Inhibition of Human Transthyretin Aggregation by Non-Steroidal Anti-Inflammatory Compounds: A Structural and Thermodynamic Analysis



Abstract
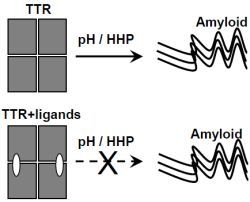
:
Abbreviations
| TTR | Transthyretin |
| FAP | familial amyloid polyneuropathy |
| SSA | senile systemic amyloidosis |
| CSF | cerebrospinal fluid |
| HBS | hormone binding site |
| CR | Congo red |
| HHP | high hydrostatic pressure |
| SEC | size exclusion chromatography |
| CNS | central nervous system |
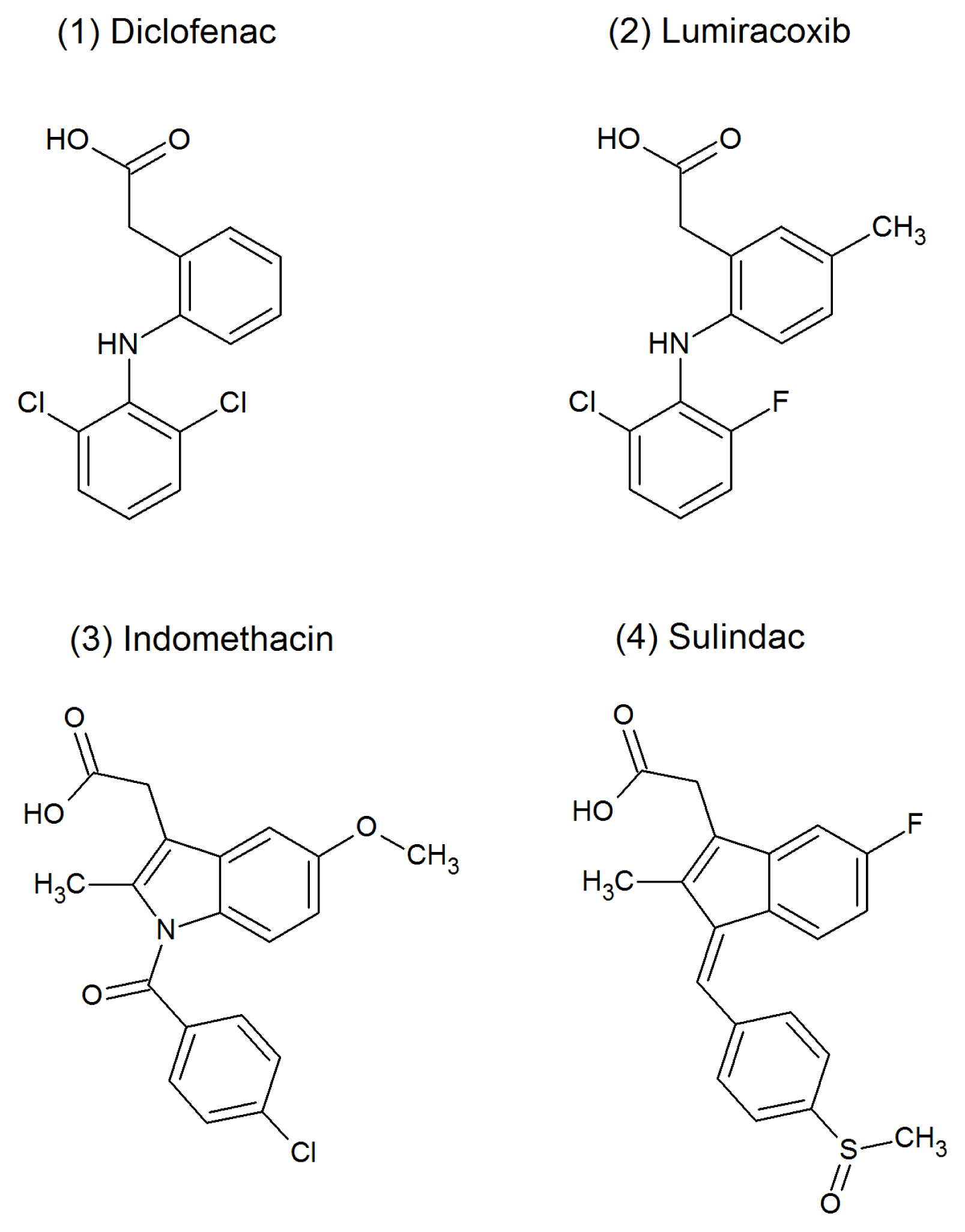
| LUM | lumiracoxib |
| IND | indomethacin |
| SUL | sulindac |
| ANS | 8-anilino-1-naphthalenesulfonic acid |
1. Introduction
2. Results and Discussion
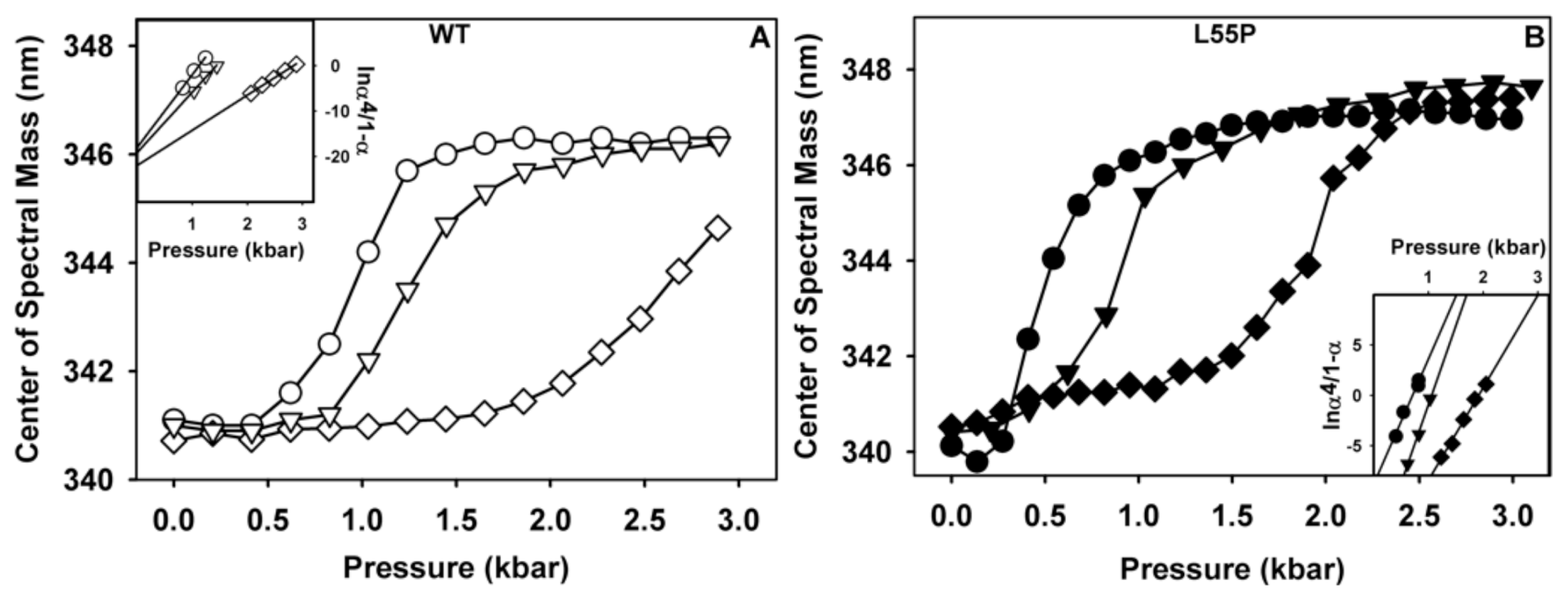
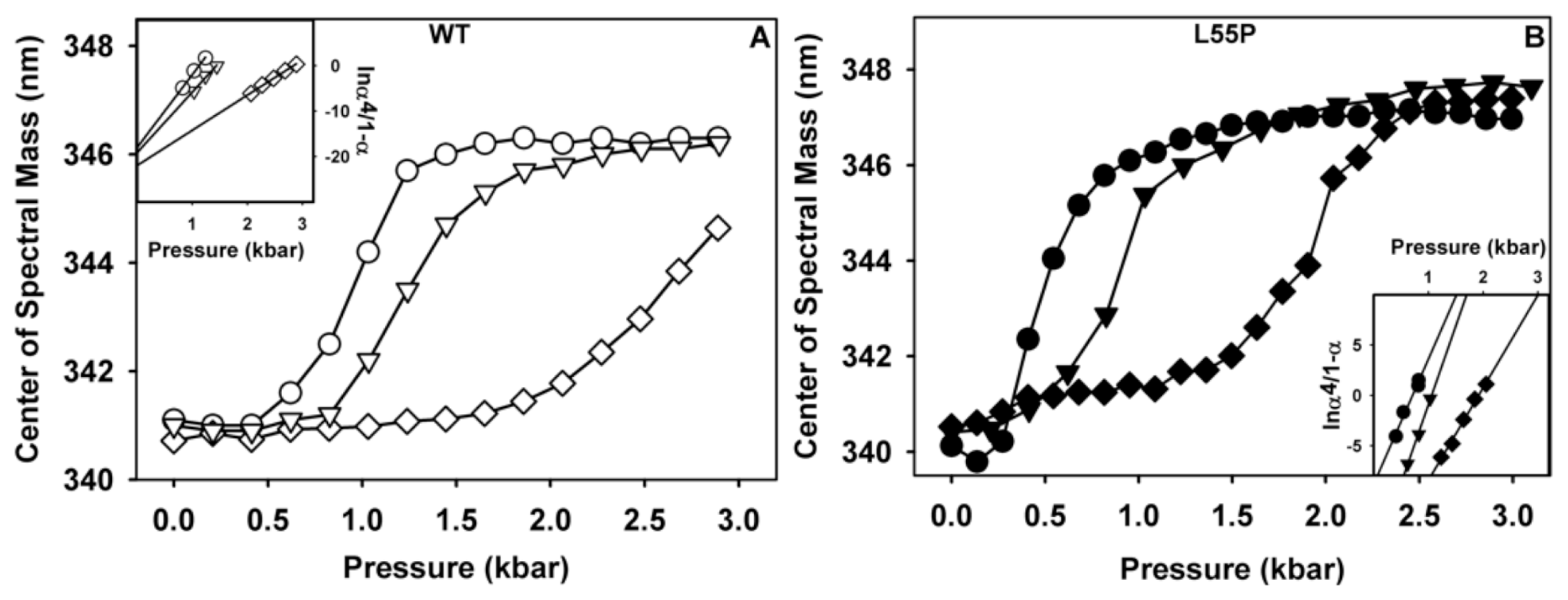
2.1. Ligand Binding Enhances Tetramer Stability
