Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer

Abstract
:
1. Introduction
2. Results and Discussion
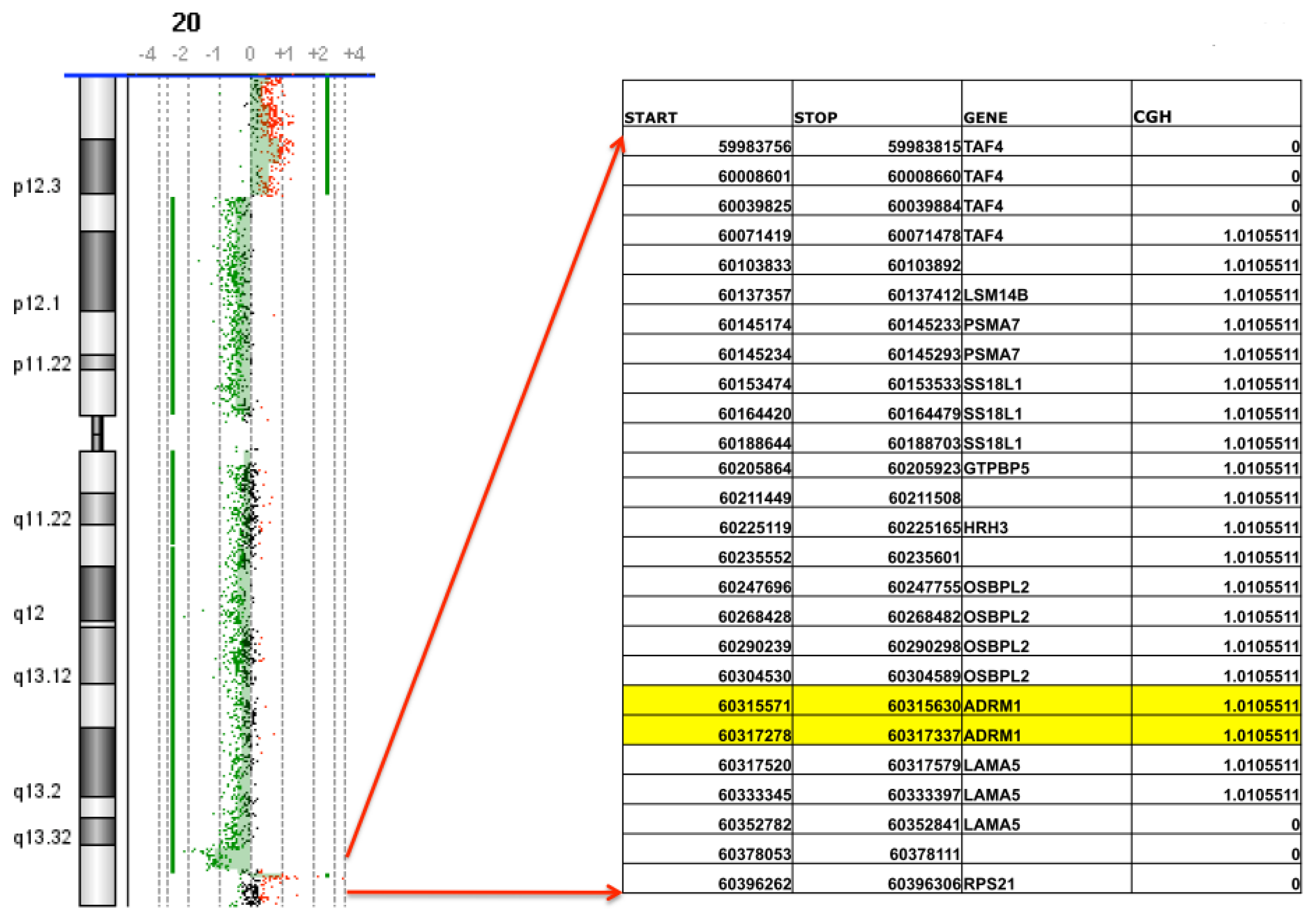
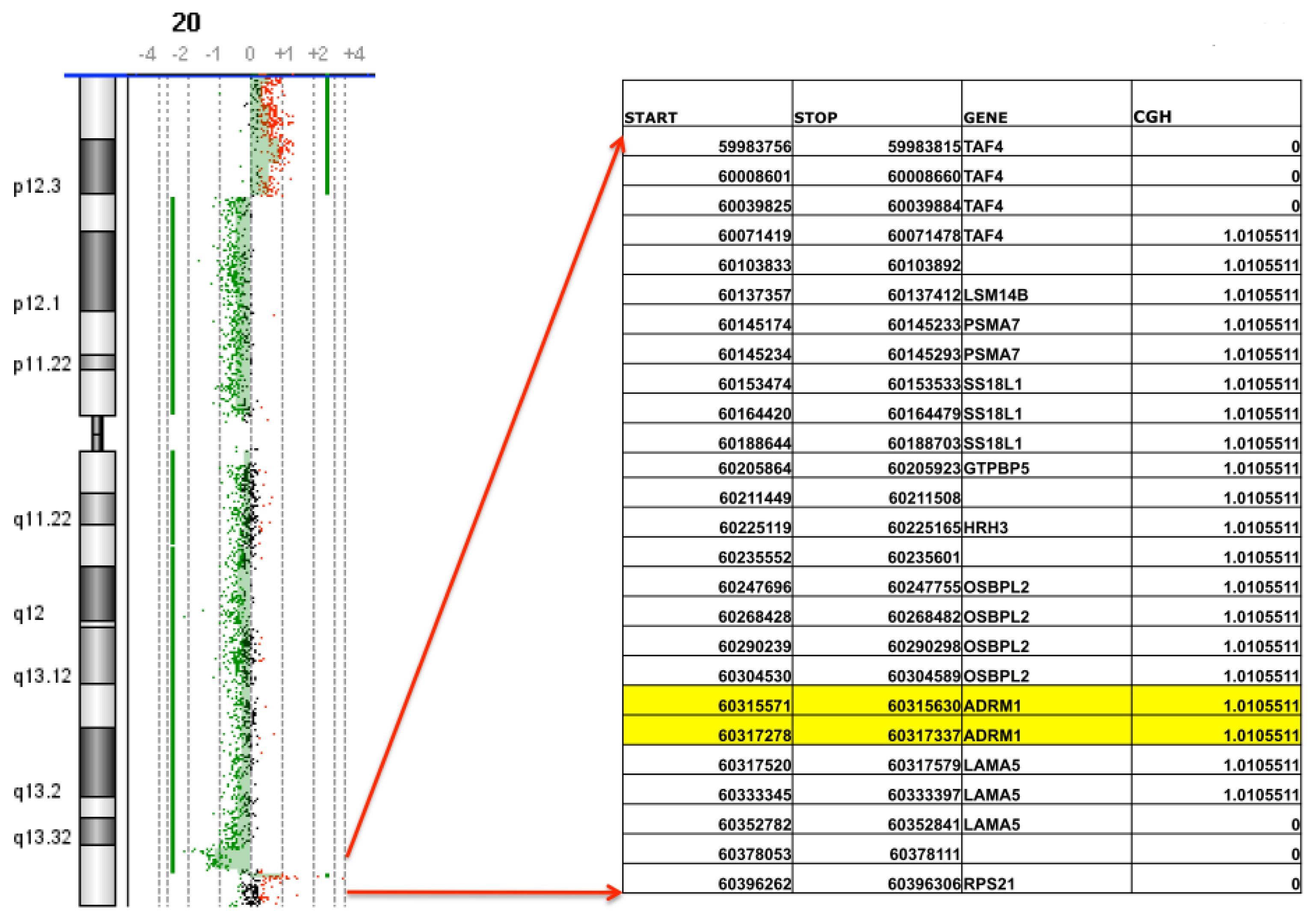
2.1. Confirmation of ADRM1 Amplification in a Second Ovarian Cancer Cohort
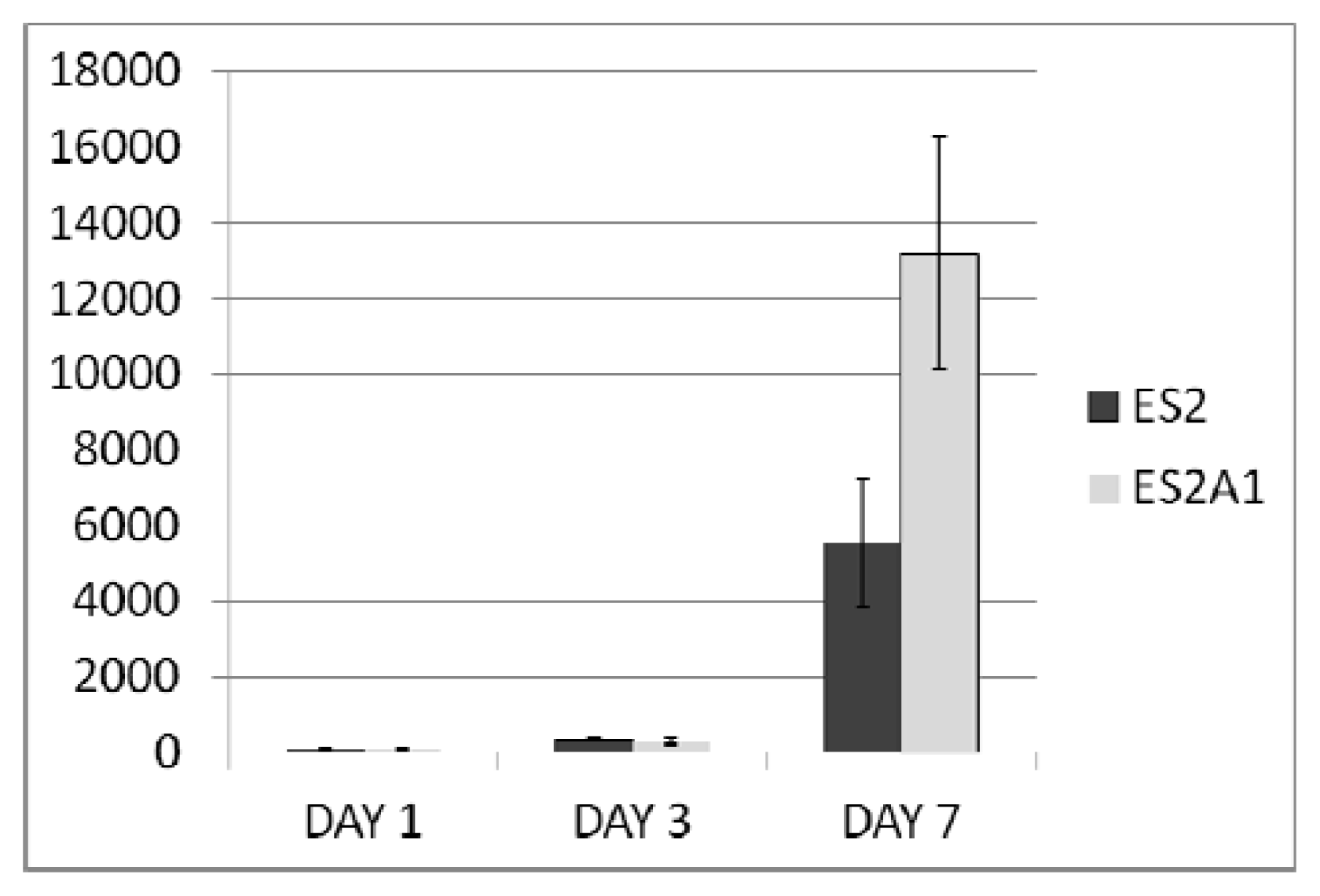
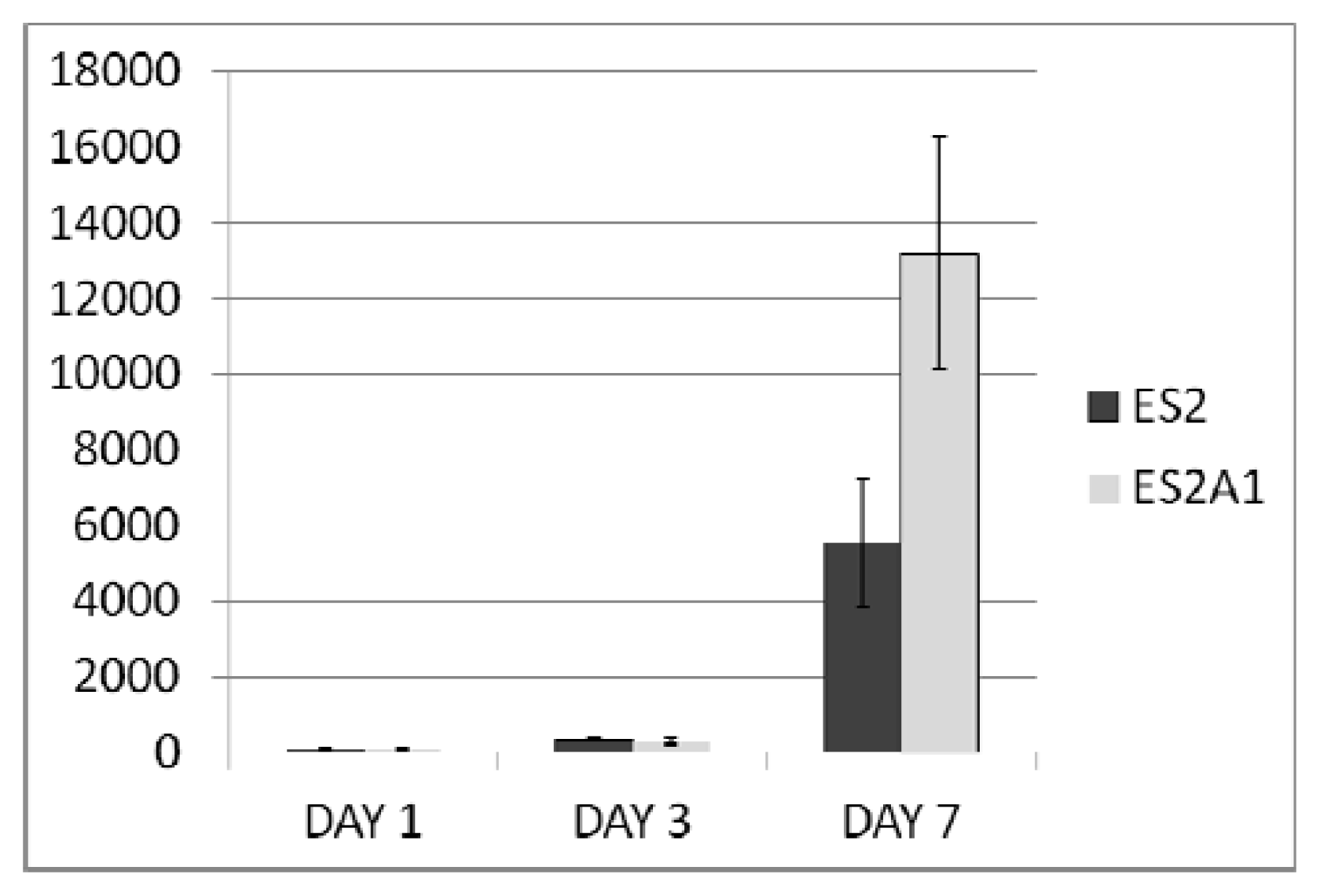
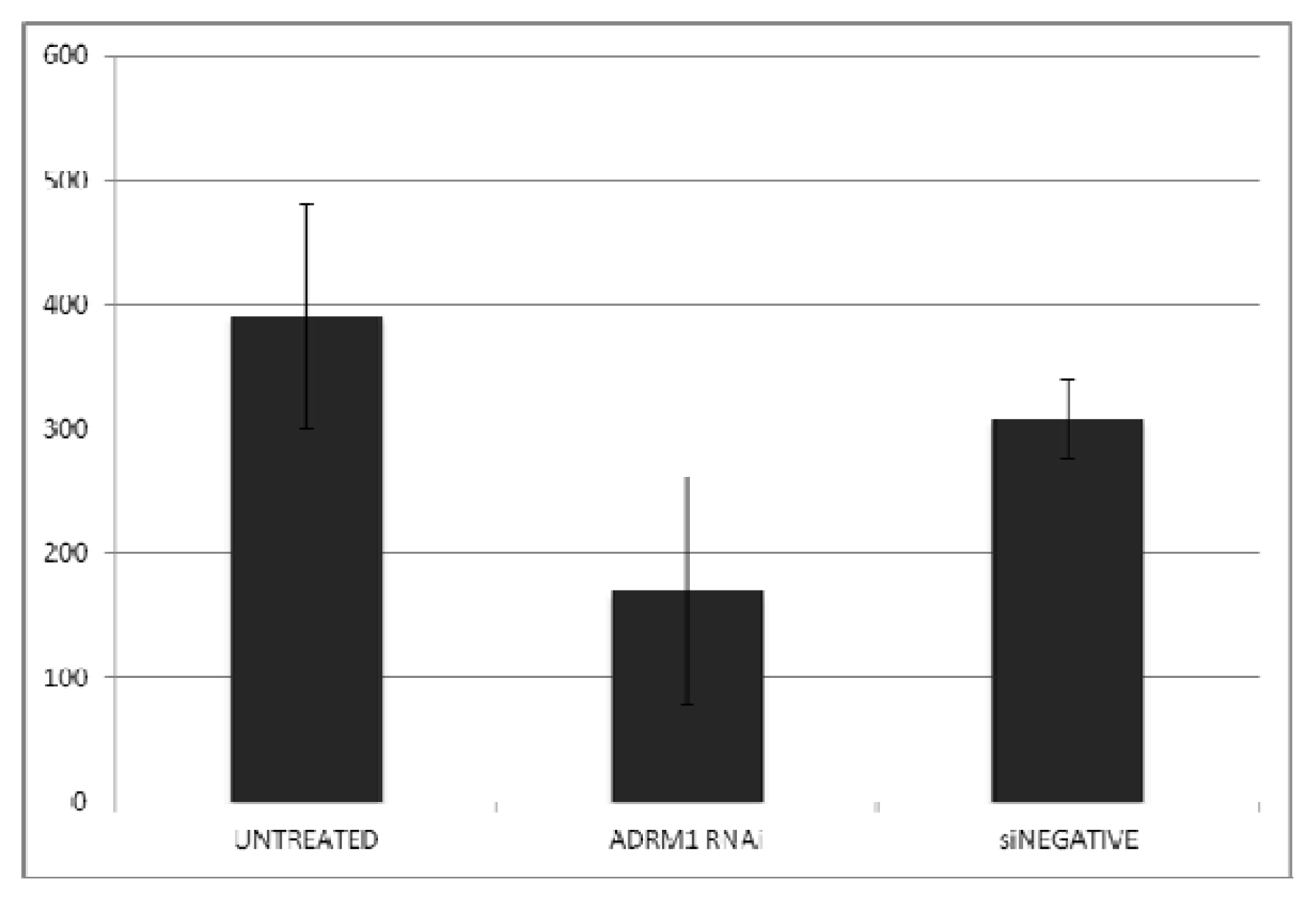
2.2. ADRM1 Over-Expression Increases Cell Proliferation and Conversely, ADRM1 Knockdown Results in Significant Growth Inhibition
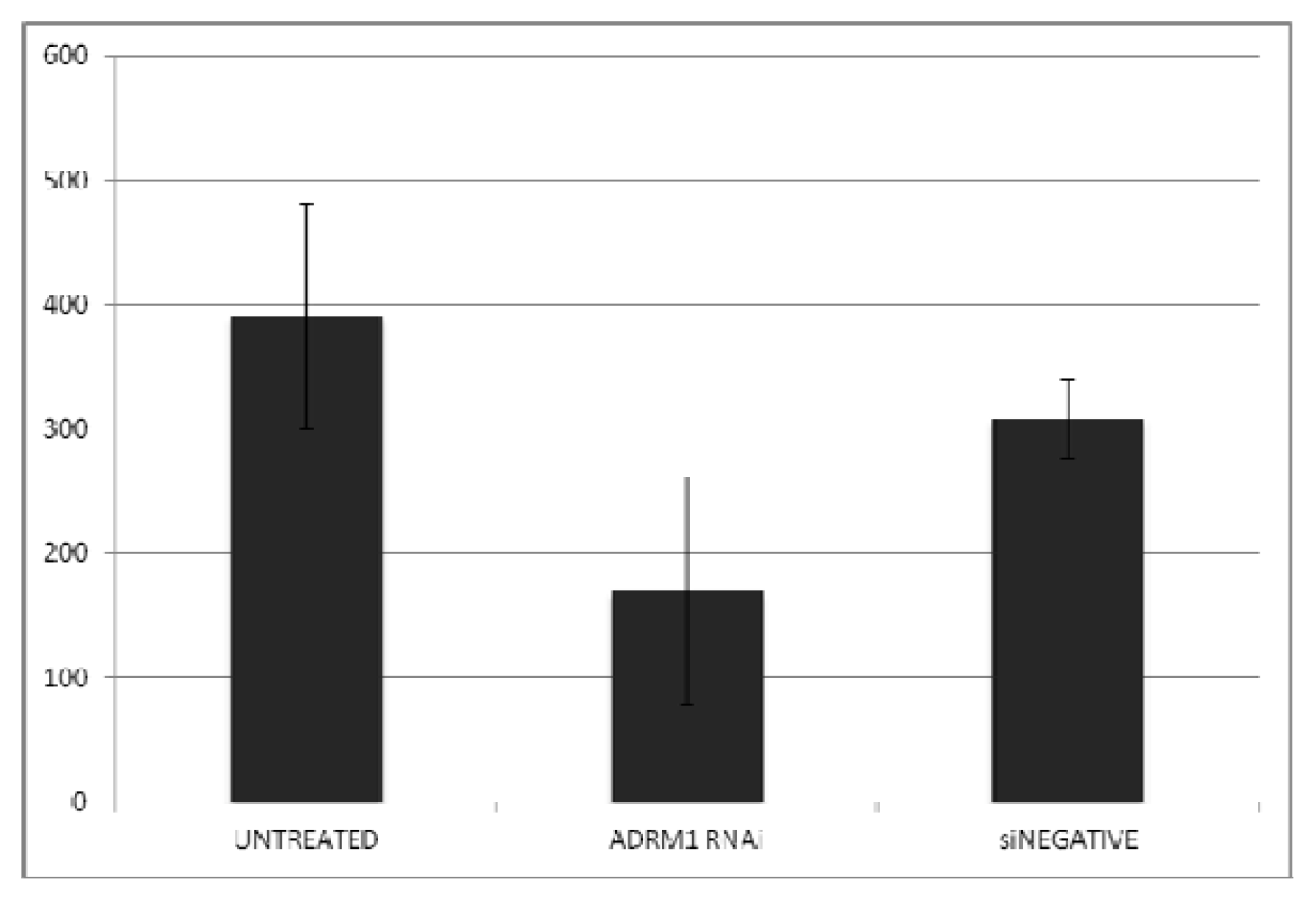
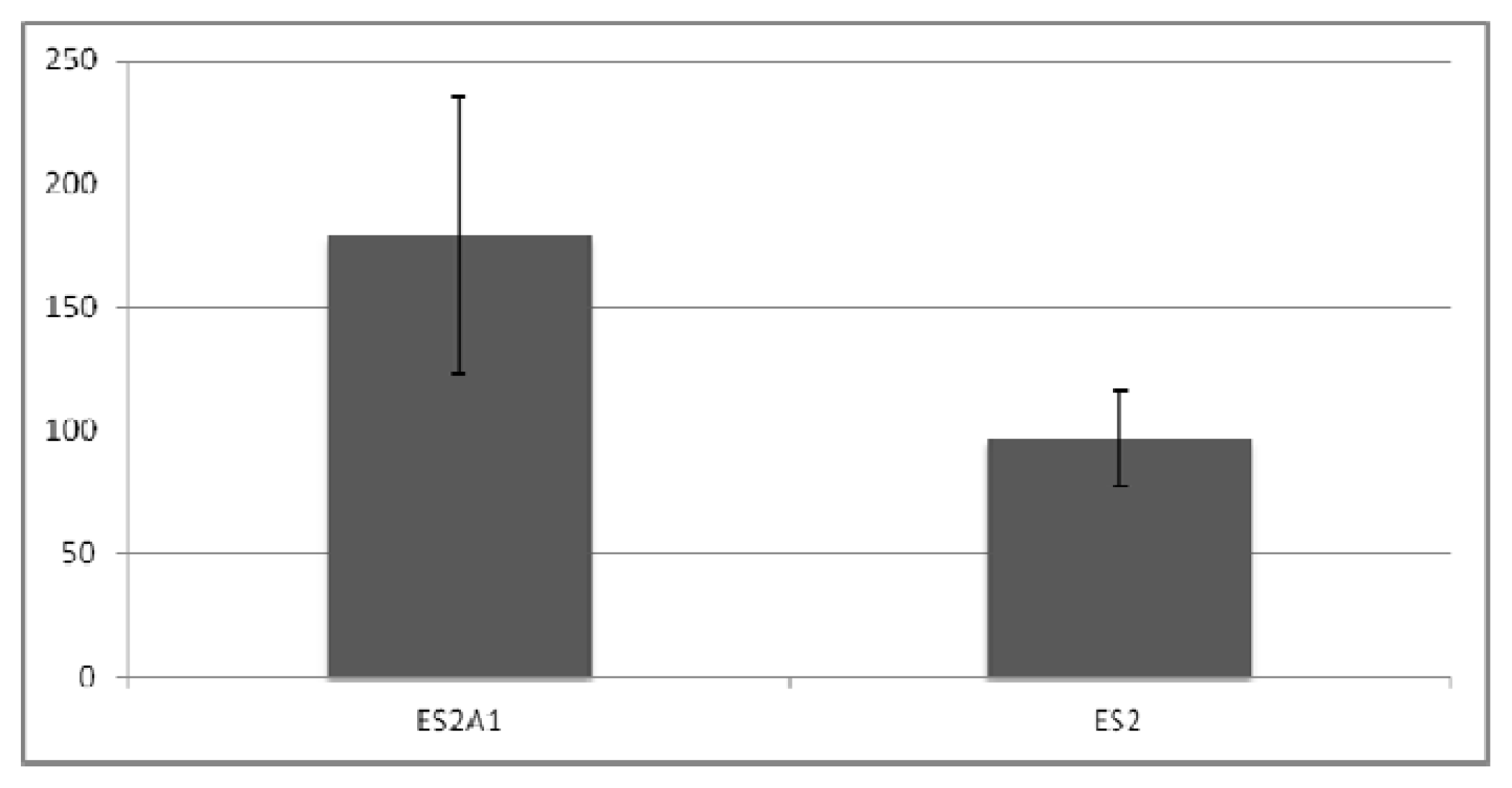
2.3. ADRM1 Over-Expression Increases Growth in Soft-Agar and Conversely, Knockdown of ADRM1 Decreases Growth in Soft Agar
2.4. ADRM1 Knockdown Results in a Decrease in Proliferation and/or Migration Using the Scratch Test
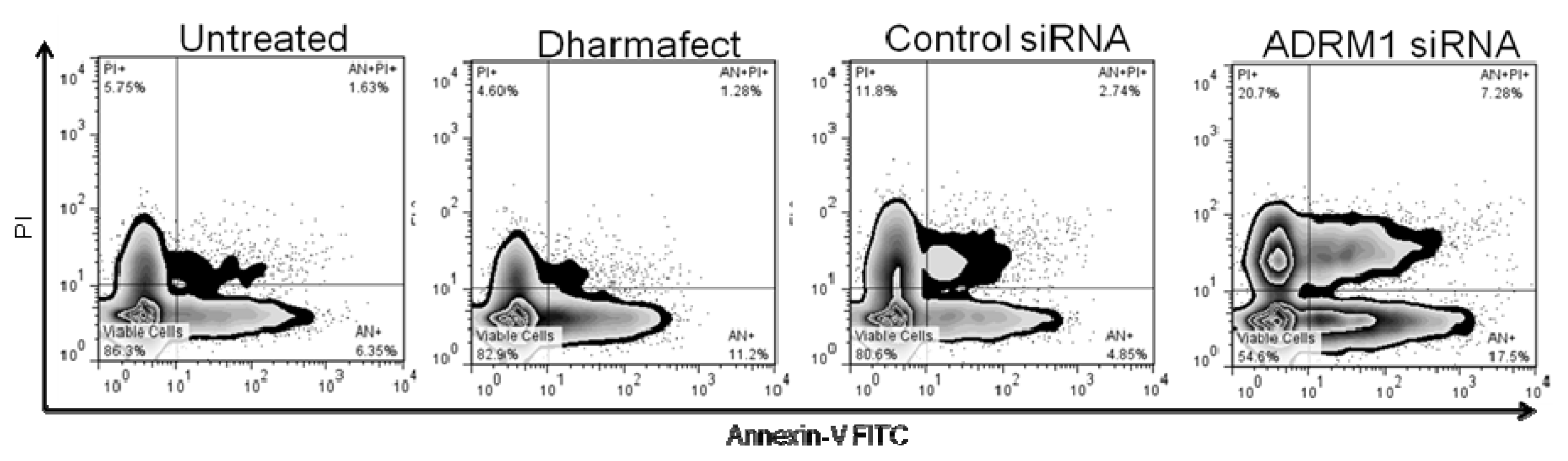
2.5. Knockdown of ADRM1 Increases Apoptosis in Ovarian Cells
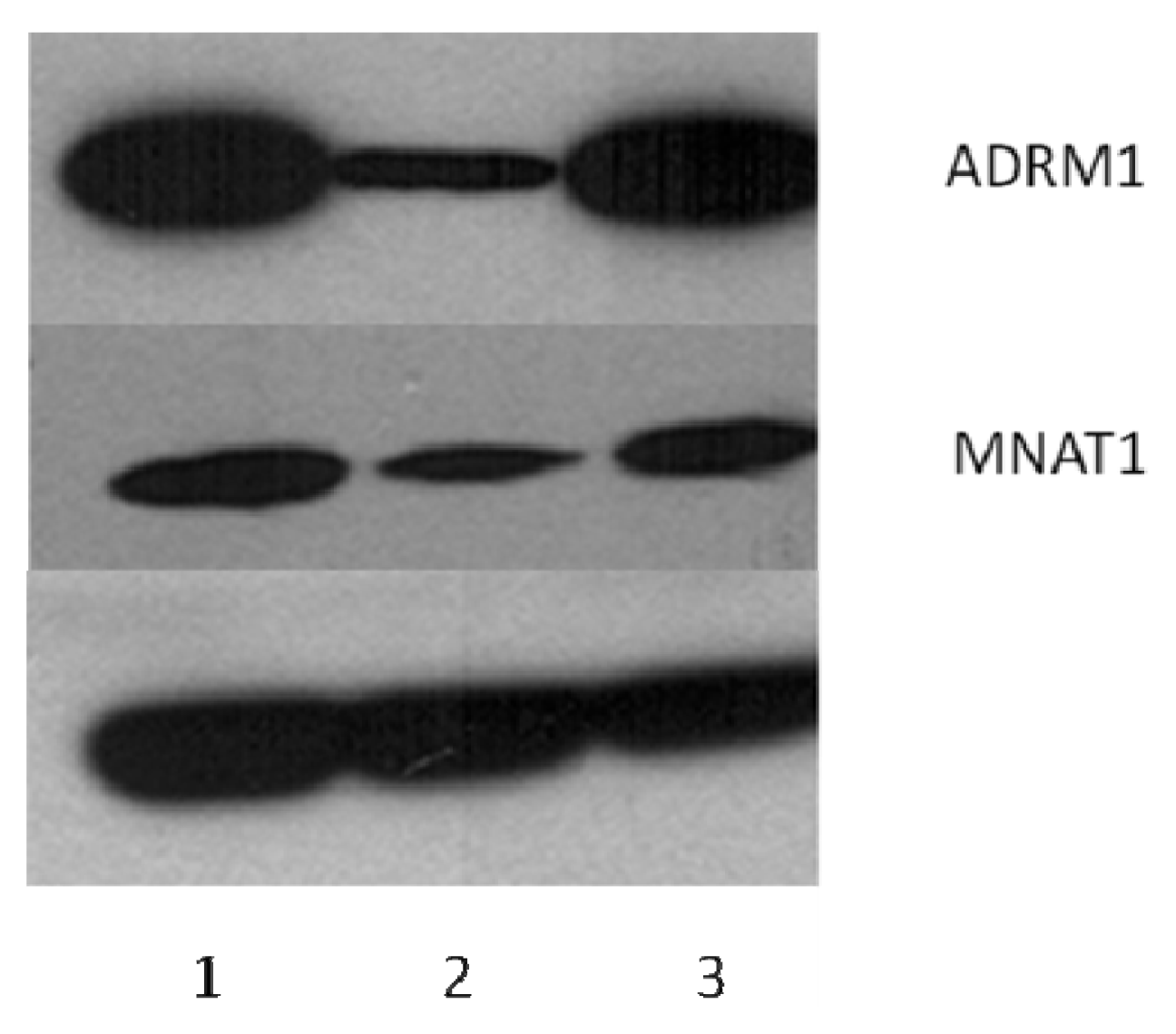
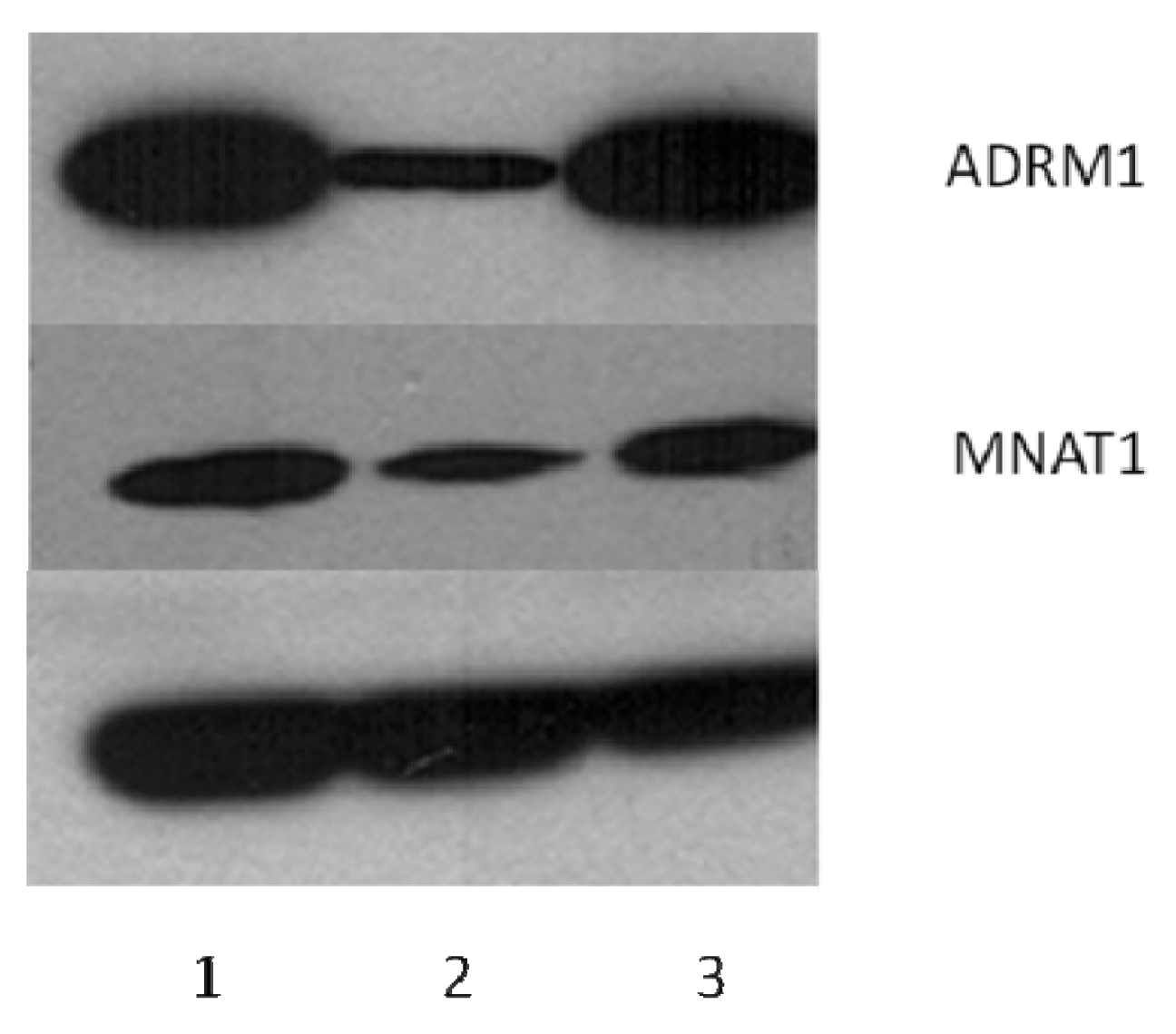
2.6. ADRM1 Knockdown Results in the Down-Regulation of 15 Proteins Including MNAT1 and the Up-Regulation of Seven Proteins
2.7. Discussion
3. Experimental Section
3.1. Tissue
3.2. Cell Lines
3.3. Array CGH
3.4. Analyses of Cell Proliferation
3.5. In Vitro Apoptosis and Cell Cycle Assays
3.6. Soft Agar Assay for Colony Formation
3.7. Antibodies and Western Blotting
3.8. RNA Interference
