From Identification to Characterization of the Multiple Sclerosis Susceptibility Gene CLEC16A

Abstract
:
1. Introduction
2. Identification of MS Risk Loci
3. CLEC16A—An Autoimmune Candidate Gene
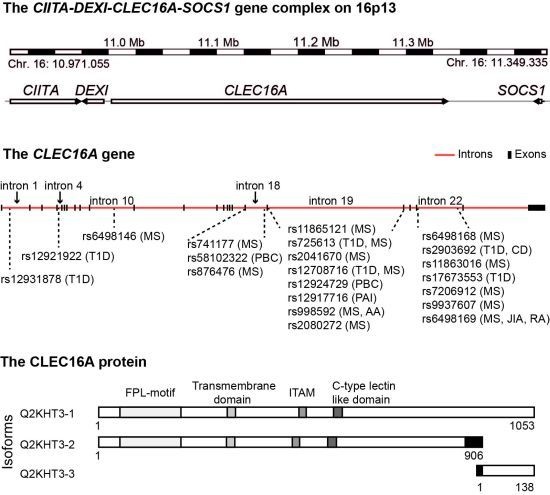
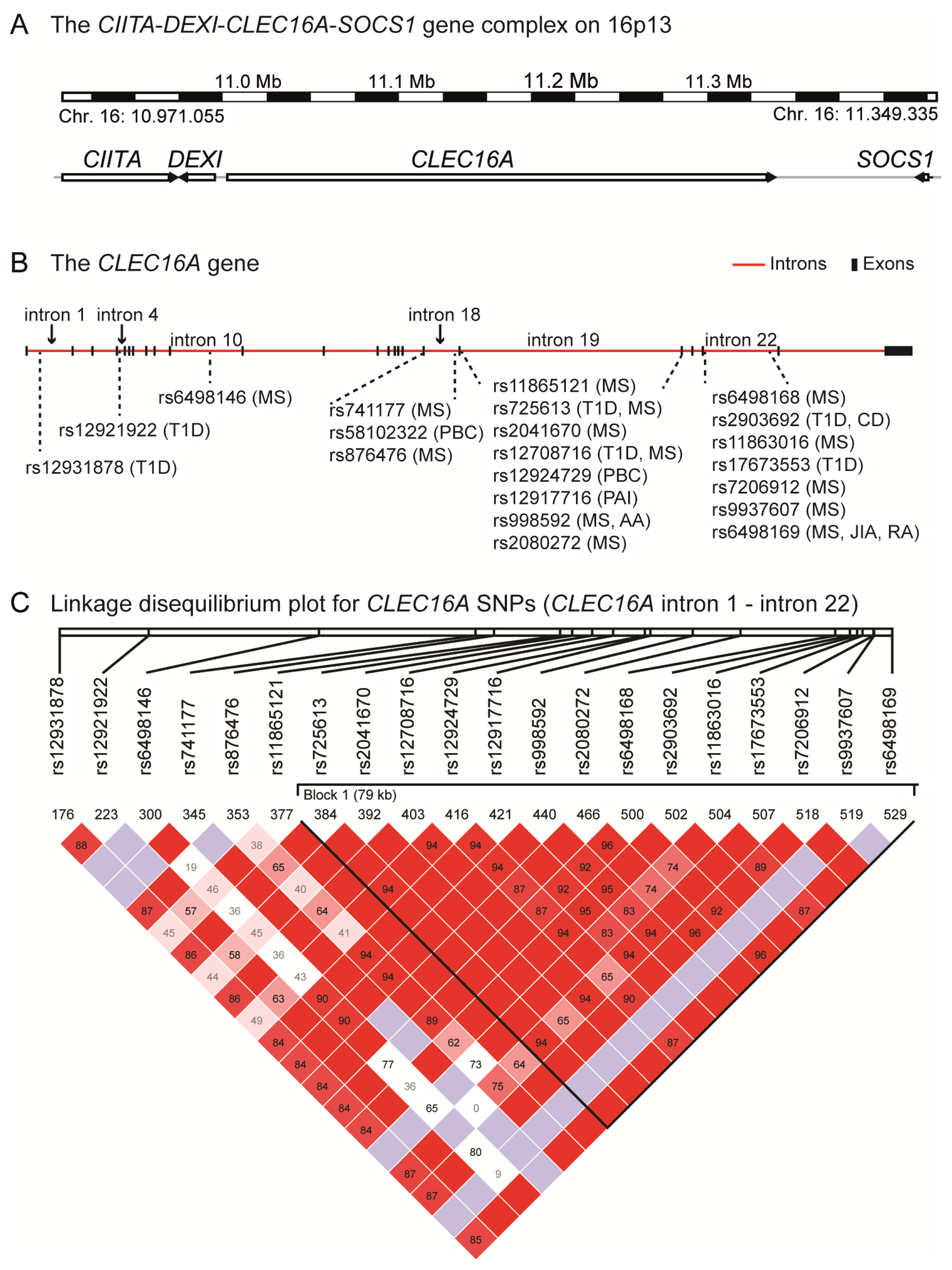
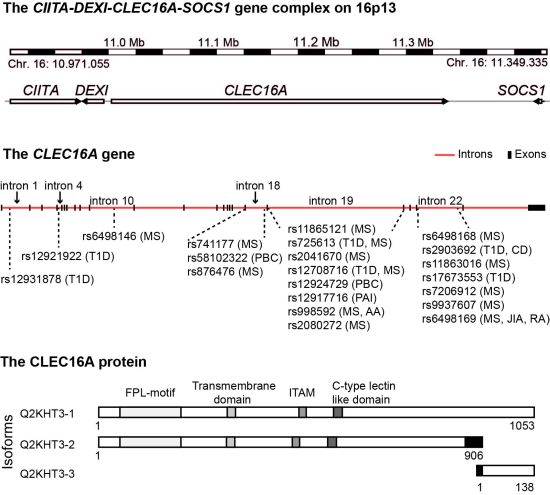
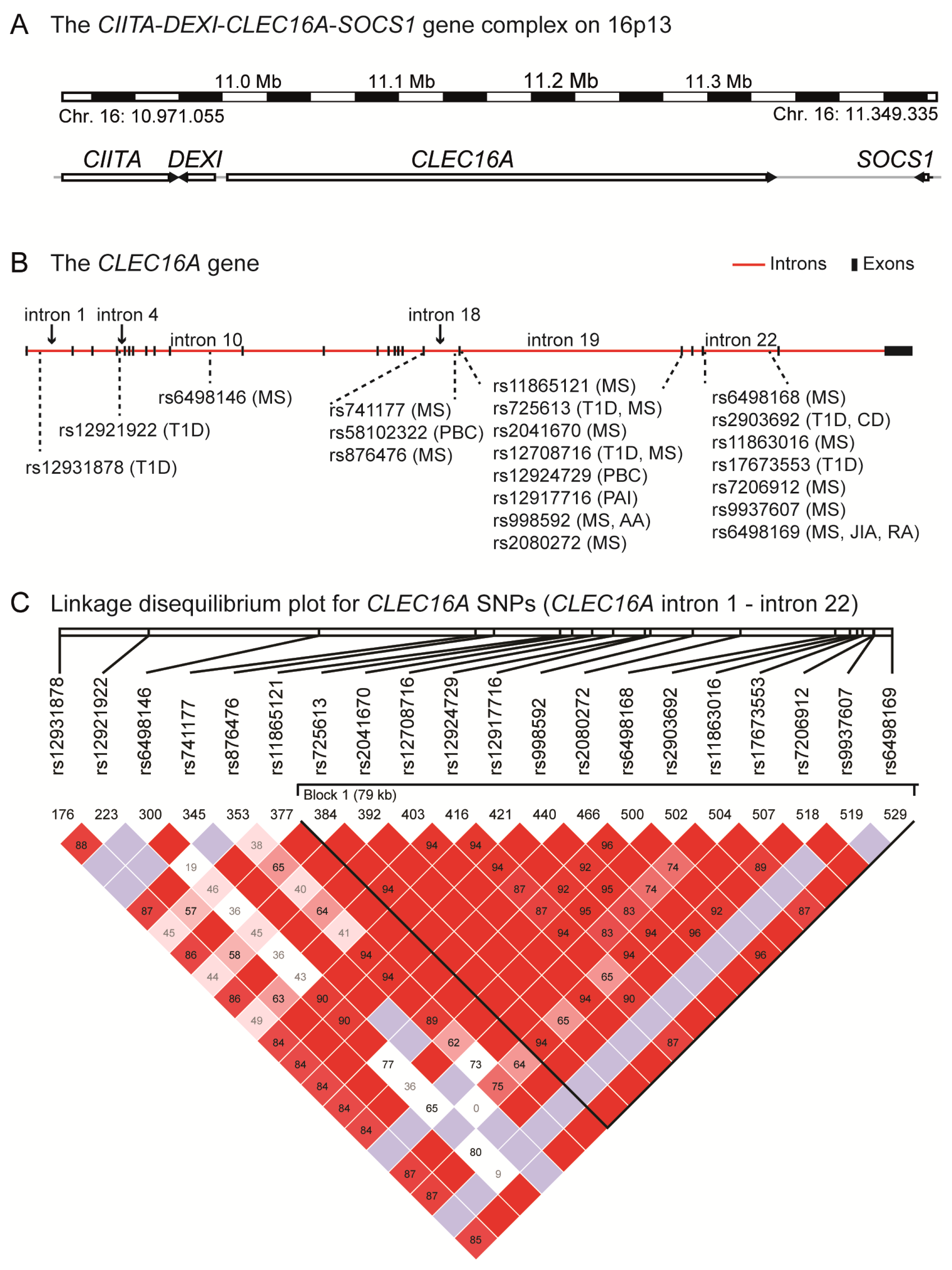
4. The CIITA-DEXI-CLEC16A-SOCS1 Gene Complex
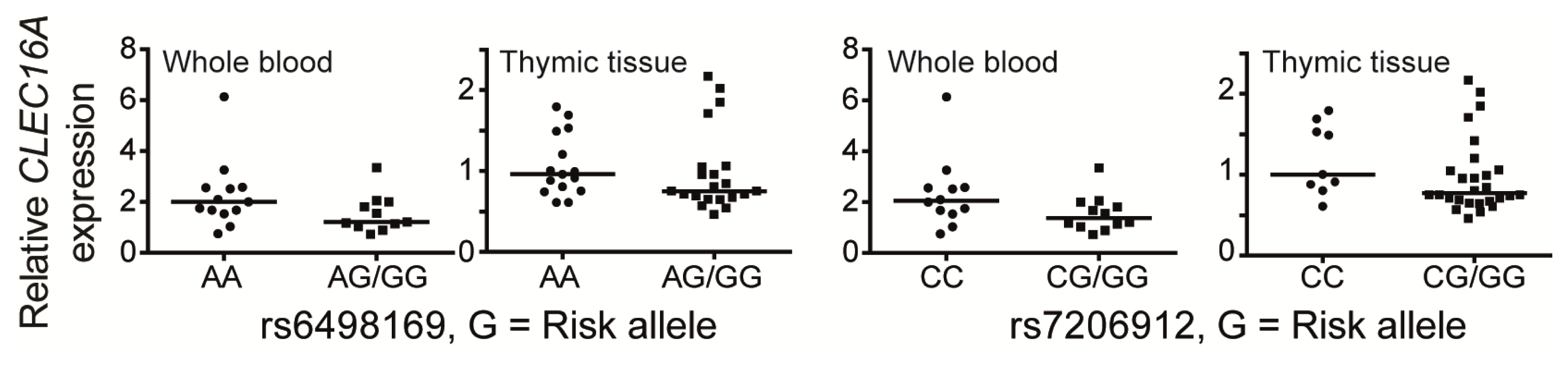
5. CLEC16A Expression
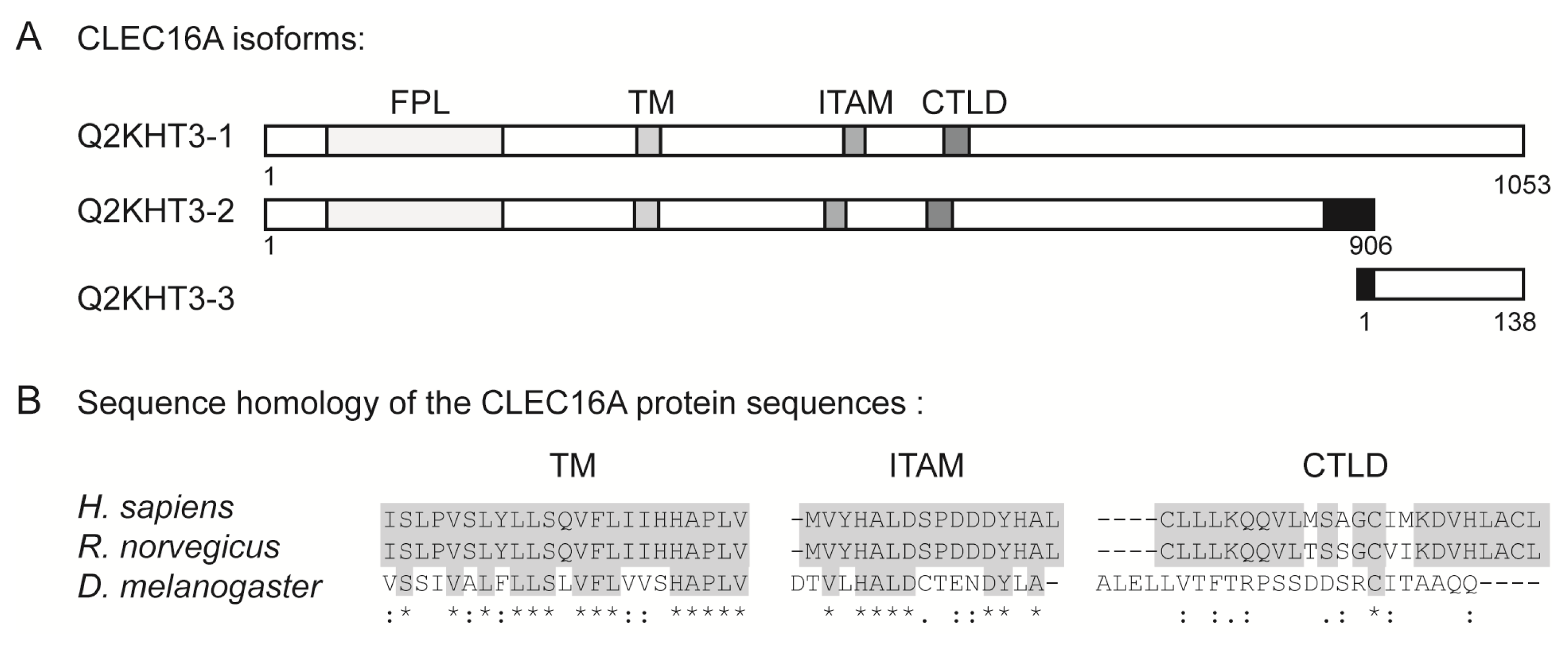
6. The Structural Domains of the CLEC16A Molecule
7. The Function of CLEC16A
8. The CLEC16 Gene Region and Disease Susceptibility
9. Conclusions
Acknowledgements
Conflict of interest
References
- Giovannoni, G.; Southam, E.; Waubant, E. Systematic review of disease-modifying therapies to assess unmet needs in multiple sclerosis: Tolerability and adherence. Mult. Scler 2012, 18, 932–946. [Google Scholar]
- Taylor, B.V.; Pearson, J.F.; Clarke, G.; Mason, D.F.; Abernethy, D.A.; Willoughby, E.; Sabel, C. MS prevalence in New Zealand, an ethnically and latitudinally diverse country. Mult. Scler 2010, 16, 1422–1431. [Google Scholar]
- Simpson, S., Jr; Blizzard, L.; Otahal, P.; van der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1132–1141. [Google Scholar]
