroviride. Bioresour. Technol 2009, 100, 1350–1357. [Google Scholar]
- Sipos, B.; Benko, Z.; Dienes, D.; Réczey, K.; Viikari, L.; Siika-aho, M. Characterisation of specific activities and hydrolytic properties of cell-wall degrading enzymes produced by Trichoderma reesei Rut C30 on different carbon sources. Appl. Biochem. Biotechnol 2010, 161, 347–364. [Google Scholar]
- Portnoy, T.; Margeot, A.; Linke, R.; Atanasova, L.; Fekete, E.; Sándor, E.; Hartl, L.; Karaffa, L.; Druzhinina, I.S.; Seiboth, B.; et al. The CRE1 carbon catabolite repressor of the fungus Trichoderma reesei: A master regulator of carbon assimilation. BMC Genomics 2011, 12, 269. [Google Scholar]
- Takashima, S.; Iikura, H.; Nakamura, A.; Masaki, H.; Uozumi, T. Analysis of Cre1 binding sites in the Trichoderma reesei cbh1 upstream region. FEMS Microbiol. Lett 1996, 145, 361–366. [Google Scholar]
- Cubero, B.; Scazzocchio, C. Two different, adjacent and divergent zinc finger binding sites are necessary for CREA-mediated carbon catabolite repression in the proline gene cluster of Aspergillus nidulans. EMBO J 1994, 13, 407–415. [Google Scholar]
- Liu, T.; Wang, TH.; Li, X.; Liu, X. Improved heterologous gene expression in Trichoderma reesei by cellobiohydrolase I gene (cbh1) promoter optimization. Acta Biochim. Biophys. Sin 2008, 40, 158–165. [Google Scholar]
- Kubicek, C.P.; Mikus, M.; Schuster, A.; Schmoll, M.; Seiboth, B. Metabolic engineering strategies for the improvement of cellulose production by Hypocrea jecorina. Biotechnol. Biofuels 2009, 2, 1–14. [Google Scholar]
- Bando, H.; Hisada, H.; Ishida, H.; Hata, Y.; Katakura, Y.; Kondo, A. Isolation of a novel promoter for efficient protein expression by Aspergillus oryzae in solid-state culture. Appl. Microbiol. Biotechnol 2011, 92, 561–569. [Google Scholar]
- Tilburn, J.; Sarkar, S.; Widdick, D.A.; Espeso, E.A.; Orejas, M.; Mungroo, J.; Peñalva, M.A. The Aspergillus PacC zinc finger transcription factor mediates regulation of both acid- and alkaline-expressed genes by ambient pH. EMBO J 1995, 14, 779–790. [Google Scholar]
- Ravagnani, A.; Gorfinkiel, L.; Langdon, T.; Diallinas, G.; Adjadj, E.; Demais, S.; Gorton, D.; Arst, H.N.; Scazzocchio, C. Subtle hydrophobic interactions between the seventh residue of the zinc finger loop and the first base of an HGATAR sequence determine promoter-specific recognition by the Aspergillus nidulans GATA factor AreA. EMBO J 1997, 16, 3974–3986. [Google Scholar]