3.9. Plasmid Constructs and Transfection
3.10. Scratch Test
3.11. Cell Growth and Protein Extraction
3.12. DIGE Labeling, Analysis and Mass Spectrometry
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Fejzo, M.S.; Dering, J.; Ginther, C.; Anderson, L.; Ramos, L.; Walsh, C.; Karlan, B.; Slamon, D.J. Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes Chromosomes Cancer 2008, 47, 873–883. [Google Scholar]
- Lamerant, N.; Kieda, C. Adhesion properties of adhesion-regulating molecule 1 protein on endothelial cells. FEBS J 2005, 272, 1833–1844. [Google Scholar]
- Cherix, N.; Froquet, R.; Charette, S.J.; Blanc, C.; Letourneur, F.; Cosson, P. A Phg2-ADRM1 pathway participates in the nutrient-controlled developmental response in Dictyostelium. Mol. Biol. Cell 2006, 17, 4982–4987. [Google Scholar]
- Hamazaki, J.; Iemura, S.; Natsume, T.; Yashiroda, H.; Tanaka, K.; Murata, S. A novel proteasome interacting protein recruits the deubiquitinating enzyme UCH37 to 26S proteasomes. EMBO J 2006, 25, 4524–4536. [Google Scholar]
- Qiu, X.B.; Ouyang, S.Y.; Li, C.J.; Miao, S.; Wang, L.; Goldberg, A.L. hRpn13/ADRM1/GP110 is a novel proteasome subunit that binds the deubiquitinating enzyme, UCH37. EMBO J 2006, 25, 5742–5753. [Google Scholar]
- Yang, J.M. Emerging roles of deubiquitinating enzymes in human cancer. Acta Pharmacol. Sin 2007, 28, 1325–1330. [Google Scholar]