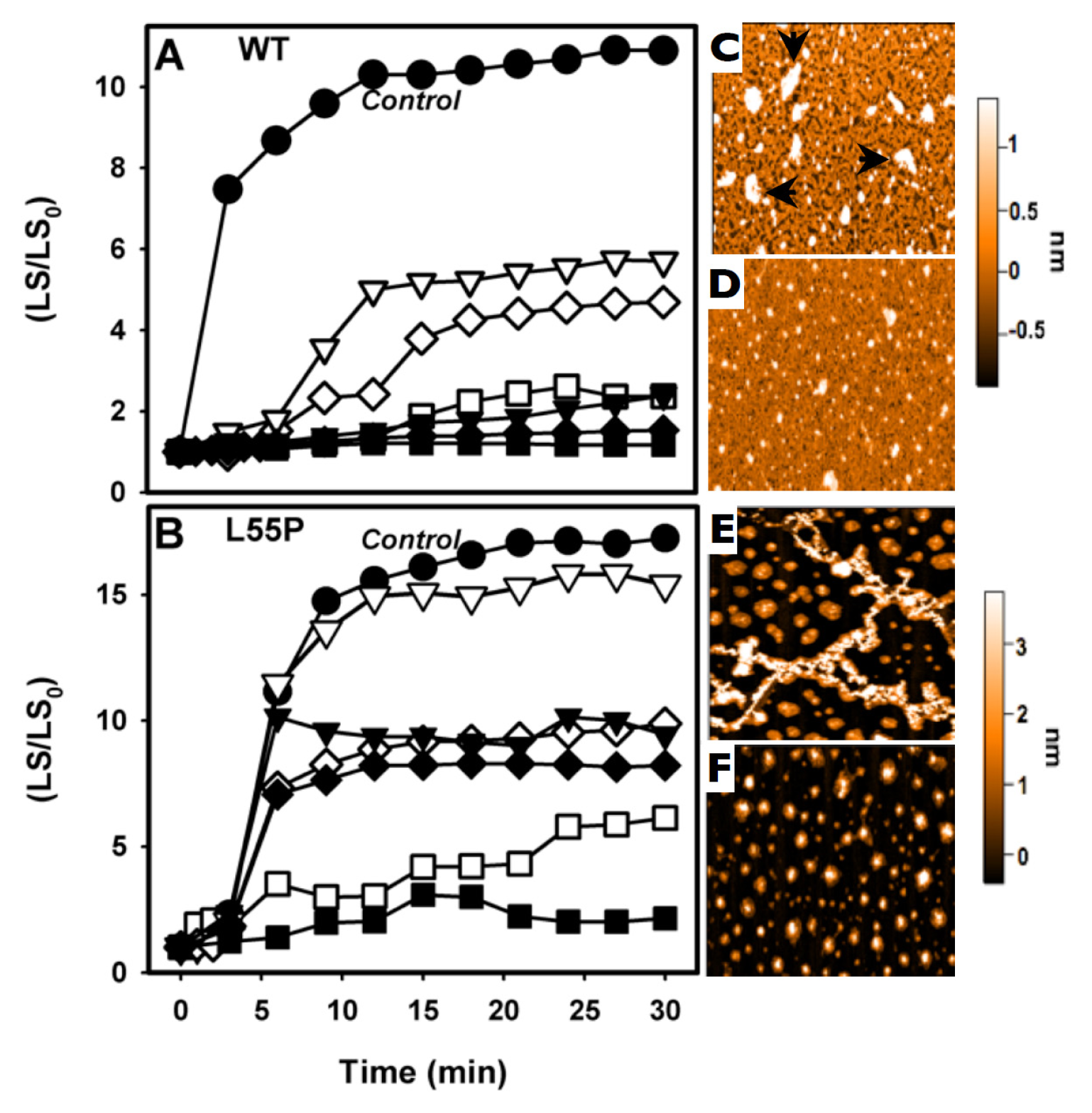
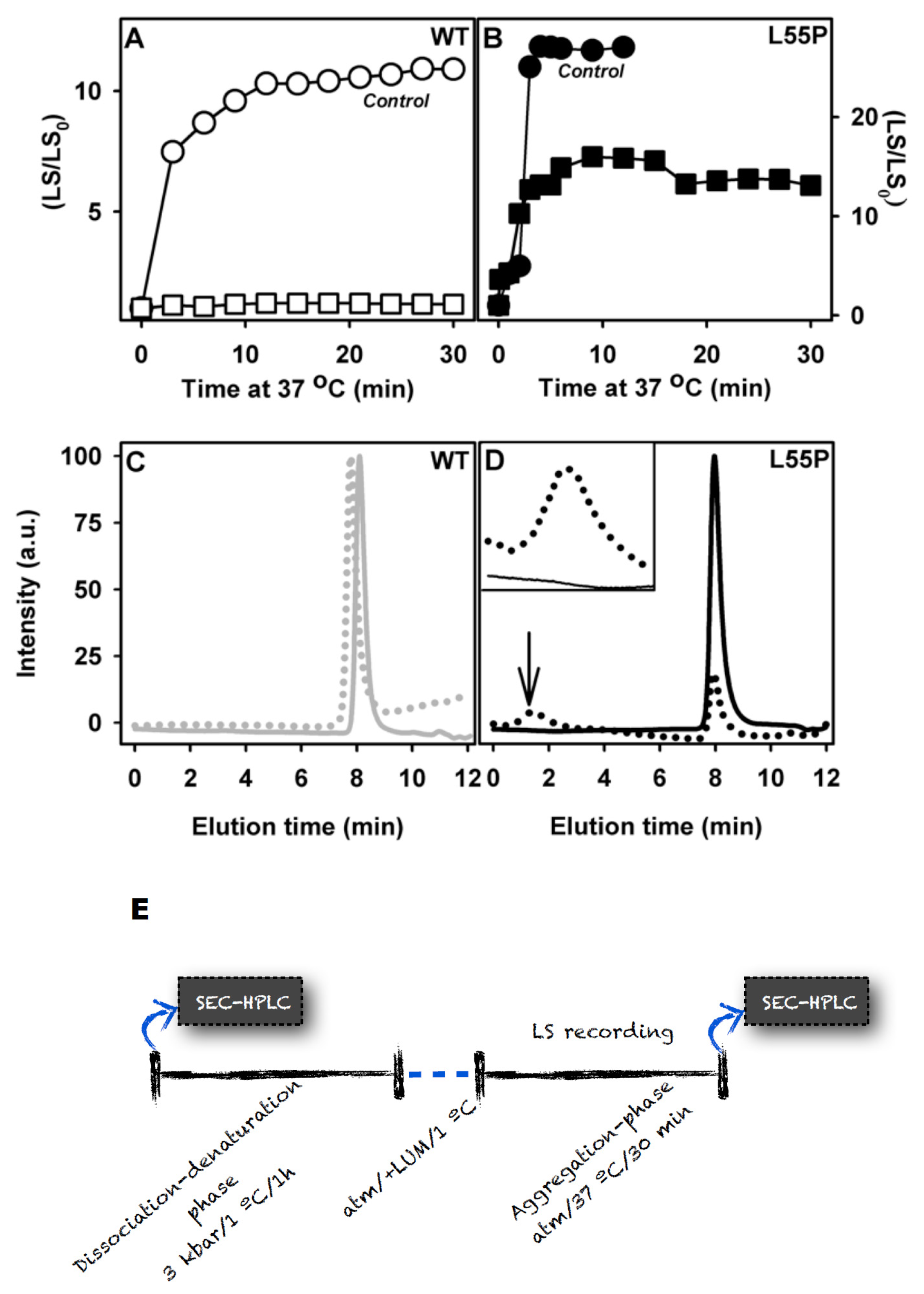
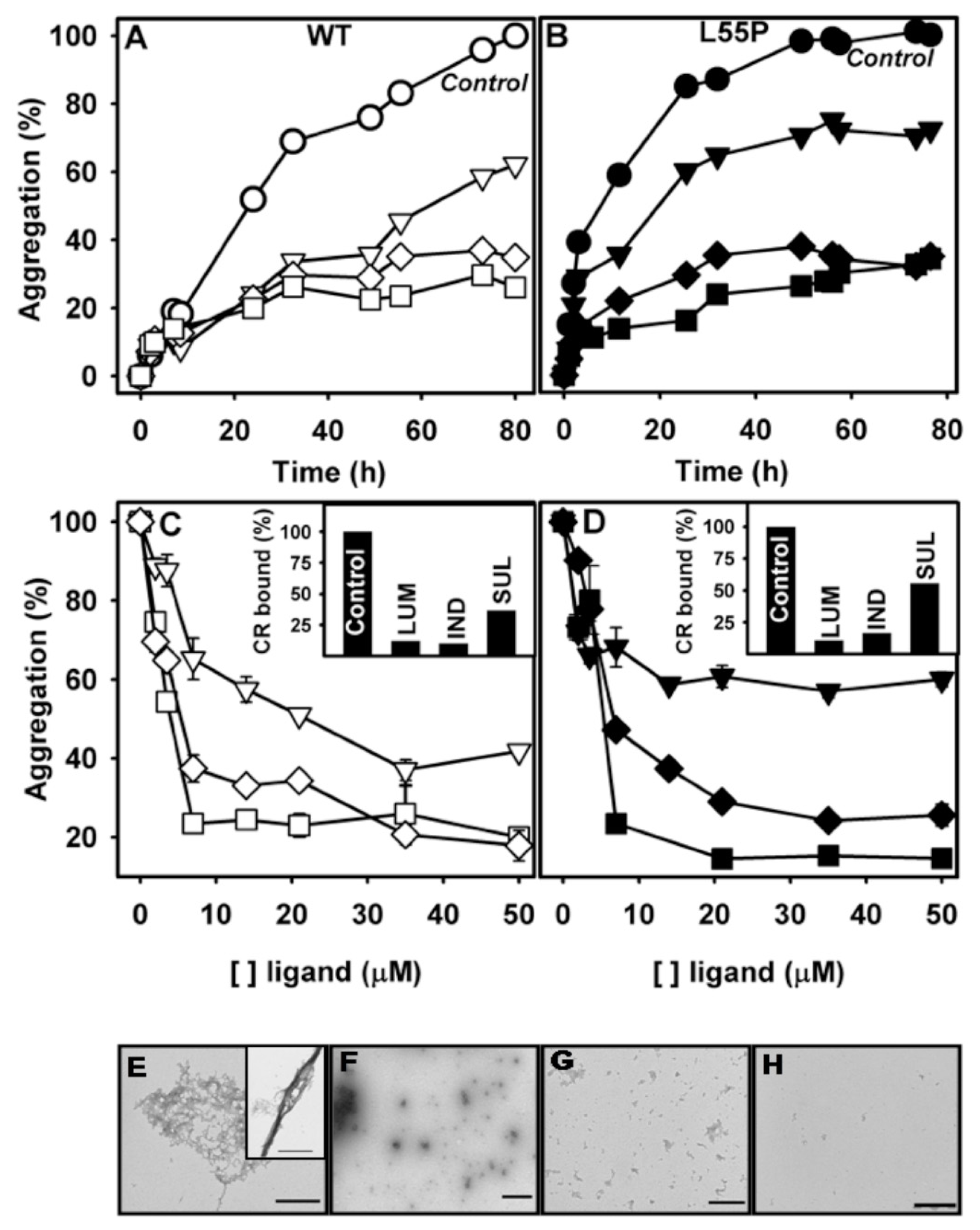
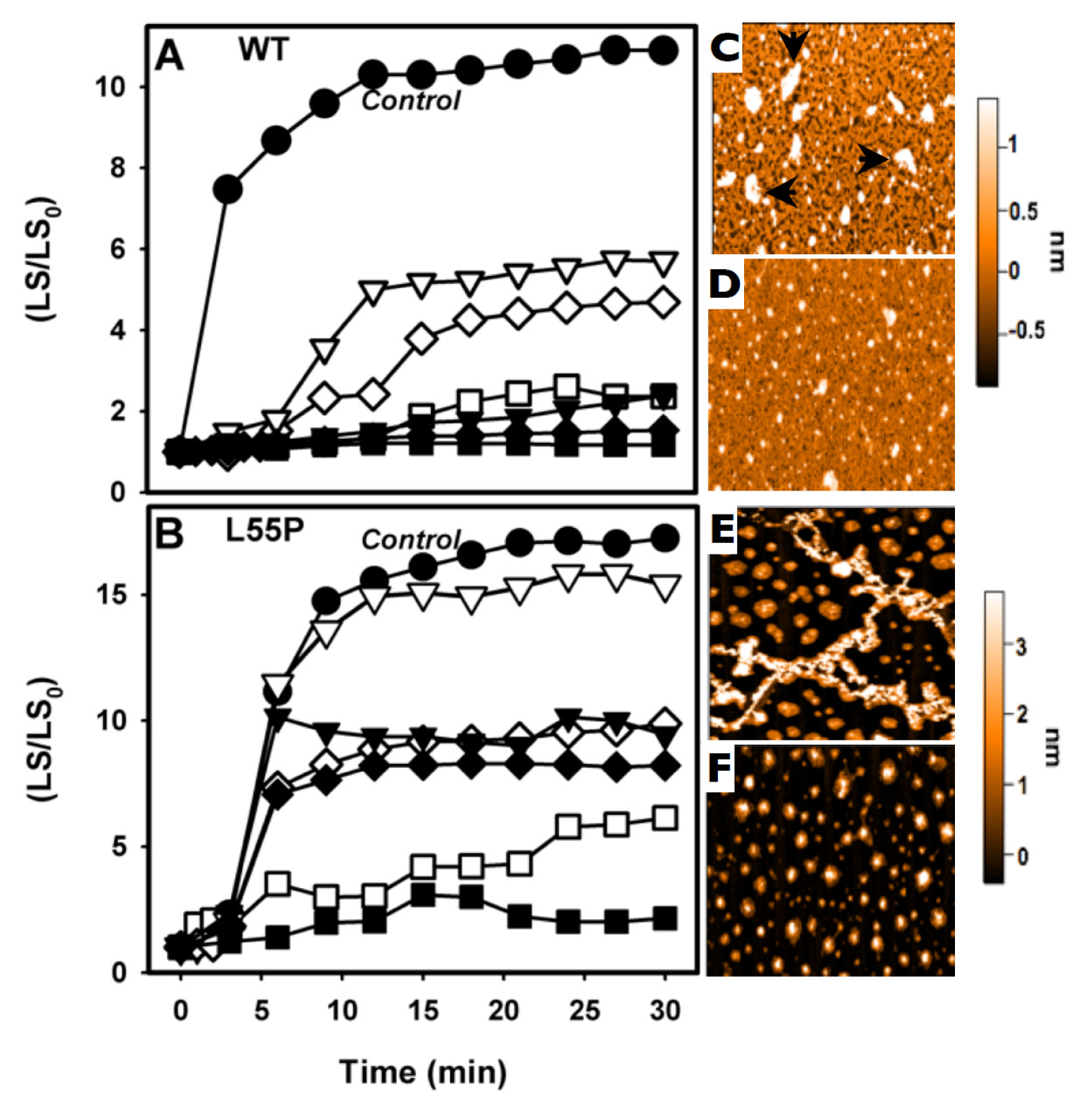
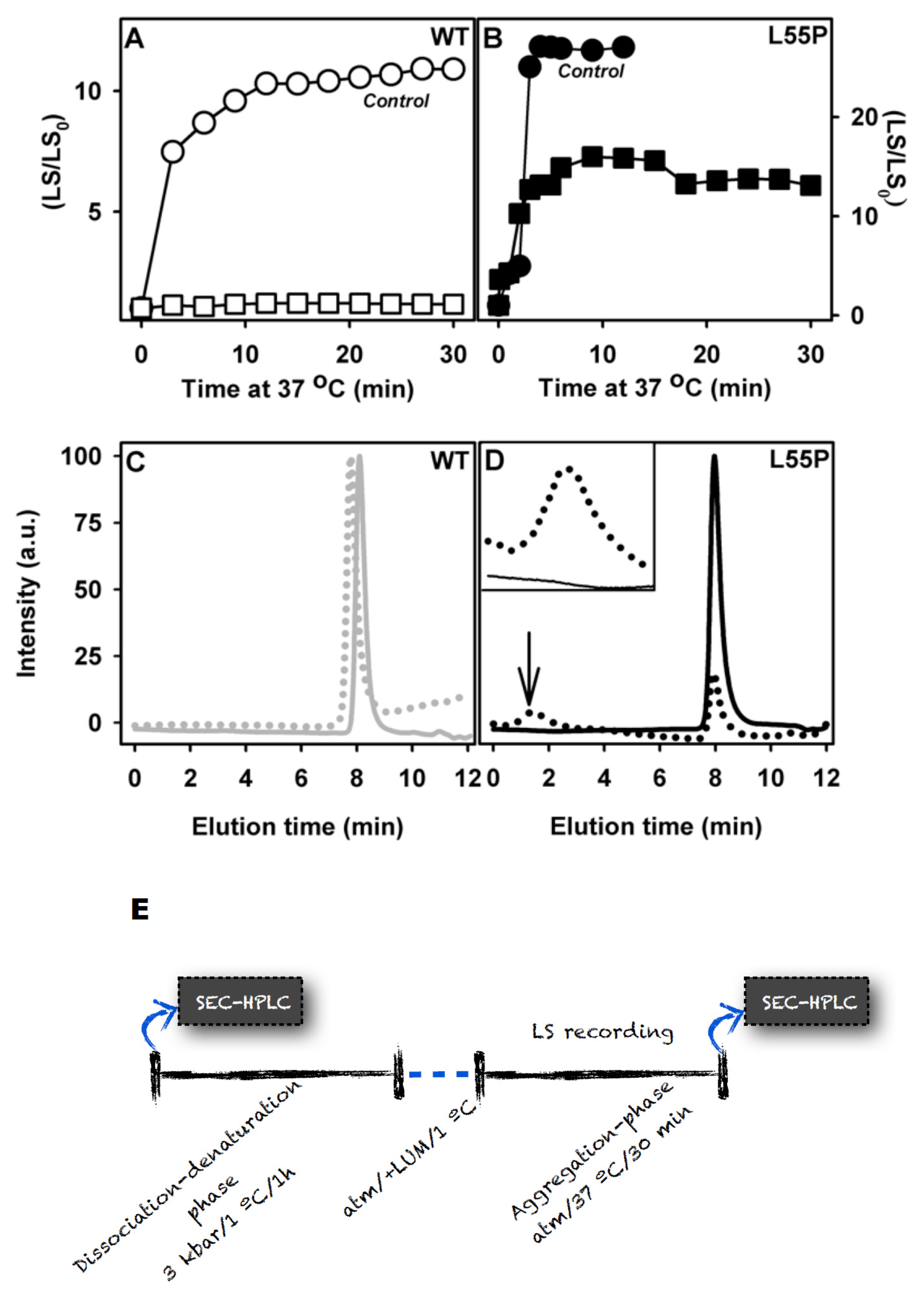
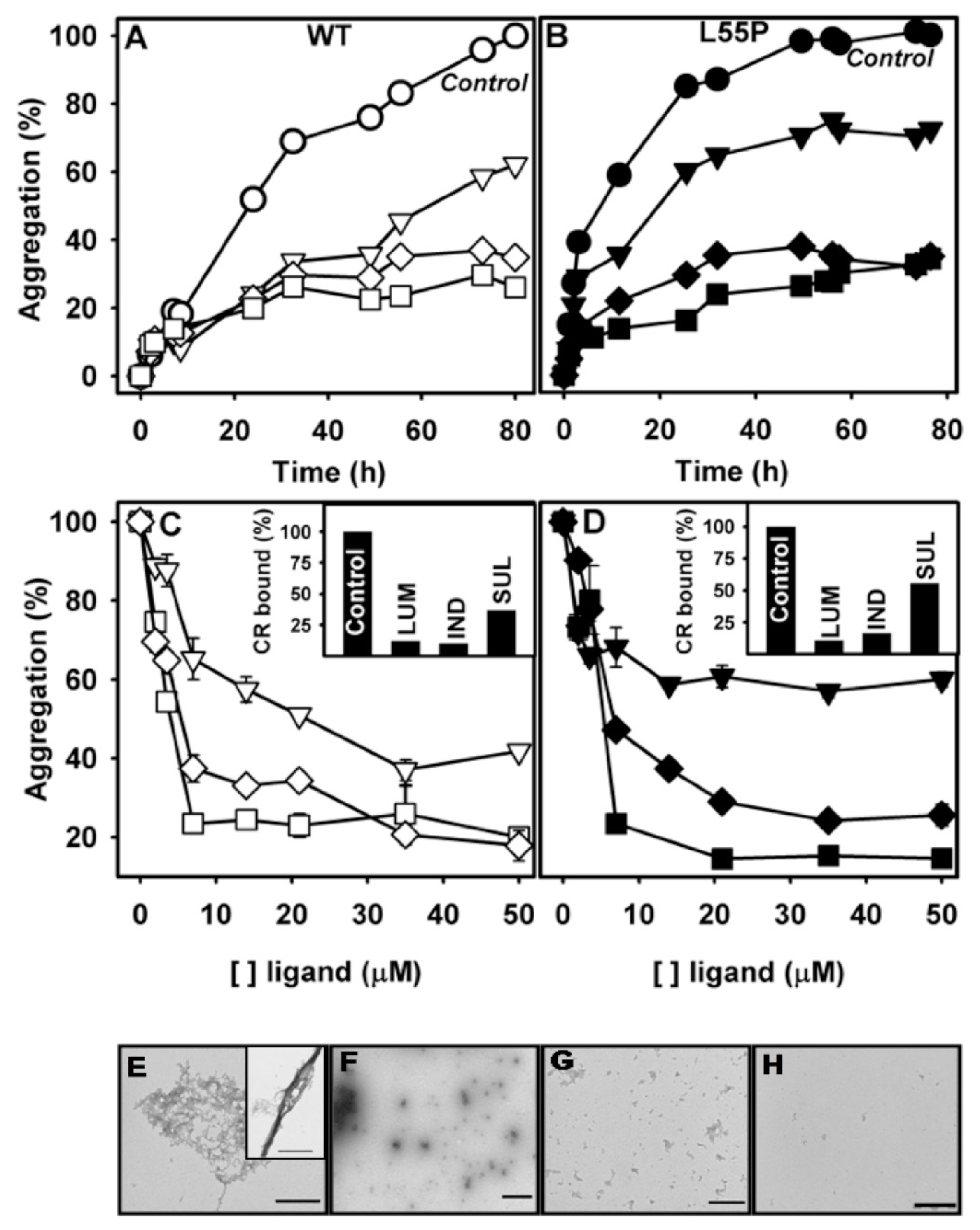
2.2. SUL, IND and LUM Inhibit the HHP-Induced Aggregation of WT-TTR and L55P
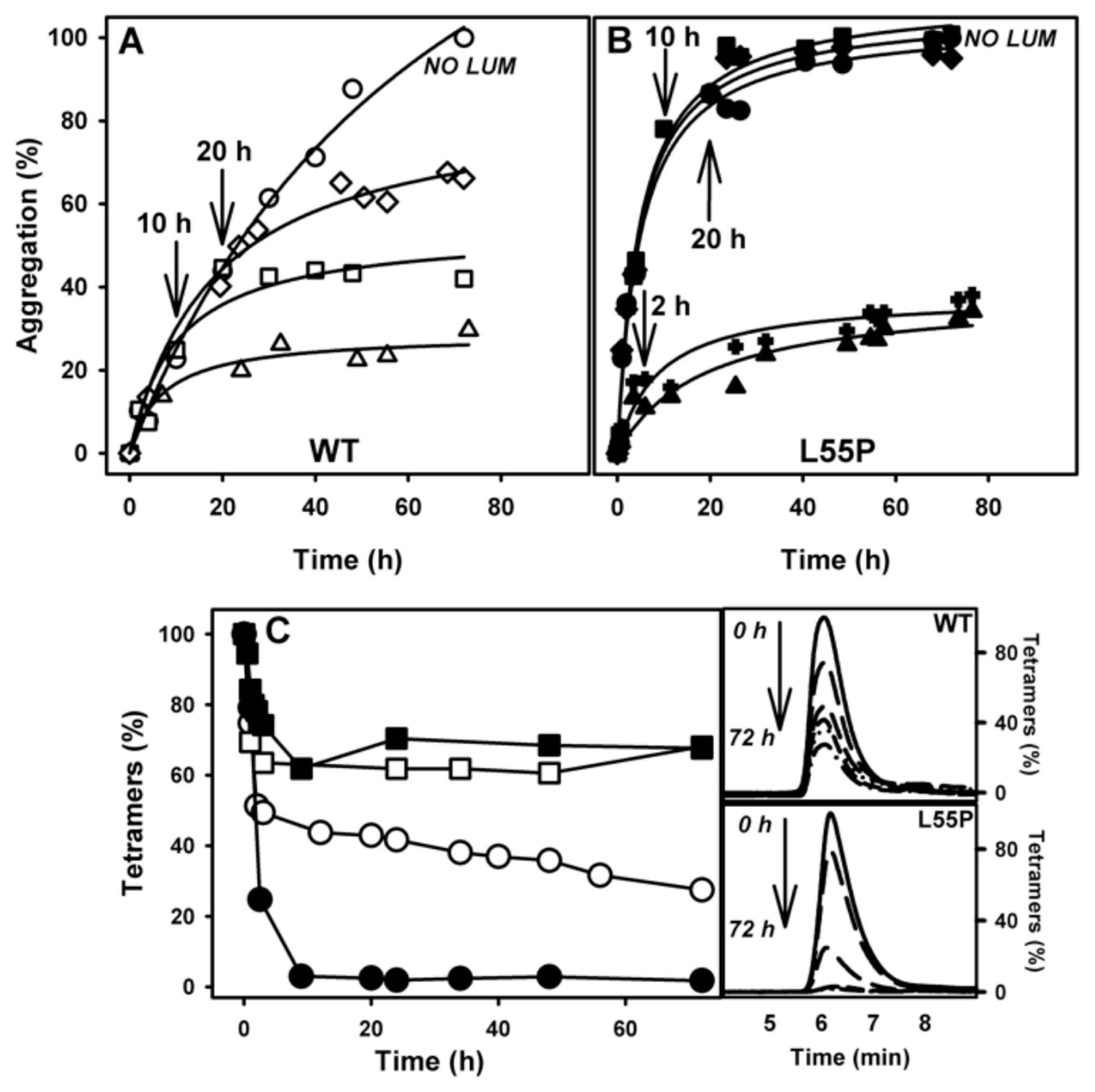