- Alonso, A.; Hernan, M.A. Temporal trends in the incidence of multiple sclerosis: A systematic review. Neurology 2008, 71, 129–135. [Google Scholar]
- O’Gorman, C.; Lucas, R.; Taylor, B. Environmental risk factors for multiple sclerosis: A review with a focus on molecular mechanisms. Int. J. Mol. Sci 2012, 13, 11718–11752. [Google Scholar]
- Kemppinen, A.; Sawcer, S.; Compston, A. Genome-wide association studies in multiple sclerosis: Lessons and future prospects. Brief. Funct. Genomics 2011, 10, 61–70. [Google Scholar]
- Gourraud, P.A.; Harbo, H.F.; Hauser, S.L.; Baranzini, S.E. The genetics of multiple sclerosis: An up-to-date review. Immunol. Rev 2012, 248, 87–103. [Google Scholar]
- Jersild, C.; Svejgaard, A.; Fog, T. HL-A antigens and multiple sclerosis. Lancet 1972, 1, 1240–1241. [Google Scholar]
- Olerup, O.; Zetterquist, H. HLA-DRB1*01 subtyping by allele-specific PCR amplification: A sensitive, specific and rapid technique. Tissue Antigens 1991, 37, 197–204. [Google Scholar]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [Green Version]
- Brynedal, B.; Duvefelt, K.; Jonasdottir, G.; Roos, I.M.; Akesson, E.; Palmgren, J.; Hillert, J. HLA-A confers an HLA-DRB1 independent influence on the risk of multiple sclerosis. PLoS One 2007, 2, e664. [Google Scholar]
- Fogdell-Hahn, A.; Ligers, A.; Gronning, M.; Hillert, J.; Olerup, O. Multiple sclerosis: A modifying influence of HLA class I genes in an HLA class II associated autoimmune disease. Tissue Antigens 2000, 55, 140–148. [Google Scholar]
- Mero, I.-L.; Gustavsen, M.W.; Sæther, H.S.; Flåm, S.T.; Berg-Hansen, P.; Søndergaard, H.B.; Jensen, P.E.H.; Berge, T.; Bjølgerud, A.; Muggerud, A.; et al. Oligoclonal band status in Scandinavian multiple sclerosis patients is associated with specific genetic risk alleles. PLoS One 2012. [Google Scholar]
- Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; de Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar]
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet 2007, 39, 1083–1091. [Google Scholar]
- Lundmark, F.; Duvefelt, K.; Iacobaeus, E.; Kockum, I.; Wallstrom, E.; Khademi, M.; Oturai, A.; Ryder, L.P.; Saarela, J.; Harbo, H.F.; et al. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet 2007, 39, 1108–1113. [Google Scholar]
- Australia and New Zealand Multiple Sclerosis Genetics Consortium. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009, 41, 824–828.