- Al-Shami, A.; Jhaver, K.G.; Vogel, P.; Wilkins, C.; Humphries, J.; Davis, J.J.; Xu, N.; Potter, D.G.; Gerhardt, B.; Mullinax, R.; et al. Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 2010, 5, e13654. [Google Scholar]
- Simins, A.B.; Weighardt, H.; Weidner, K.M.; Weidle, U.H.; Holzmann, B. Functional cloning of ARM-1, an adhesion-regulating molecule upregulated in metastatic tumor cells. Clin. Exp. Metastasis 1999, 17, 641–648. [Google Scholar]
- Pilarsky, C.; Wenzig, M.; Specht, T.; Saeger, H.D.; Grützmann, R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 2004, 6, 744–750. [Google Scholar]
- Chen, W.; Hu, X.T.; Shi, Q.L.; Zhang, F.B.; He, C. Silencing of ADRM1 by RNA interference suppresses proliferation of colorectal cancer cells. Zhonghua ZhongLiu ZaZhi 2009, 31, 815–819. [Google Scholar]
- Chen, W.; Hu, X.T.; Shi, Q.L.; Zhang, F.B.; He, C. Knockdown of the novel proteasome subunit ADRM1 located on the 20q13 amplicon inhibits colorectal cancer cell migration, survival and tumorigenicity. Oncol. Rep 2009, 21, 531–537. [Google Scholar]
- Fejzo, M.S.; Ginther, C.; Dering, J.; Anderson, L.; Venkatessen, N.; Karlan, B.; Slamon, D.J. Knock-down of ovarian cancer amplification target ADRM1 leads to down regulation of GIPC1 and up-regulation of RECK. Genes Chromosomes Cancer 2011, 50, 434–441. [Google Scholar]
- Jiao, Y.; Ou, W.; Meng, F.; Zhou, H.; Wang, A. Targeting HSP90 in ovarian cancers with multiple receptor tyrosine kinase coactivation. Mol. Cancer 2011, 10, 125. [Google Scholar]