2.3. SUL, IND and LUM Inhibit Acid-Induced Aggregation of WT-TTR and L55P
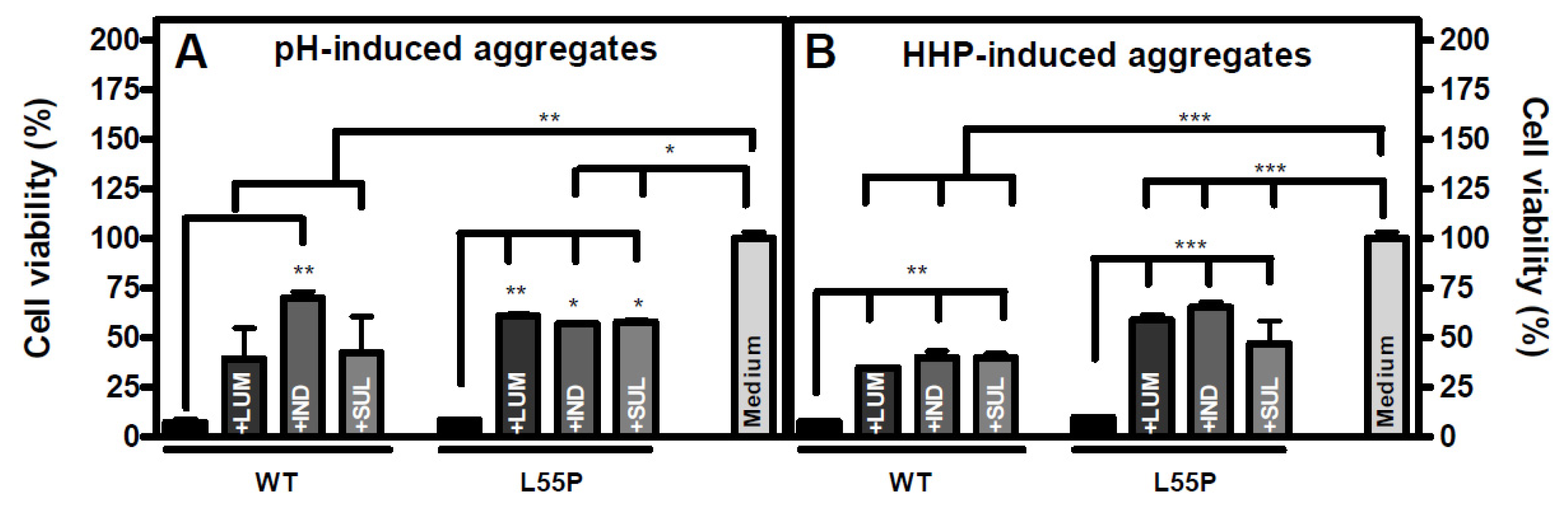
2.4. The Species Formed When Aggregation Is Performed in the Presence of LUM, IND or SUL Are Not Toxic to Cells in Culture
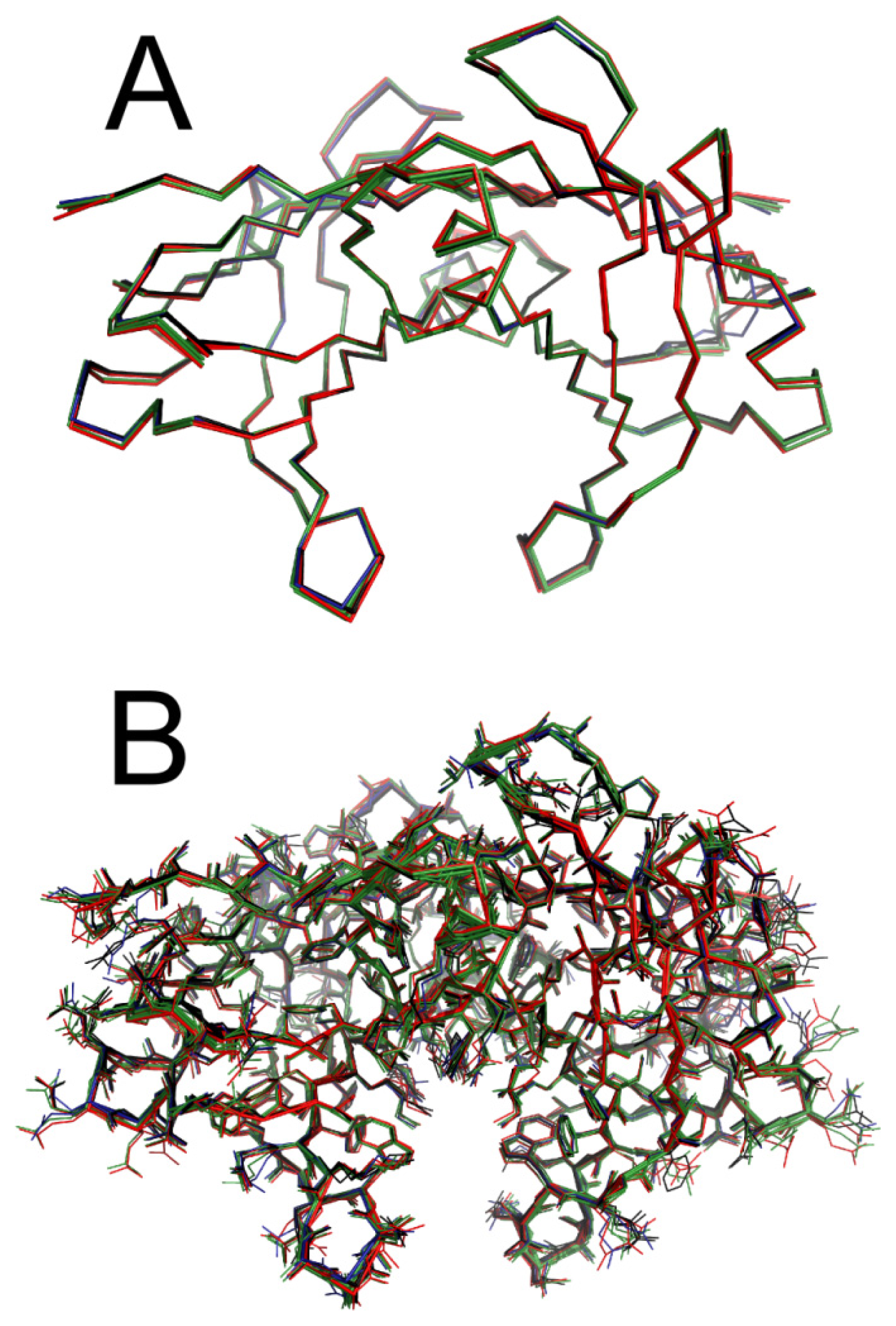
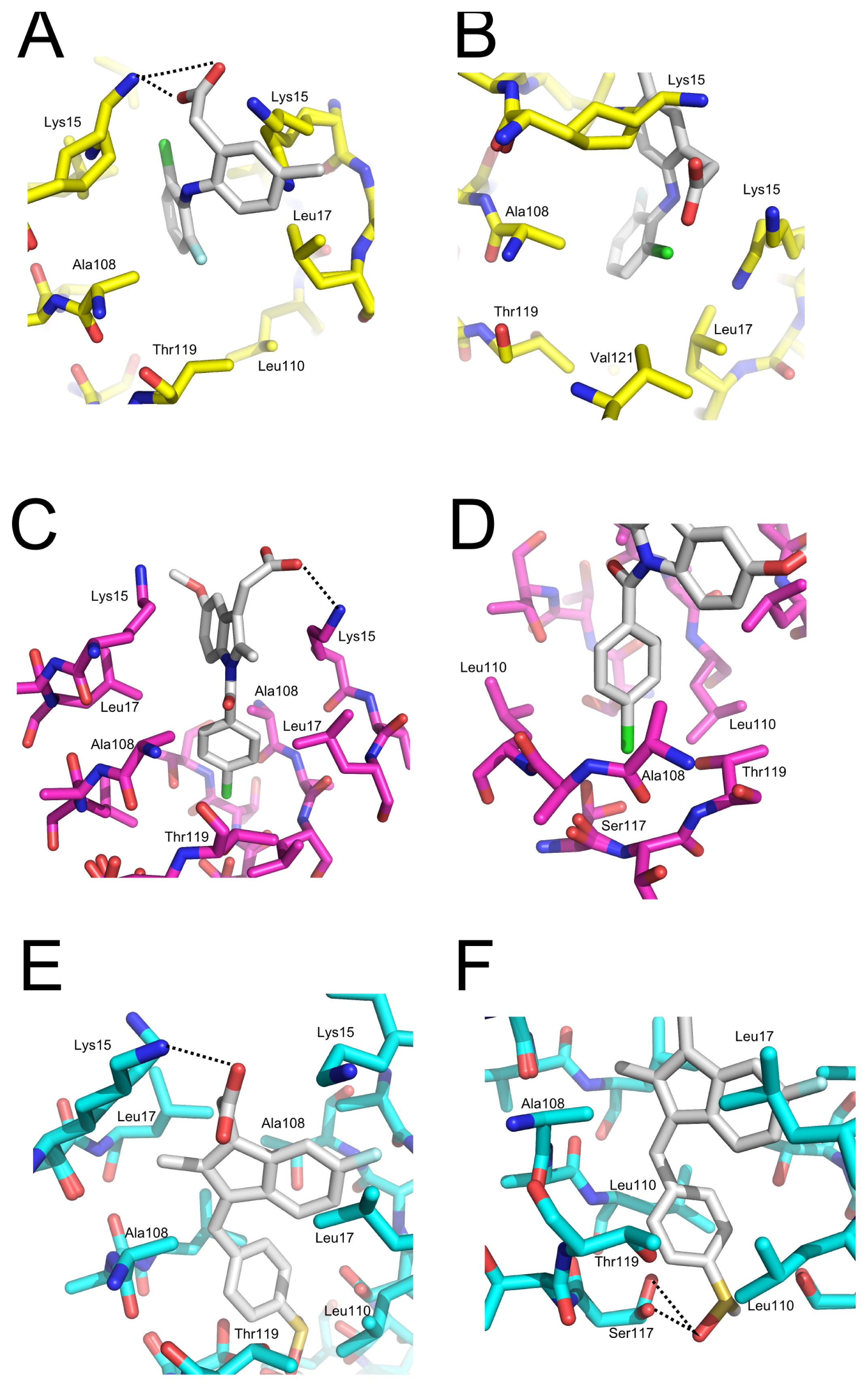
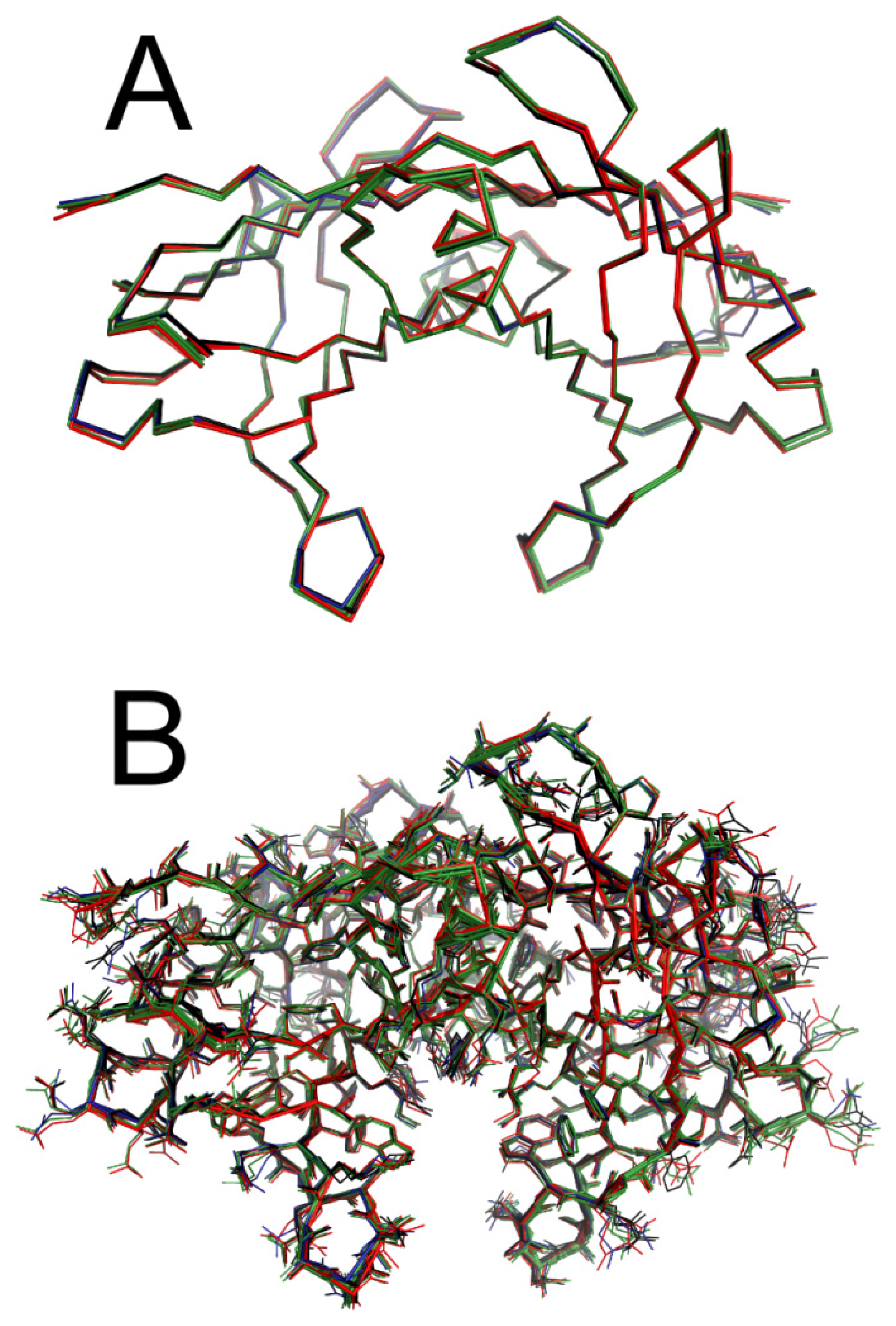
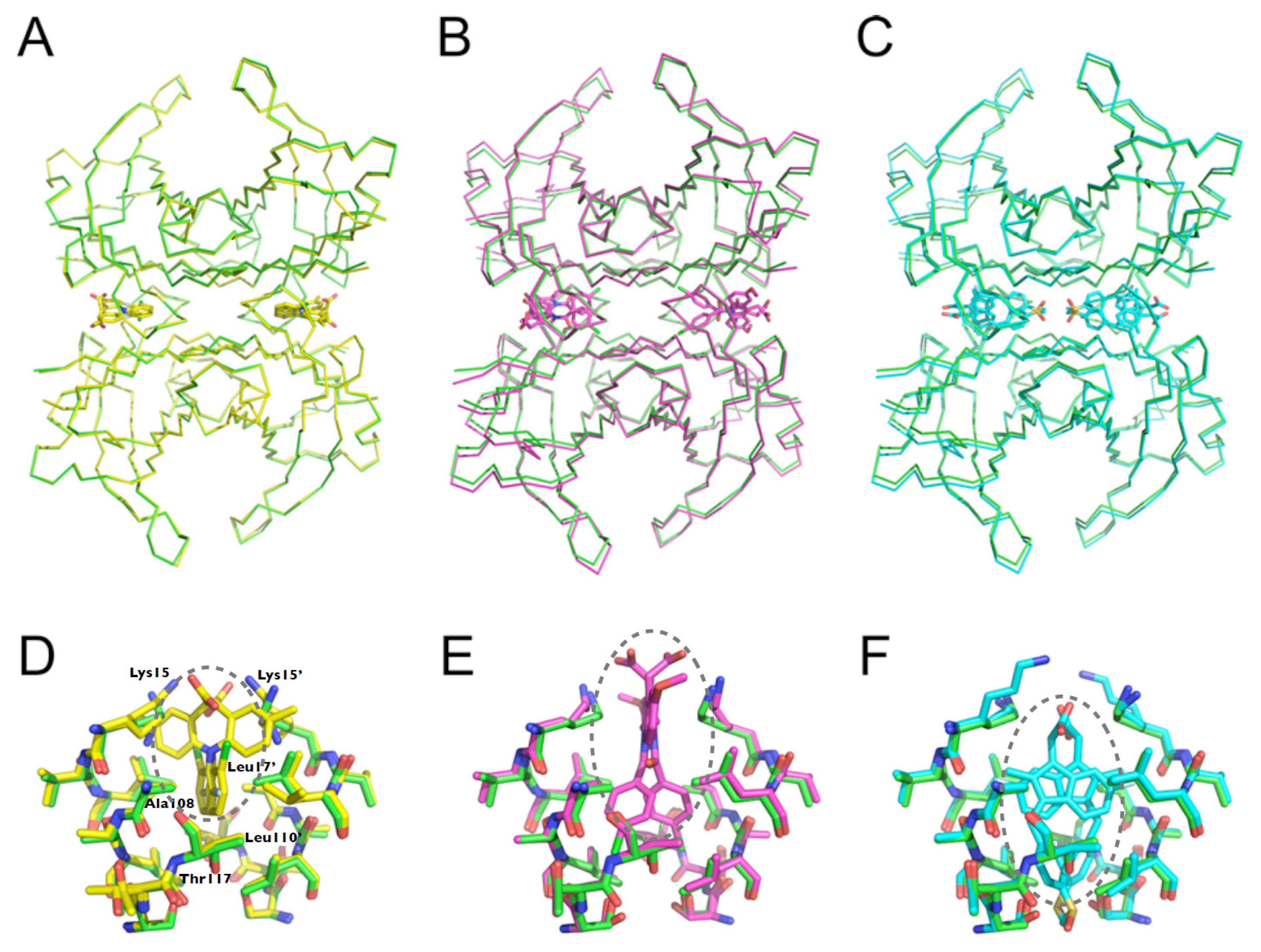