- Jakkula, E.; Leppa, V.; Sulonen, A.M.; Varilo, T.; Kallio, S.; Kemppinen, A.; Purcell, S.; Koivisto, K.; Tienari, P.; Sumelahti, M.L.; et al. Genome-wide association study in a high-risk isolate for multiple sclerosis reveals associated variants in STAT3 gene. Am. J. Hum. Genet 2010, 86, 285–291. [Google Scholar]
- Sanna, S.; Pitzalis, M.; Zoledziewska, M.; Zara, I.; Sidore, C.; Murru, R.; Whalen, M.B.; Busonero, F.; Maschio, A.; Costa, G.; et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat. Genet 2010, 42, 495–497. [Google Scholar]
- Nischwitz, S.; Cepok, S.; Kroner, A.; Wolf, C.; Knop, M.; Muller-Sarnowski, F.; Pfister, H.; Roeske, D.; Rieckmann, P.; Hemmer, B.; et al. Evidence for VAV2 and ZNF433 as susceptibility genes for multiple sclerosis. J. Neuroimmunol 2010, 227, 162–166. [Google Scholar]
- Briggs, F.B.; Bartlett, S.E.; Goldstein, B.A.; Wang, J.; McCauley, J.L.; Zuvich, R.L.; de Jager, P.L.; Rioux, J.D.; Ivinson, A.J.; Compston, A.; et al. Evidence for CRHR1 in multiple sclerosis using supervised machine learning and meta-analysis in 12,566 individuals. Hum. Mol. Genet 2010, 19, 4286–4295. [Google Scholar]
- De Jager, P.L.; Baecher-Allan, C.; Maier, L.M.; Arthur, A.T.; Ottoboni, L.; Barcellos, L.; McCauley, J.L.; Sawcer, S.; Goris, A.; Saarela, J.; et al. The role of the CD58 locus in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 5264–5269. [Google Scholar] [Green Version]
- De Jager, P.L.; Jia, X.; Wang, J.; de Bakker, P.I.; Ottoboni, L.; Aggarwal, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet 2009, 41, 776–782. [Google Scholar]
- Baranzini, S.E.; Wang, J.; Gibson, R.A.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.W.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet 2009, 18, 767–778. [Google Scholar]
- Comabella, M.; Craig, D.W.; Camina-Tato, M.; Morcillo, C.; Lopez, C.; Navarro, A.; Rio, J.; Biomarker, M.S.S.G.; Montalban, X.; Martin, R. Identification of a novel risk locus for multiple sclerosis at 13q31.3 by a pooled genome-wide scan of 500,000 single nucleotide polymorphisms. PLoS One 2008, 3, e3490. [Google Scholar]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; et al. Wellcome Trust Case Control Consortium, Australo-Anglo-American Spondylitis Consortium. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar]
- Esposito, F.; Reischl, J.; Lehr, S.; Bauer, D.; et al. Bayer Pharma MS Genetics Working Group, Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium, GeneMSA; International Multiple Sclerosis Genetics Consortium. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar]
- Qiu, Z.X.; Zhang, K.; Qiu, X.S.; Zhou, M.; Li, W.M. CD226 Gly307Ser association with multiple autoimmune diseases: A meta-analysis. Hum. Immunol 2012, 74, 249–255. [Google Scholar]
- Hoppenbrouwers, I.A.; Aulchenko, Y.S.; Janssens, A.C.; Ramagopalan, S.V.; Broer, L.; Kayser, M.; Ebers, G.C.; Oostra, B.A.; van Duijn, C.M.; Hintzen, R.Q. Replication of CD58 and CLEC16A as genome-wide significant risk genes for multiple sclerosis. J. Hum. Genet 2009, 54, 676–680. [Google Scholar]
- Rubio, J.P.; Stankovich, J.; Field, J.; Tubridy, N.; Marriott, M.; Chapman, C.; Bahlo, M.; Perera, D.; Johnson, L.J.; Tait, B.D.; et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun 2008, 9, 624–630. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 2009, 10, 11–14.
- Mero, I.L.; Ban, M.; Lorentzen, A.R.; Smestad, C.; Celius, E.G.; Saether, H.; Saeedi, H.; Viken, M.K.; Skinningsrud, B.; Undlien, D.E.; et al. Exploring the CLEC16A gene reveals a MS-associated variant with correlation to the relative expression of CLEC16A isoforms in thymus. Genes Immun 2011, 12, 191–198. [Google Scholar]
- Zoledziewska, M.; Costa, G.; Pitzalis, M.; Cocco, E.; Melis, C.; Moi, L.; Zavattari, P.; Murru, R.; Lampis, R.; Morelli, L.; et al. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun 2009, 10, 15–17. [Google Scholar]
- Perera, D.; Stankovich, J.; Butzkueven, H.; Taylor, B.V.; Foote, S.J.; Kilpatrick, T.J.; Rubio, J.P. Fine mapping of multiple sclerosis susceptibility genes provides evidence of allelic heterogeneity at the IL2RA locus. J. Neuroimmunol 2009, 211, 105–109. [Google Scholar]
- Martinez, A.; Perdigones, N.; Cenit, M.C.; Espino, L.; Varade, J.; Lamas, J.R.; Santiago, J.L.; Fernandez-Arquero, M.; de la Calle, H.; Arroyo, R.; et al. Chromosomal region 16p13: Further evidence of increased predisposition to immune diseases. Ann. Rheum. Dis 2010, 69, 309–311. [Google Scholar]
- D’Netto, M.J.; Ward, H.; Morrison, K.M.; Ramagopalan, S.V.; Dyment, D.A.; DeLuca, G.C.; Handunnetthi, L.; Sadovnick, A.D.; Ebers, G.C. Risk alleles for multiple sclerosis in multiplex families. Neurology 2009, 72, 1984–1988. [Google Scholar]
- Johnson, B.A.; Wang, J.; Taylor, E.M.; Caillier, S.J.; Herbert, J.; Khan, O.A.; Cross, A.H.; de Jager, P.L.; Gourraud, P.A.; Cree, B.C.; et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun 2010, 11, 343–350. [Google Scholar]
- BioGPS Gene Annotation Portal. Available online: http://biogps.org accessed on 15 December 2012.