- Peyrat, J.F.; Messaoudi, S.; Brion, J.D.; Alami, M. Inhibitors of the heat shock protein 90: From cancer clinical trials to neurodegenerative diseases. Atlas Genet. Cytogenet. Oncol. Haematol. 2010. Available online: http://AtlasGeneticsOncology.org/Deep/HSP90inCancerTreatmentID20086.html (accessed on 25 January 2013.
- Mikhailova, O.N.; Gulyaeva, L.F.; Prudnikov, A.V.; Gerasimov, A.V.; Krasilnikov, S.E. Estrogen-metabolizing gene polymorphisms in the assessment of female hormone-dependent cancer risk. Pharmacogenomics J 2006, 6, 189–193. [Google Scholar]
- Gulyaeva, L.F.; Mikhailova, O.N.; PustyInyak, V.O.; Kim, I.V., IV; Gerasimov, A.V.; Krasilnikov, S.E.; Filipenko, M.L.; Pechkovsky, E.V. Comparative analysis of SNP in estrogen-metabolizing enzymes for ovarian, endometrial, and breast cancers in Novosibirsk, Russia. Adv. Exp. Med. Biol. 2008, 617, 359–366. [Google Scholar]
- Suzuki, Y.; Demoliere, C.; Kitamura, D.; Takeshita, H.; Deuschle, U.; Watanabe, T. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol 1997, 158, 2736–2744. [Google Scholar]
- Han, Y.; Chen, Y.S.; Liu, Z.; Bodyak, N.; Rigor, D.; Bisping, E.; Pu, W.T.; Kang, P.M. Overexpression of HAX-1 protects cardiac myocytes from apoptosis through caspase-9 inhibition. Circ. Res 2006, 99, 415–423. [Google Scholar]