2.5. Crystal Structures of WT-TTR in Complex with LUM, SUL, and IND
3. Experimental Section
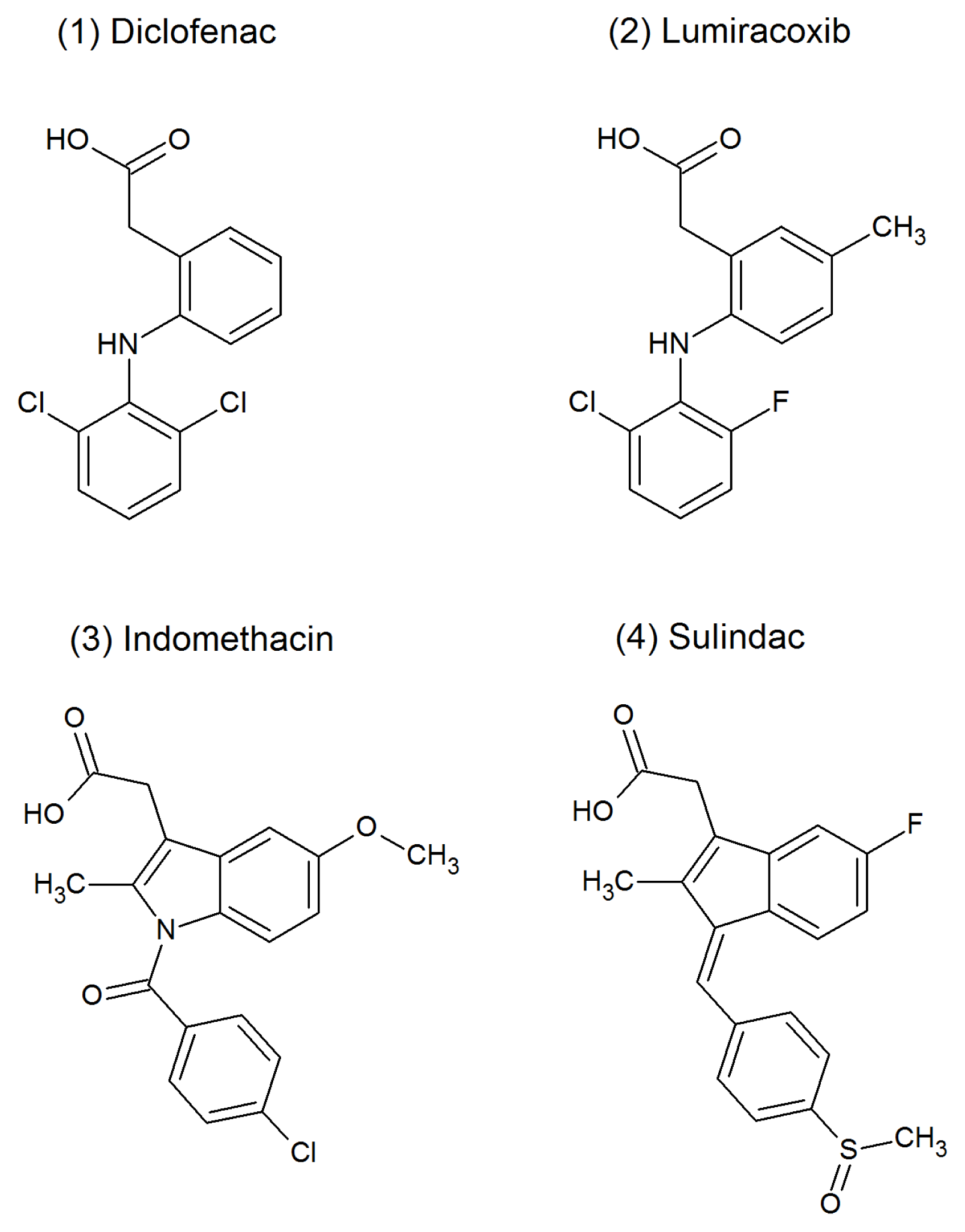
3.1. Chemicals
3.2. Protein Purification
3.3. Congo Red (CR) Binding Assay
3.4. Atomic Force Microscopy
3.5. Aggregation Measurements
- Acid-induced aggregation: 3.5 μM of WT-TTR and L55P tetramers were left to aggregate in 200 mM sodium acetate and 100 mM KCl at pH 4.4 and 37 °C in staticsolutions. At specific time points (legend of each figure) or after 72 h, turbidity at 400 nm was measured to evaluate the extent of aggregation. The compounds were added at the beginning of the aggregation reaction or at intermediate time points as stated in the legend of each figure. The efficacy of each compound was calculated as 100% of the turbidity of the sample aggregated in the absence of any addition.