- National Center for Biotechnology Information, reference sequence NM_015226.2 (Homo sapiens C-type lectin domain family 16, member A (CLEC16A)). Available online: http://www.ncbi.nlm.nih.gov/nuccore/NM_015226.2 accessed on 15 December 2012.
- Hakonarson, H.; Grant, S.F.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 2007, 448, 591–594. [Google Scholar]
- Skinningsrud, B.; Husebye, E.S.; Pearce, S.H.; McDonald, D.O.; Brandal, K.; Wolff, A.B.; Lovas, K.; Egeland, T.; Undlien, D.E. Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J. Clin. Endocrinol. Metab 2008, 93, 3310–3317. [Google Scholar]
- Marquez, A.; Varade, J.; Robledo, G.; Martinez, A.; Mendoza, J.L.; Taxonera, C.; Fernandez-Arquero, M.; Diaz-Rubio, M.; Gomez-Garcia, M.; Lopez-Nevot, M.A.; et al. Specific association of a CLEC16A/KIAA0350 polymorphism with NOD2/CARD15(−) Crohn’s disease patients. Eur. J. Hum. Genet 2009, 17, 1304–1308. [Google Scholar]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, R.P.; et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun 2012, 13, 328–335. [Google Scholar]
- Skinningsrud, B.; Lie, B.A.; Husebye, E.S.; Kvien, T.K.; Forre, O.; Flato, B.; Stormyr, A.; Joner, G.; Njolstad, P.R.; Egeland, T.; et al. A CLEC16A variant confers risk for juvenile idiopathic arthritis and anti-cyclic citrullinated peptide antibody negative rheumatoid arthritis. Ann. Rheum. Dis 2010, 69, 1471–1474. [Google Scholar]
- Jagielska, D.; Redler, S.; Brockschmidt, F.F.; Herold, C.; Pasternack, S.M.; Garcia Bartels, N.; Hanneken, S.; Eigelshoven, S.; Refke, M.; Barth, S.; et al. Follow-up study of the first genome-wide association scan in alopecia areata: IL13 and KIAA0350 as susceptibility loci supported with genome-wide significance. J. Invest. Dermatol 2012, 132, 2192–2197. [Google Scholar]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar]
- Nischwitz, S.; Cepok, S.; Kroner, A.; Wolf, C.; Knop, M.; Muller-Sarnowski, F.; Pfister, H.; Rieckmann, P.; Hemmer, B.; Ising, M.; et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol. Scand 2011, 123, 400–406. [Google Scholar]
- Pandit, L.; Ban, M.; Sawcer, S.; Singhal, B.; Nair, S.; Radhakrishnan, K.; Shetty, R.; Misri, Z.; Hegde, S.; Bhat, I.G. Evaluation of the established non-MHC multiple sclerosis loci in an Indian population. Mult. Scler 2011, 17, 139–143. [Google Scholar]
- Zuvich, R.L.; Bush, W.S.; McCauley, J.L.; Beecham, A.H.; De Jager, P.L.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; Hauser, S.L.; Sawcer, S.J.; et al. Interrogating the complex role of chromosome 16p13.13 in multiple sclerosis susceptibility: Independent genetic signals in the CIITA-CLEC16A-SOCS1 gene complex. Hum. Mol. Genet 2011, 20, 3517–3524. [Google Scholar]
- Todd, J.A.; Walker, N.M.; Cooper, J.D.; Smyth, D.J.; Downes, K.; Plagnol, V.; Bailey, R.; Nejentsev, S.; Field, S.F.; Payne, F.; et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet 2007, 39, 857–864. [Google Scholar]
- Awata, T.; Kawasaki, E.; Tanaka, S.; Ikegami, H.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Kobayashi, T.; Iizuka, H.; Uga, M.; et al. Association of type 1 diabetes with two Loci on 12q13 and 16p13 and the influence coexisting thyroid autoimmunity in Japanese. J. Clin. Endocrinol. Metab 2009, 94, 231–235. [Google Scholar]
- Wu, X.; Zhu, X.; Wang, X.; Ma, J.; Zhu, S.; Li, J.; Liu, Y. Intron polymorphism in the KIAA0350 gene is reproducibly associated with susceptibility to type 1 diabetes (T1D) in the Han Chinese population. Clin. Endocrinol 2009, 71, 46–49. [Google Scholar]
- Sang, Y.; Zong, W.; Yan, J.; Liu, M. The correlation between the CLEC16A gene and genetic susceptibility to type 1 diabetes in Chinese children. Int. J. Endocrinol 2012, 2012, 245384. [Google Scholar]
- Howson, J.M.; Rosinger, S.; Smyth, D.J.; Boehm, B.O.; Group, A.-E.S.; Todd, J.A. Genetic analysis of adult-onset autoimmune diabetes. Diabetes 2011, 60, 2645–2653. [Google Scholar]
- Yamashita, H.; Awata, T.; Kawasaki, E.; Ikegami, H.; Tanaka, S.; Maruyama, T.; Shimada, A.; Nakanishi, K.; Takahashi, K.; Kobayashi, T.; et al. Analysis of the HLA and non-HLA susceptibility loci in Japanese type 1 diabetes. Diabetes/Metab. Res. Rev 2011, 27, 844–848. [Google Scholar]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet 2011, 43, 329–332. [Google Scholar]
- Chang, C.H.; Flavell, R.A. Class II transactivator regulates the expression of multiple genes involved in antigen presentation. J. Exp. Med 1995, 181, 765–767. [Google Scholar]
- Chang, C.H.; Guerder, S.; Hong, S.C.; van Ewijk, W.; Flavell, R.A. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity 1996, 4, 167–178. [Google Scholar]