- Radhika, V.; Onesime, D.; Ha, J.H.; Dhanasekaran, N. Galpha13 stimulates cell migration through cortactin-interacting protein Hax-1. J. Biol. Chem 2004, 279, 49406–49413. [Google Scholar]
- Ramsay, A.G.; Keppler, M.D.; Jazayeri, M.; Thomas, G.J.; Parsons, M.; Violette, S.; Weinreb, P.; Hart, I.R.; Marshall, J.F. HS1-associated protein X-1 regulates carcinoma cell migration and invasion via clathrin-mediated endocytosis of integrin alphavbeta6. Cancer Res 2007, 67, 5275–5284. [Google Scholar]
- Lolli, G.; Johnson, L.N. CAK-Cyclin-dependent activating kinase: A key kinase in cell cycle control and a target for drugs? Cell Cycle 2005, 4, 572–577. [Google Scholar]
- Long, J.; Luo, G.; Liu, C.; Cui, X.; Satoh, K.; Xiao, Z.; Zhang, B.; Xu, J.; Ni, Q.; Li, M.; et al. Development of a unique mouse model for pancreatic cancer lymphatic metastasis. Int. J. Oncol 2012, 41, 1662–1668. [Google Scholar]
- Liu, J.P.; Yuan, S.Z.; Zhang, S.N. Experimental study of MAT1 gene silencing mediated by siRNA in pancreatic cancer. Zhonghua Yi Xue Za Zhi 2007, 87, 2719–2723. [Google Scholar]
- Sawiris, G.P.; Sherman-Baust, C.A.; Becker, K.G.; Cheadle, C.; Teichberg, D.; Morin, P.J. Development of a highly specialized cDNA array for the study and diagnosis of epithelial ovarian cancer. Cancer Res 2002, 62, 2923–2928. [Google Scholar]