- Marzluf, G.A. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev 1997, 61, 17–32. [Google Scholar]
- Delgado-Jarana, J.; Rincón, A.M.; Benítez, T. Aspartyl protease from Trichoderma harzianum CECT 2413: Cloning and characterization. Microbiology 2002, 148, 1305–1315. [Google Scholar]
- Lee, L.Y.; Gelvin, S.B. T-DNA binary vectors and systems. Plant Physiol 2008, 146, 325–332. [Google Scholar]
- Michielse, C.B.; Hooykaas, P.J.J.; van den Hondel, C.; Ram, A.F.J. Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr. Genet 2005, 48, 1–17. [Google Scholar]
- Mullins, E.D.; Chen, X.; Romaine, P.; Raina, R.; Geiser, D.M.; Kang, S. Agrobacterium-mediated transformation of Fusarium oxysporum: An efficient tool for insertional mutagenesis and gene transfer. Phytopathology 2001, 91, 173–180. [Google Scholar]
- de Groot, M.J.; Bundock, P.; Hooykaas, P.J.; Beijersbergen, A.G. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat. Biotechnol 1998, 16, 839–842. [Google Scholar]
- Bundock, P.; den Dulk-Ras, A.; Beijersbergen, A.; Hooykaas, P.J. Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J 1995, 14, 3206–3214. [Google Scholar]
- Reddy, P.S.; Mahanty, S.; Kaul, T.; Nair, S.; Sopory, S.K.; Reddy, M.K. A high-throughput genome-walking method and its use for cloning unknown flanking sequences. Anal. Biochem 2008, 381, 248–253. [Google Scholar]
- Leoni, C.; Volpicella, M.; Leo, F.D.; Gallerani, R.; Ceci, L.R. Genome walking in eukaryotes. FEBS J 2011, 278, 3953–3977. [Google Scholar]
- Penttilä, M.; Nevalainen, H.; Rättö, M.; Salminen, E.; Knowles, J. A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 1987, 61, 155–164. [Google Scholar]
- Ghose, T.K. Measurement of cellulase activities. Pure. Appl. Chem 1987, 59, 257–268. [Google Scholar]
- Saha, B.C. Production, purification and properties of endoglucanase from a newly isolated strain of Mucor circinelloides. Process. Biochem 2004, 39, 1871–1876. [Google Scholar]
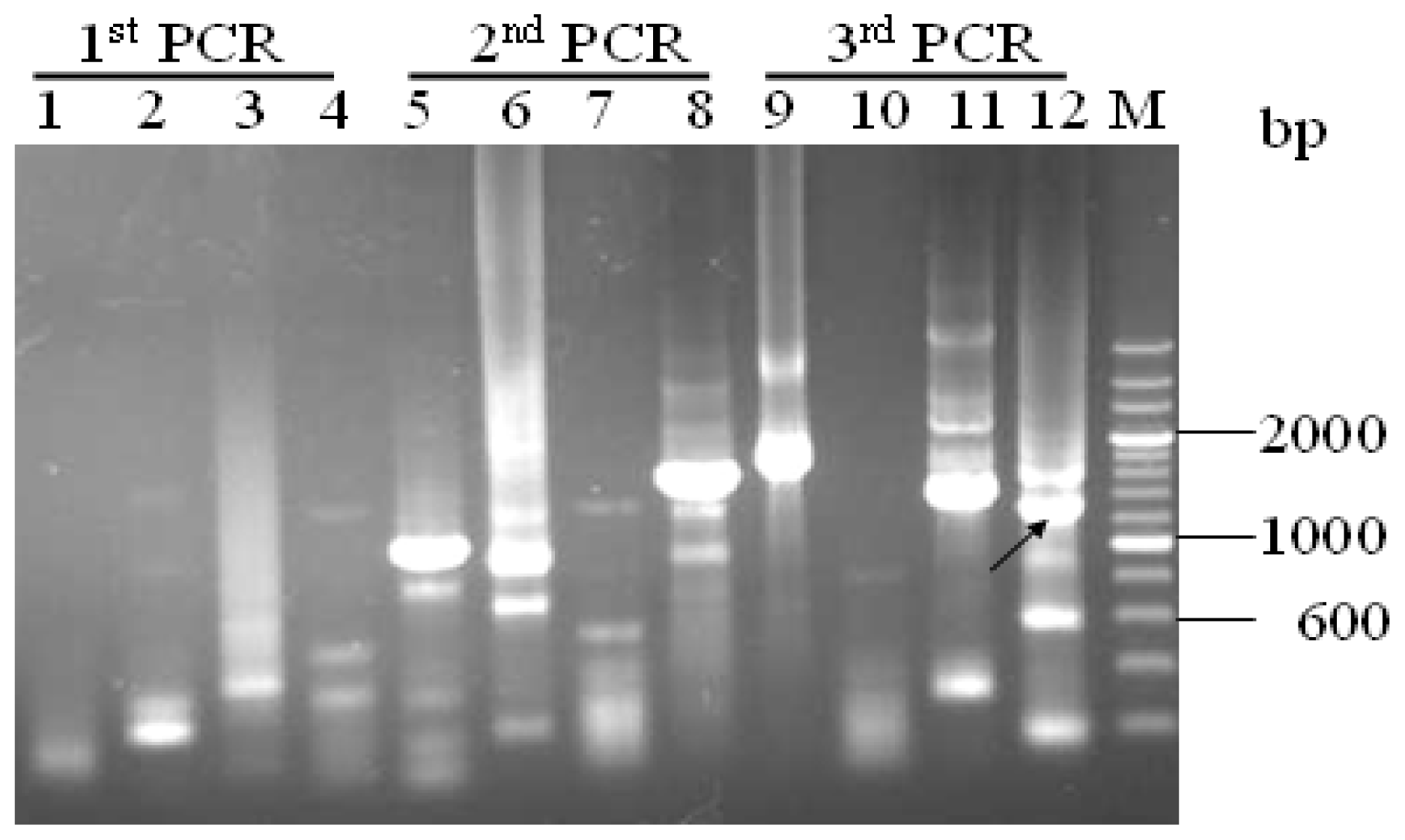
 ”) involved in carbon, nitrogen, and pH regulations, respectively. Arrow underline the sequences indicated the reverse primer SP3. M = A or C; S = G or C; Y = C or T; R = A or G; H = A, C or T.