- Rasmussen, H.B.; Kelly, M.A.; Clausen, J. Genetic susceptibility to multiple sclerosis: Detection of polymorphic nucleotides and an intron in the 3′ untranslated region of the major histocompatibility complex class II transactivator gene. Hum. Immunol 2001, 62, 371–377. [Google Scholar]
- Swanberg, M.; Lidman, O.; Padyukov, L.; Eriksson, P.; Akesson, E.; Jagodic, M.; Lobell, A.; Khademi, M.; Borjesson, O.; Lindgren, C.M.; et al. MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat. Genet 2005, 37, 486–494. [Google Scholar]
- Bronson, P.G.; Caillier, S.; Ramsay, P.P.; McCauley, J.L.; Zuvich, R.L.; De Jager, P.L.; Rioux, J.D.; Ivinson, A.J.; Compston, A.; Hafler, D.A.; et al. CIITA variation in the presence of HLA-DRB1*1501 increases risk for multiple sclerosis. Hum. Mol. Genet 2010, 19, 2331–2340. [Google Scholar]
- Gyllenberg, A.; Asad, S.; Piehl, F.; Swanberg, M.; Padyukov, L.; Van Yserloo, B.; Rutledge, E.A.; McNeney, B.; Graham, J.; Orho-Melander, M.; et al. Age-dependent variation of genotypes in MHC II transactivator gene (CIITA) in controls and association to type 1 diabetes. Genes Immun 2012, 13, 632–640. [Google Scholar]
- Eike, M.C.; Skinningsrud, B.; Ronninger, M.; Stormyr, A.; Kvien, T.K.; Joner, G.; Njolstad, P.R.; Forre, O.; Flato, B.; Alfredsson, L.; et al. CIITA gene variants are associated with rheumatoid arthritis in Scandinavian populations. Genes Immun 2012, 13, 431–436. [Google Scholar]
- Bronson, P.G.; Goldstein, B.A.; Ramsay, P.P.; Beckman, K.B.; Noble, J.A.; Lane, J.A.; Seldin, M.F.; Kelly, J.A.; Harley, J.B.; Moser, K.L.; et al. The rs4774 CIITA missense variant is associated with risk of systemic lupus erythematosus. Genes Immun 2011, 12, 667–671. [Google Scholar]
- Ghaderi, M.; Gambelunghe, G.; Tortoioli, C.; Brozzetti, A.; Jatta, K.; Gharizadeh, B.; de Bellis, A.; Pecori Giraldi, F.; Terzolo, M.; Betterle, C.; et al. MHC2TA single nucleotide polymorphism and genetic risk for autoimmune adrenal insufficiency. J. Clin. Endocrinol. Metab 2006, 91, 4107–4111. [Google Scholar]
- Vandenbroeck, K.; Alvarez, J.; Swaminathan, B.; Alloza, I.; Matesanz, F.; Urcelay, E.; Comabella, M.; Alcina, A.; Fedetz, M.; Ortiz, M.A.; et al. A cytokine gene screen uncovers SOCS1 as genetic risk factor for multiple sclerosis. Genes Immun 2012, 13, 21–28. [Google Scholar]
- Fenner, J.E.; Starr, R.; Cornish, A.L.; Zhang, J.G.; Metcalf, D.; Schreiber, R.D.; Sheehan, K.; Hilton, D.J.; Alexander, W.S.; Hertzog, P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol 2006, 7, 33–39. [Google Scholar]
- Aubin, C.; Cross, A.H.; Piccio, L.; Aggarwal, N.T.; et al. International Multiple Sclerosis Genetics, C. Genetic variation in the IL7RA/IL7 pathway increases multiple sclerosis susceptibility. Hum. Genet 2010, 127, 525–535. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Comprehensive follow-up of the first genome-wide association study of multiple sclerosis identifies KIF21B and TMEM39A as susceptibility loci. Hum. Mol. Genet. 2010, 19, 953–962.
- Leikfoss, I.S.; Mero, I.L.; Dahle, M.K.; Lie, B.A.; Harbo, H.F.; Spurkland, A.; Berge, T. Multiple sclerosis-associated single-nucleotide polymorphisms in CLEC16A correlate with reduced SOCS1 and DEXI expression in the thymus. Genes Immun 2013, 14, 62–66. [Google Scholar]
- Davison, L.J.; Wallace, C.; Cooper, J.D.; Cope, N.F.; Wilson, N.K.; Smyth, D.J.; Howson, J.M.; Saleh, N.; Al-Jeffery, A.; Angus, K.L.; et al. Long-range DNA looping and gene expression analyses identify DEXI as an autoimmune disease candidate gene. Hum. Mol. Genet 2012, 21, 322–333. [Google Scholar]
- Nica, A.C.; Montgomery, S.B.; Dimas, A.S.; Stranger, B.E.; Beazley, C.; Barroso, I.; Dermitzakis, E.T. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet 2010, 6, e1000895. [Google Scholar]
- Edgar, A.J.; Birks, E.J.; Yacoub, M.H.; Polak, J.M. Cloning of dexamethasone-induced transcript: A novel glucocorticoid-induced gene that is upregulated in emphysema. Am. J. Respir. Cell Mol. Biol 2001, 25, 119–124. [Google Scholar]
- Cortes, A.; Brown, M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther 2011, 13, 101. [Google Scholar]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet 2012, 44, 1336–1340. [Google Scholar]
- Cooper, J.D.; Simmonds, M.J.; Walker, N.M.; Burren, O.; Brand, O.J.; Guo, H.; Wallace, C.; Stevens, H.; Coleman, G. Wellcome Trust Case Control Consortium, et al. Seven newly identified loci for autoimmune thyroid disease. Hum. Mol. Genet. 2012, 21, 5202–5208. [Google Scholar]
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, Inflammation, and autoimmunity. Front. Immunol 2012, 3, 20. [Google Scholar]
- Friese, M.A.; Jones, E.Y.; Fugger, L. MHC II molecules in inflammatory diseases: Interplay of qualities and quantities. Trends Immunol 2005, 26, 559–561. [Google Scholar]
- UCSC Genome Bioinformatics. Available online: http://genome.ucsc.edu accessed on 15 December 2012.