- Darb-Esfahani, S.; Wirtz, R.M.; Sinn, B.V.; Budczies, J.; Noske, A.; Weichert, W.; Faggad, A.; Scharff, S.; Sehouli, J.; Oskay-Ozcelik, G.; et al. Estrogen receptor 1 mRNA is a prognostic factor in ovarian carcinoma: Determination by kinetic PCR in formalin-fixed paraffin-embedded tissue. Endocr. Relat. Cancer 2009, 16, 1229–1239. [Google Scholar]
- Prisco, M.G.; Zannoni, G.F.; de Stefano, I.; Vellone, V.G.; Tortorella, L.; Fagotti, A.; Mereu, L.; Scambia, G.; Gallo, D. Prognostic role of metastasis tumor antigen 1 in patients with ovarian cancer: A clinical study. Hum. Pathol 2012, 43, 282–288. [Google Scholar]
- Jung, E.J.; Avliyakulov, N.K.; Boontheung, P.; Loo, J.A.; Nel, A.E. Pro-oxidative DEP chemicals induce heat shock proteins and an unfolding protein response in a bronchial epithelial cell line as determined by DIGE analysis. Proteomics 2007, 3, 3906–3918. [Google Scholar]
- Mosessian, S.; Avliyakulov, N.K.; Mulholland, D.J.; Boontheung, P.; Loo, J.A.; Wu, H. Analysis of PTEN complex assembly and identification of heterogeneous nuclear ribonucleoprotein C as a component of the PTEN-associated complex. J. Biol. Chem 2009, 284, 30159–30166. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Av. Ratio | t-test | ID | Protein | pI | MW |
|---|---|---|---|---|---|
| 1.43 | 0.0019 | LAMB1_HUMAN | Laminin subunit beta-1 | 4.83 | 205150 |
| 1.41 | 0.00043 | LAMC1_HUMAN | Laminin subunit gamma-1 | 5.01 | 183191 |
| 1.28 | 0.00016 | ITAV_HUMAN | Integrin alpha-V | 5.45 | 117048 |