- HHP-induced aggregation: WT-TTR at 3.5 μM and L55P at 1 μM were pressurized at 3 kbar for 1 h in MES 100 mM and KCl 100 mM at pH 5 and 1 °C (dissociation-denaturation phase). After this time, the pressure was released, and the temperature was quickly increased to 37 °C, at which time aggregation was triggered (aggregation-phase). Then, light scattering (LS) was measured over time to evaluate the extent of aggregation. Each LS measurement was normalized to the LS value before compression (LS/LS0). In several experiments, LUM, IND and SUL were added at the beginning of the experiment. In another protocol, LUM was added after the dissociation-denaturation phase, keeping the high-pressure cuvette on ice to avoid aggregation. Then, the temperature was increased to 37 °C to trigger aggregation, now in the presence of LUM.
3.6. Electron Microscopy
3.7. Inhibitor Binding Measured by ANS Displacement
3.8. Size Exclusion Chromatography (SEC)
3.9. Neuroblastoma Cell Culture and Viability Assay (N2a)
3.10. Stability Studies under HHP
3.11. Thermodynamic Parameters
3.12. WT-TTR Crystallization, Data Diffraction and Structure Solution
- We grew crystals from 2 μL drops containing 5 mg/mL TTR, 11% peg 400, 50 mM Tris and 50 mM KCl, equilibrated at 18 °C against 22% peg 400, 100 mM Tris pH 7.4 and 100 mM KCl, and collected data with CuKα radiation (1.5418 Å) generated by a Rigaku UltraX 18 rotating anode operated at 50 kV and 90 mA, equipped with Osmic confocal Max-Flux optics and recorded on a MAR 345dtb image plate mounted over a mar dtb (MAR Research);
- We grew crystals from 2 μL drops containing 5 mg/mL TTR, 14% v/v peg 400, 50 mM Hepes sodium pH 7.5 and 100 mM CaCl2, equilibrated at 20 °C against 28% v/v peg 400, 100 mM Hepes sodium pH 7.5 and 200 mM CaCl2 (directly from Hampton’s Crystal Screen screening kit, condition #14) and collected data with the D03-MX1 beamline from LNLS (National Synchrotron Light Laboratory, LNLS, Campinas, Brazil; [63], where the wavelength was set to 1.427 Å and the diffraction intensities were measured using a MARCCD detector mounted over a mar345dtb (MAR Research).
4. Conclusions
Supplementary Information
ijms-14-05284-s001.docxAcknowledgments
Conflict of Interest
References
- Gonzalez, G.; Offord, R.E. The subunit structure of prealbumin. Biochem. J 1971, 125, 309–317. [Google Scholar]
- Blake, C.C.F.; Geisow, M.J.; Oatley, S.J.; Rérat, B.; Rérat, C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol 1978, 121, 339–356. [Google Scholar]
- Palaninathan, S.K. Nearly 200 X-ray crystal structures of transthyretin: What do they tell us about this protein and the design of drugs for TTR amyloidoses? Curr. Med. Chem 2012, 19, 2324–2342. [Google Scholar]
- Nilsson, S.F.; Rask, L.; Peterson, P.A. Studies on thyroid hormone-binding proteins. II. Binding of thyroid hormones, retinol-binding protein, and fluorescent probes to prealbumin and effects of thyroxine on prealbumin subunit self association. J. Biol. Chem 1975, 250, 8554–8563. [Google Scholar]
- Miroy, G.J.; Lai, Z.; Lashuel, H.A.; Peterson, S.A.; Strang, C.; Kelly, J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. USA 1996, 93, 15051–15056. [Google Scholar]
- Peterson, S.A.; Klabunde, T.; Lashuel, H.A.; Purkey, H.; Sacchettini, J.C.; Kelly, J.W. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1998, 95, 12956–12960. [Google Scholar]
- Klabunde, T.; Petrassi, H.M.; Oza, V.B.; Raman, P.; Kelly, J.W.; Sacchettini, J.C. Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol 2000, 7, 312–321. [Google Scholar]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Invest 2004, 84, 545–552. [Google Scholar]
- Tojo, K.; Sekijima, Y.; Kelly, J.W.; Ikeda, S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci. Res 2006, 56, 441–449. [Google Scholar]
- Mairal, T.; Nieto, J.; Pinto, M.; Almeida, M.R.; Gales, L.; Ballesteros, A.; Barluenga, J.; Pérez, J.J.; Vázquez, J.T.; Centeno, N.B.; et al. Iodine atoms: A new molecular feature for the design of potent transthyretin fibrillogenesis inhibitors. PLoS One 2009, 4, e4124. [Google Scholar]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formation. FEBS Lett 2011, 585, 2424–2430. [Google Scholar]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Natural polyphenols as modulators of TTR amyloidogenesis: In vitro and in vivo evidences towards therapy. Amyloid 2012, 19, 39–42. [Google Scholar]
- Ferreira, N.; Santos, S.A.O.; Domingues, M.R.M.; Saraiva, M.J.; Almeida, M.R. Dietary curcumin counteracts extracellular transthyretin deposition: Insights on the mechanism of amyloid inhibition. Biochim. Biophys. Acta 2013, 1832, 39–45. [Google Scholar]
- Ramirez-Alvarado, M.; Kelly, J.W.; Dobson, C.M. Protein Misfolding Diseases: Current and Emerging Principles and Therapies; Ramirez-Alvarado, M., Kelly, J.W., Dobson, C.M., Eds.; Wiley Series in Protein and Peptide Science; Wiley: Malden, MA, USA, 2011. [Google Scholar]
- Saraiva, M.J.; Magalhaes, J.; Ferreira, N.; Almeida, M.R. Transthyretin deposition in familial amyloidotic polyneuropathy. Curr. Med. Chem 2012, 19, 2304–2311. [Google Scholar]
- Cornwell, G.G.; Sletten, K.; Johansson, B.; Westermark, P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem. Biophys. Res. Commun 1988, 154, 648–653. [Google Scholar]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar]
- Saraiva, M.J.; Costa, P.P.; Birken, S.; Goodman, D.S. Presence of an abnormal transthyretin (prealbumin) in Portuguese patients with familial amyloidotic polyneuropathy. Trans. Assoc. Am. Physicians 1983, 96, 261–270. [Google Scholar]
- Hou, X.; Aguilar, M.-I.; Small, D.H. Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J 2007, 274, 1637–1650. [Google Scholar]
- Jacobson, D.R.; McFarlin, D.E.; Kane, I.; Buxbaum, J.N. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum. Genet 1992, 89, 353–356. [Google Scholar]
- Saraiva, M.J. Transthyretin mutations in health and disease. Hum. Mutat 1995, 5, 191–196. [Google Scholar]
- Hammarström, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA 2002, 99, 16427–16432. [Google Scholar]
- Lai, Z.; Colón, W.; Kelly, J.W. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 1996, 35, 6470–6482. [Google Scholar]
- Quintas, A.; Saraiva, M.J.; Brito, R.M. The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J. Biol. Chem 1999, 274, 32943–32949. [Google Scholar]
- Jiang, X.; Smith, C.S.; Petrassi, H.M.; Hammarström, P.; White, J.T.; Sacchettini, J.C.; Kelly, J.W. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 2001, 40, 11442–11452. [Google Scholar]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.; Brito, R.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem 2001, 276, 27207–27213. [Google Scholar]
- Ferrão-Gonzales, A.D.; Palmieri, L.; Valory, M.; Silva, J.L.; Lashuel, H.; Kelly, J.W.; Foguel, D. Hydration and packing are crucial to amyloidogenesis as revealed by pressure studies on transthyretin variants that either protect or worsen amyloid disease. J. Mol. Biol 2003, 328, 963–974. [Google Scholar]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarström, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar]
- Foss, T.R.; Wiseman, R.L.; Kelly, J.W. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 2005, 44, 15525–15533. [Google Scholar]
- Palaninathan, S.K.; Mohamedmohaideen, N.N.; Snee, W.C.; Kelly, J.W.; Sacchettini, J.C. Structural insight into pH-induced conformational changes within the native human transthyretin tetramer. J. Mol. Biol 2008, 382, 1157–1167. [Google Scholar]
- Ferrão-Gonzales, A.D.; Souto, S.O.; Silva, J.L.; Foguel, D. The preaggregated state of an amyloidogenic protein: Hydrostatic pressure converts native transthyretin into the amyloidogenic state. Proc. Natl. Acad. Sci. USA 2000, 97, 6445–6450. [Google Scholar]
- Foguel, D. High pressure studies on transthyretin. Protein Pept. Lett 2005, 12, 245–249. [Google Scholar]
- Holmgren, G.; Ericzon, B.G.; Groth, C.G.; Steen, L.; Suhr, O.; Andersen, O.; Wallin, B.G.; Seymour, A.; Richardson, S.; Hawkins, P.N. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993, 341, 1113–1116. [Google Scholar]
- Herlenius, G.; Wilczek, H.E.; Larsson, M.; Ericzon, B.-G. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: Results from the familial amyloidotic polyneuropathy world transplant registry. Transplantation 2004, 77, 64–71. [Google Scholar]