- Nagase, T.; Ishikawa, K.; Nakajima, D.; Ohira, M.; Seki, N.; Miyajima, N.; Tanaka, A.; Kotani, H.; Nomura, N.; Ohara, O. Prediction of the coding sequences of unidentified human genes. VII. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res 1997, 4, 141–150. [Google Scholar]
- Wu, X.; Li, J.; Chen, C.; Yan, Y.; Jiang, S.; Wu, X.; Shao, B.; Xu, J.; Kang, L.; Huang, Y.; et al. Involvement of CLEC16A in activation of astrocytes after LPS treated. Neurochem. Res 2012, 37, 5–14. [Google Scholar]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet 2007, 8, 749–761. [Google Scholar]
- Bates, E.E.; Fournier, N.; Garcia, E.; Valladeau, J.; Durand, I.; Pin, J.J.; Zurawski, S.M.; Patel, S.; Abrams, J.S.; Lebecque, S.; et al. APCs express DCIR, a novel C-type lectin surface receptor containing an immunoreceptor tyrosine-based inhibitory motif. J. Immunol 1999, 163, 1973–1983. [Google Scholar]
- Fujikado, N.; Saijo, S.; Yonezawa, T.; Shimamori, K.; Ishii, A.; Sugai, S.; Kotaki, H.; Sudo, K.; Nose, M.; Iwakura, Y. Dcir deficiency causes development of autoimmune diseases in mice due to excess expansion of dendritic cells. Nat. Med 2008, 14, 176–180. [Google Scholar]
- McGreal, E.P.; Martinez-Pomares, L.; Gordon, S. Divergent roles for C-type lectins expressed by cells of the innate immune system. Mol. Immunol 2004, 41, 1109–1121. [Google Scholar]
- Prosite—Database of Protein Domains, Families and Functional Sites. Available online: http://prosite.expasy.org accessed on 15 December 2012.
- Zelensky, A.N.; Gready, J.E. The C-type lectin-like domain superfamily. FEBS J 2005, 272, 6179–6217. [Google Scholar]
- Cummings, R.D.; McEver, R.P. C-Type Lectins. In Essentials of Glycobiology, 2nd ed; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor: New York, NY, USA, 2009. [Google Scholar]
- Kingeter, L.M.; Lin, X. C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell. Mol. Immunol 2012, 9, 105–112. [Google Scholar]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites—Update 2011. Nucleic Acids Res 2011, 39, D261–D267. [Google Scholar]
- The Eukaryotic Linear Motif Resource for Functional Sites in Proteins. Available online: http://elm.eu.org accessed on 15 December 2012.
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res 2009, 37, D205–D210. [Google Scholar]
- Swiss EMBnet Node Server. Available online http://www.ch.embnet.org accessed on 15 December 2012.
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov accessed on 15 December 2012.
- UniProt—A resource of Protein Sequence and Functional Information. Available online: www.uniprot.org accessed on 15 December 2012.
- Kim, S.; Wairkar, Y.P.; Daniels, R.W.; DiAntonio, A. The novel endosomal membrane protein Ema interacts with the class C Vps-HOPS complex to promote endosomal maturation. J. Cell Biol 2010, 188, 717–734. [Google Scholar]
- Kim, S.; Diantonio, A. A role for the membrane Golgi protein Ema in autophagy. Autophagy 2012, 8, 1269–1270. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. In Curr. Prot. Bioinform.; 2002; pp. 2.3.1–2.3.22. [Google Scholar]
- Seto, E.S.; Bellen, H.J.; Lloyd, T.E. When cell biology meets development: Endocytic regulation of signaling pathways. Genes Dev 2002, 16, 1314–1336. [Google Scholar]
- German, C.L.; Sauer, B.M.; Howe, C.L. The STAT3 beacon: IL-6 recurrently activates STAT 3 from endosomal structures. Exp. Cell Res 2011, 317, 1955–1969. [Google Scholar]
- Payelle-Brogard, B.; Pellegrini, S. Biochemical monitoring of the early endocytic traffic of the type I interferon receptor. J. Interferon Cytokine Res 2010, 30, 89–98. [Google Scholar]
- Westlake, C.J.; Junutula, J.R.; Simon, G.C.; Pilli, M.; Prekeris, R.; Scheller, R.H.; Jackson, P.K.; Eldridge, A.G. Identification of Rab11 as a small GTPase binding protein for the Evi5 oncogene. Proc. Natl. Acad. Sci. USA 2007, 104, 1236–1241. [Google Scholar]
- Hehnly, H.; Chen, C.T.; Powers, C.M.; Liu, H.L.; Doxsey, S. The centrosome regulates the rab11-dependent recycling endosome pathway at appendages of the mother centriole. Curr. Biol 2012, 22, 1944–1950. [Google Scholar]
- Pierdominici, M.; Vomero, M.; Barbati, C.; Colasanti, T.; Maselli, A.; Vacirca, D.; Giovannetti, A.; Malorni, W.; Ortona, E. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J 2012, 26, 1400–1412. [Google Scholar]
- Zhou, X.J.; Lu, X.L.; Lv, J.C.; Yang, H.Z.; Qin, L.X.; Zhao, M.H.; Su, Y.; Li, Z.G.; Zhang, H. Genetic association of PRDM1-ATG5 intergenic region and autophagy with systemic lupus erythematosus in a Chinese population. Ann. Rheumatic Dis 2011, 70, 1330–1337. [Google Scholar]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis 2011, 17, 1392–1397. [Google Scholar]
- Shin, D.M.; Yuk, J.M.; Lee, H.M.; Lee, S.H.; Son, J.W.; Harding, C.V.; Kim, J.M.; Modlin, R.L.; Jo, E.K. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell. Microbiol 2010, 12, 1648–1665. [Google Scholar]