| 1.22 | 0.00011 | IDHC_HUMAN | Isocitrate dehydrogenase (NADP) cytoplasmic | 6.53 | 46915 |
| 1.21 | 0.00017 | PLST_HUMAN | Plastin-3 | 5.41 | 71279 |
| 1.21 | 0.00024 | ST1A3_HUMAN | Sulfotransferase 1A3/1A4 | 5.68 | 34288 |
| 1.2 | 2.30E−05 | DC1I2_HUMAN | Cytoplasmic dynein 1 intermediate chain 2 | 5.08 | 71811 |
| −1.17 | 0.00012 | PURA2_HUMAN | Adenylosuccinate synthetase isozyme 2 | 6.13 | 50465 |
| −1.18 | 0.0053 | CAPZB_HUMAN | F-actin-capping protein subunit beta | 5.36 | 31616 |
| −1.2 | 0.00059 | CSDE1_HUMAN | Cold shock domain-containing protein E1 | 5.88 | 89684 |
| −1.21 | 0.00021 | KAP0_HUMAN | cAMP-dependent protein kinase type I-alpha regulatory subunit | 5.27 | 43183 |
| −1.25 | 8.20E−05 | SUGT1_HUMAN | Suppressor of G2 allele of SKP1 homolog | 5.07 | 41284 |
| −1.26 | 0.0083 | HS90B_HUMAN | Heat shock protein HSP 90-beta | 4.97 | 83554 |
| −1.31 | 0.033 | HS90A_HUMAN | Heat shock protein HSP 90-alpha | 4.94 | 85006 |
| −1.33 | 0.031 | H90B3_HUMAN | Putative heat shock protein HSP 90-beta-3 | 4.71 | 68624 |
| −1.33 | 0.0038 | H90B3_HUMAN | Putative heat shock protein HSP 90-beta-3 | 4.71 | 68624 |
| −1.39 | 0.0098 | HS90A_HUMAN | Heat shock protein HSP 90-alpha | 4.94 | 85006 |
| −1.44 | 0.0024 | H90B3_HUMAN | Putative heat shock protein HSP 90-beta-3 | 4.71 | 68626 |
| −1.47 | 0.00026 | H90B3_HUMAN | Putative heat shock protein HSP 90-beta-3 | 4.71 | 68626 |