”) involved in carbon, nitrogen, and pH regulations, respectively. Arrow underline the sequences indicated the reverse primer SP3. M = A or C; S = G or C; Y = C or T; R = A or G; H = A, C or T.
”) involved in carbon, nitrogen, and pH regulations, respectively. Arrow underline the sequences indicated the reverse primer SP3. M = A or C; S = G or C; Y = C or T; R = A or G; H = A, C or T.
”) involved in carbon, nitrogen, and pH regulations, respectively. Arrow underline the sequences indicated the reverse primer SP3. M = A or C; S = G or C; Y = C or T; R = A or G; H = A, C or T.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer name | Primer sequence (5′–3′) |
|---|---|
| SP1-Rb | AGACGGAGGTGATGTACTCCGAATC |
| SP2-Rb | CTTCTTGGCCTTGAAGAGAGCAGTAG |
| SP3-Rb | CGAGGAAGGAAACGAGAAAAGCTC |
| M13-Fa | CGCCAGGGTTTTCCCAGTCACGAC |
| M13-Rb | GAGCGGATAACAATTTCACACAGG |
| PproA-Fa | CTAGTCTAGAAGTAGCAAAGGCTAACGGATTAG |
| PproA-Rb | CCGCTCGAGCTTGAATCTCGGAGAGGGCTG |
| Bgl1-Fa | CCGCTCGAGATGCGTTACCGAACAGCAG |
| Bgl1-Rb | CCGGAATTCCTACGCTACCGACAGAGTGCT |
| TtrpC-Fa | CCGGAATTCAGTAGATGCCGACCGCG |
| TtrpC-Rb | CCCAAGCTTAAGAAGGATTACCTCTAAACAAG |
| hph-Fa | CGACAGCGTCTCCGACCTGA |
| hph-Rb | CGCCCAAGCTGCATCATCGAA |
| BGLYG-Fa | ATCACCTACCCGCCTTCA |
| BGLYG-Rb | TCTCGTCGTCGGATGTTG |
| 18s-Fa | AGGCGCGCAAATTACCCAATCC |
| 18s-Rb | GCCCTCCAATTGTTCCTCGTTAAG |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Long, C.; Cheng, Y.; Gan, L.; Liu, J.; Long, M. Identification of a Genomic Region Containing a Novel Promoter Resistant to Glucose Repression and Over-Expression of β-Glucosidase Gene in Hypocrea orientalis EU7-22. Int. J. Mol. Sci. 2013, 14, 8479-8490. https://doi.org/10.3390/ijms14048479
Long C, Cheng Y, Gan L, Liu J, Long M. Identification of a Genomic Region Containing a Novel Promoter Resistant to Glucose Repression and Over-Expression of β-Glucosidase Gene in Hypocrea orientalis EU7-22. International Journal of Molecular Sciences. 2013; 14(4):8479-8490. https://doi.org/10.3390/ijms14048479
Chicago/Turabian StyleLong, Chuannan, Yijin Cheng, Lihui Gan, Jian Liu, and Minnan Long. 2013. "Identification of a Genomic Region Containing a Novel Promoter Resistant to Glucose Repression and Over-Expression of β-Glucosidase Gene in Hypocrea orientalis EU7-22" International Journal of Molecular Sciences 14, no. 4: 8479-8490. https://doi.org/10.3390/ijms14048479