- Portillo, J.A.; Okenka, G.; Reed, E.; Subauste, A.; van Grol, J.; Gentil, K.; Komatsu, M.; Tanaka, K.; Landreth, G.; Levine, B.; et al. The CD40-autophagy pathway is needed for host protection despite IFN-Gamma-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS One 2010, 5, e14472. [Google Scholar]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci 2008, 65, 2702–2720. [Google Scholar]
- Cookson, W.; Liang, L.; Abecasis, G.; Moffatt, M.; Lathrop, M. Mapping complex disease traits with global gene expression. Nat. Rev. Genet 2009, 10, 184–194. [Google Scholar]
- Nicolae, D.L.; Gamazon, E.; Zhang, W.; Duan, S.; Dolan, M.E.; Cox, N.J. Trait-associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genet 2010, 6, e1000888. [Google Scholar]
- Consortium, E.P.; Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res 2012, 22, 1748–1759. [Google Scholar]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | SNP | Intron | Cases/Controls/Trios | Subject cohort | Reference |
|---|---|---|---|---|---|
| Multiple Sclerosis | rs6498168 | 22 | 2322/5418/1540 | UK, USA | [14] |
| rs6498169 | 22 | 1146/1309 | Australia | [30] | |
| rs725613 | 19 | 1498/1706 | Sardinia | [33] | |
| rs12708716 | 19 | 5737/10296/2369 | Aus., Bel., Nor., Swe., UK, USA | [31] | |
| rs6498169 | 19 | ||||
| rs6498146 | 10 | ||||
| rs741177 | 18 | 1146/1309 | Europe | [34] | |
| rs876476 | 18 | ||||
| rs11863016 | 22 | ||||
| rs9937607 | 22 | ||||
| rs11865121 | 19 | 2624/7220 | UK, USA | [23] | |
| rs6498169 | 22 | 435/550 | Spain | [35] | |
| rs6498169 | 22 | 211/182 (+521 multiplex controls) | UK | [36] | |
| rs6498169 | 22 | 1853/2128 | Holland, Can. | [29] | |
| rs12708716 | 19 | 918/656 | USA (afro American) | [37] | |
| rs6498169 | 22 | ||||
| rs2080272 | 19 | ||||
| rs2041670 | 19 | 603/825 | Europe | [47] | |
| rs998592 | 19 | ||||
| rs12708716 | 19 | 197/197 | India | [48] | |
| rs12708716 | 19 | ||||
| rs7206912 | 22 | 3102/5047/1113 | Nor., UK | [32] | |
| rs6498169 | 22 | ||||
| rs7184083 | 22 | 1343/1379 | UK, US | [49] | |
| Type 1 Diabetes | rs12708716 | 19 | 2000/3000 | UK | [50] |
| rs725613 | 19 | ||||
| rs2903692 | 22 | 1896/1146/873 | Eur., USA, Aus., Can. | [40] | |
| rs17673553 | 22 | ||||
| rs12708716 | 19 | 4000/5000/2997 | UK, USA, Fin., Nor., Rom. | [26] | |
| rs725613 | 19 | 1037/1706 | Italy (Sardinia) | [33] | |
| rs2903692 | 22 | 735/621 | Japan | [51] | |
| rs725613 | 19 | 205/422 | China (Han) | [52] | |
| rs6498169 | 22 | 316/550 | Spain | [35] | |
| rs12921922 | 4 | 131/121 | China | [53] | |
| rs12931878 | 1 | ||||
| rs12708716 | 19 | 1212/2513 | Germany | [54] | |
| rs2903692 | 22 | 1743/790 | Japan | [55] | |
| Rheumatoid arthritis | rs6498169 | 22 | 600/550 | Spain | [35] |
| rs6498169 | 22 | 1318/2149 | Norway | [44] | |
| rs6498169 | 22 | 600/550 | Spain | [35] | |
| Primary biliary cirrhosis | rs58102322 | 18 | 1450/2967 | Europe | [43] |
| rs12924129 | 19 | ||||
| rs12924729 | 19 | 2460/7677 | UK | [56] | |
| Crohn’s | rs2903692 | 22 | 1264/890 | Spain | [42] |
| Addisons | rs12917716 | 19 | 542/1220 | Norway, UK | [41] |
| A. Areata | rs998592 | 19 | 1702/1723 | Europe | [45] |
| Disease | SNP | SNP localization | Cases/Controls/Trios | Subject cohort | References |
|---|---|---|---|---|---|
| Multiple sclerosis | rs4774 1 | Exon 11 | 1320/1363 | Europe | [61] |
| rs4774 | Exon 11 | 1343/1379 | UK, US | [49] | |
| rs3087456 | Promoter | 548/528 | Scandinavia | [59,60] | |
| Type 1 Diabetes | rs11074932 | Promoter | 5 cohorts | Sweden | [62] |
| rs3087456 | Promoter | ||||
| Rheumatoid arthritis | rs3087456 | Promoter | 1288/709 | Scandinavia | [60] |
| rs3087456/ rs4774 2 | Promoter/ exon 11 | 580/454 | Spain | [35] | |
| rs8048002 | Intron 3 | 3551/4827 | Norway, Sweden | [63] | |
| rs3087456 | Promoter | ||||
| SLE | rs4774 | Exon 11 | 1342/4301 | Europe | [64] |
| Addisons disease | rs8048002 | Intron 3 | 542/1220 | Norway, UK | [41] |
| rs3087465 | Promoter | 128/406 | Italy | [65] | |
| Disease | SNP | SNP location | Cases/Controls | Subject cohort | Reference |
|---|---|---|---|---|---|
| Multiple sclerosis | rs1640923 | Downstream | 1343/1379 | UK, US | [49] |
| rs12922733 | Upstream | ||||
| rs243324 | Promoter | 3919/4003 | Europe, USA | [66] | |
| Primary biliary cirrhosis | rs243325 | Promoter | 1450/2967 | Europe | [43] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Berge, T.; Leikfoss, I.S.; Harbo, H.F. From Identification to Characterization of the Multiple Sclerosis Susceptibility Gene CLEC16A. Int. J. Mol. Sci. 2013, 14, 4476-4497. https://doi.org/10.3390/ijms14034476
Berge T, Leikfoss IS, Harbo HF. From Identification to Characterization of the Multiple Sclerosis Susceptibility Gene CLEC16A. International Journal of Molecular Sciences. 2013; 14(3):4476-4497. https://doi.org/10.3390/ijms14034476
Chicago/Turabian StyleBerge, Tone, Ingvild Sørum Leikfoss, and Hanne F. Harbo. 2013. "From Identification to Characterization of the Multiple Sclerosis Susceptibility Gene CLEC16A" International Journal of Molecular Sciences 14, no. 3: 4476-4497. https://doi.org/10.3390/ijms14034476