| −1.48 | 1.50E−06 | CALU_HUMAN | Calumenin | 4.47 | 37198 |
| −1.5 | 0.0053 | HAX1_HUMAN | HCLS1-associated protein X-1 | 4.76 | 31601 |
| −1.59 | 1.30E−06 | MAT1_HUMAN | CDK-activating kinase assembly factor MAT1 | 5.79 | 36256 |
| −1.78 | 4.00E−07 | ADRM1_HUMAN | Proteasomal ubiquitin receptor ADRM1 | 4.96 | 42412 |
| −2.11 | 1.40E−09 | ADRM1_HUMAN | Proteasomal ubiquitin receptor ADRM1 | 4.96 | 42412 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fejzo, M.S.; Anderson, L.; Von Euw, E.M.; Kalous, O.; Avliyakulov, N.K.; Haykinson, M.J.; Konecny, G.E.; Finn, R.S.; Slamon, D.J. Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer. Int. J. Mol. Sci. 2013, 14, 3094-3109. https://doi.org/10.3390/ijms14023094
Fejzo MS, Anderson L, Von Euw EM, Kalous O, Avliyakulov NK, Haykinson MJ, Konecny GE, Finn RS, Slamon DJ. Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer. International Journal of Molecular Sciences. 2013; 14(2):3094-3109. https://doi.org/10.3390/ijms14023094
Chicago/Turabian StyleFejzo, Marlena S., Lee Anderson, Erika M. Von Euw, Ondrej Kalous, Nuraly K. Avliyakulov, Michael J. Haykinson, Gottfried E. Konecny, Richard S. Finn, and Dennis J. Slamon. 2013. "Amplification Target ADRM1: Role as an Oncogene and Therapeutic Target for Ovarian Cancer" International Journal of Molecular Sciences 14, no. 2: 3094-3109. https://doi.org/10.3390/ijms14023094