- Sekijima, Y.; Hammarström, P.; Matsumura, M.; Shimizu, Y.; Iwata, M.; Tokuda, T.; Ikeda, S.-I.; Kelly, J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Invest 2003, 83, 409–417. [Google Scholar]
- Azevedo, E.P.C.; Pereira, H.M.; Garratt, R.C.; Kelly, J.W.; Foguel, D.; Palhano, F.L. Dissecting the structure, thermodynamic stability, and aggregation properties of the A25T transthyretin (A25T-TTR) variant involved in leptomeningeal amyloidosis: identifying protein partners that co-aggregate during A25T-TTR fibrillogenesis in cerebrospi. Biochemistry 2011, 50, 11070–11083. [Google Scholar]
- Hammarström, P.; Sekijima, Y.; White, J.T.; Wiseman, R.L.; Lim, A.; Costello, C.E.; Altland, K.; Garzuly, F.; Budka, H.; Kelly, J.W. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: A prescription for central nervous system amyloidosis? Biochemistry 2003, 42, 6656–6663. [Google Scholar]
- Bulawa, C.E.; Connelly, S.; Devit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar]
- Johnson, S.M.; Connelly, S.; Fearns, C.; Powers, E.T.; Kelly, J.W. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol 2012, 421, 185–203. [Google Scholar]
- Baures, P.W.; Oza, V.B.; Peterson, S.A.; Kelly, J.W. Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg. Med. Chem 1999, 7, 1339–1347. [Google Scholar]
- McCammon, M.G.; Scott, D.J.; Keetch, C.A.; Greene, L.H.; Purkey, H.E.; Petrassi, H.M.; Kelly, J.W.; Robinson, C.V. Screening transthyretin amyloid fibril inhibitors. Structure 2002, 10, 851–863. [Google Scholar]
- Oza, V.B.; Smith, C.; Raman, P.; Koepf, E.K.; Lashuel, H.A.; Petrassi, H.M.; Chiang, K.P.; Powers, E.T.; Sachettinni, J.; Kelly, J.W. Synthesis, structure, and activity of diclofenac analogues as transthyretin amyloid fibril formation inhibitors. J. Med. Chem 2002, 45, 321–332. [Google Scholar]
- Lima, L.M.T.R.; de Silva, V.A.; de Palmieri, L.C.; Oliveira, M.C.B.R.; Foguel, D.; Polikarpov, I. Identification of a novel ligand binding motif in the transthyretin channel. Bioorg. Med. Chem 2010, 18, 100–110. [Google Scholar]
- Almeida, M.R.; Macedo, B.; Cardoso, I.; Alves, I.; Valencia, G.; Arsequell, G.; Planas, A.; Saraiva, M.J. Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J 2004, 381, 351–356. [Google Scholar]
- Johnson, S.M.; Connelly, S.; Wilson, I.A.; Kelly, J.W. Toward optimization of the linker substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J. Med. Chem 2008, 51, 6348–6358. [Google Scholar]
- Paladini, A.A.; Weber, G. Pressure-induced reversible dissociation of enolase. Biochemistry 1981, 20, 2587–2593. [Google Scholar]
- Silva, J.L. Current topics new insights into the mechanisms of protein misfolding and aggregation in amyloidogenic diseases derived from pressure studies. Biochemistry 2004, 43, 11361–11370. [Google Scholar]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2006; p. 980. [Google Scholar]
- Royer, C.A. Probing protein folding and conformational transitions with fluorescence. Chem. Rev 2006, 106, 1769–1784. [Google Scholar]
- Cordeiro, Y.; Kraineva, J.; Suarez, M.C.; Tempesta, A.G.; Kelly, J.W.; Silva, J.L.; Winter, R.; Foguel, D. Fourier transform infrared spectroscopy provides a fingerprint for the tetramer and for the aggregates of transthyretin. Biophys. J 2006, 91, 957–967. [Google Scholar]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004, 43, 7365–7381. [Google Scholar]
- Taylor, B.M.; Sarver, R.W.; Fici, G.; Poorman, R.A.; Lutzke, B.S.; Molinari, A.; Kawabe, T.; Kappenman, K.; Buhl, A.E.; Epps, D.E. Spontaneous aggregation and cytotoxicity of the beta-amyloid Abeta1–40: A kinetic model. J. Protein Chem 2003, 22, 31–40. [Google Scholar]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimarães, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol 2001, 159, 1993–2000. [Google Scholar]
- Reixach, N.; Deechongkit, S.; Jiang, X.; Kelly, J.W.; Buxbaum, J.N. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. USA 2004, 101, 2817–2822. [Google Scholar]
- Andersson, K.; Olofsson, A.; Nielsen, E.H.; Svehag, S.-E.; Lundgren, E. Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem. Biophys. Res. Commun 2002, 294, 309–314. [Google Scholar]
- Glenner, G.G.; Eanes, E.D.; Page, D.L. The relation of the properties of congo red-stained amyloid fibrils to the conformation. J. Histochem. Cytochem 1972, 20, 821–826. [Google Scholar]
- Klunk, W.E.; Jacob, R.F.; Mason, R.P. Quantifying amyloid by congo red spectral shift assay. Methods Enzymol 1999, 309, 285–305. [Google Scholar]
- Malmo, C.; Vilasi, S.; Iannuzzi, C.; Tacchi, S.; Cametti, C.; Irace, G.; Sirangelo, I. Tetracycline inhibits W7FW14F apomyoglobin fibril extension and keeps the amyloid protein in a pre-fibrillar, highly cytotoxic state. FASEB J 2006, 20, 346–347. [Google Scholar]
- Vilasi, S.; Sarcina, R.; Maritato, R.; De Simone, A.; Irace, G.; Sirangelo, I. Heparin induces harmless fibril formation in amyloidogenic W7FW14F apomyoglobin and amyloid aggregation in wild-type protein in vitro. PLoS One 2011, 6, e22076. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Gupta, S.; Chhibber, M.; Sinha, S.; Surolia, A. Design of mechanism-based inhibitors of transthyretin amyloidosis: studies with biphenyl ethers and new structural templates. J. Med. Chem 2007, 50, 5589–5599. [Google Scholar]
- Sirangelo, I.; Irace, G. Inhibition of aggregate formation as therapeutic target in protein misfolding diseases: Effect of tetracycline and trehalose. Expert Opin. Ther. Targets 2010, 14, 1311–1321. [Google Scholar]
- Polikarpov, I.; Perles, L.A.; De Oliveira, R.T.; Oliva, G.; Castellano, E.E.; Garratt, R.C.; Craievich, A. Set-up and experimental parameters of the protein crystallography beamline at the Brazilian National Synchrotron Laboratory. J. Synchrotron Radiat 1998, 5, 72–76. [Google Scholar]
- Furnham, N.; Blundell, T.L.; DePristo, M.A.; Terwilliger, T.C. Is one solution good enough? Nat. Struct. Mol. Biol. 2006, 13, 184–185, discussion 185. [Google Scholar]
- Hörnberg, A.; Eneqvist, T.; Olofsson, A.; Lundgren, E.; Sauer-Eriksson, A.E. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol 2000, 302, 649–669. [Google Scholar]
- Potterton, L.; McNicholas, S.; Krissinel, E.; Gruber, J.; Cowtan, K.; Emsley, P.; Murshudov, G.N.; Cohen, S.; Perrakis, A.; Noble, M. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 2288–2294. [Google Scholar]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr 1997, 30, 1022–1025. [Google Scholar]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr 2011, 67, 355–367. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 486–501. [Google Scholar]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr 2004, 60, 1355–1363. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr 1993, 26, 283–291. [Google Scholar]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Adams, P.D.; Read, R.J.; Zwart, P.H.; Hung, L.-W. Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr. D Biol. Crystallogr 2008, 64, 515–524. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | L55P | |||||
|---|---|---|---|---|---|---|
| Apo | SUL | IND | Apo | SUL | IND | |
| ΔCM (nm) | 5.2 | 5.2 | 3.9 | 6.8 | 6.0 | 6.3 |
| p1/2 (bar) | 980 | 1240 | 2607 | 514 | 932 | 1824 |
| ΔGf (kcal/mol) | −16.01 ± 0.3 | −17.98 ± 0.25 | −21.90 ± 0.35 | −24.60 ± 0.6 | −32.40 ± 0.75 | −35.30 ± 0.8 |
| ΔΔGf (kcal/mol) | - | 1.96 ± 0.2 | 5.89 ± 0.05 | - | 7.8 ± 0.15 | 10.7 ± 0.2 |
| ΔVf (mL/mol) | 175.5 ± 15.56 | 241.0 ± 19.8 | 264.40 ± 9.89 | 275.10 ± 14.85 | 326.0 ± 12.02 | 407.0 ± 12.73 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sant'Anna, R.O.; Braga, C.A.; Polikarpov, I.; Ventura, S.; Lima, L.M.T.R.; Foguel, D. Inhibition of Human Transthyretin Aggregation by Non-Steroidal Anti-Inflammatory Compounds: A Structural and Thermodynamic Analysis. Int. J. Mol. Sci. 2013, 14, 5284-5311. https://doi.org/10.3390/ijms14035284
Sant'Anna RO, Braga CA, Polikarpov I, Ventura S, Lima LMTR, Foguel D. Inhibition of Human Transthyretin Aggregation by Non-Steroidal Anti-Inflammatory Compounds: A Structural and Thermodynamic Analysis. International Journal of Molecular Sciences. 2013; 14(3):5284-5311. https://doi.org/10.3390/ijms14035284
Chicago/Turabian StyleSant'Anna, Ricardo O., Carolina A. Braga, Igor Polikarpov, Salvador Ventura, Luis Mauricio T. R. Lima, and Debora Foguel. 2013. "Inhibition of Human Transthyretin Aggregation by Non-Steroidal Anti-Inflammatory Compounds: A Structural and Thermodynamic Analysis" International Journal of Molecular Sciences 14, no. 3: 5284-5311. https://doi.org/10.3390/ijms14035284