Compound K, a Ginsenoside Metabolite, Inhibits Colon Cancer Growth via Multiple Pathways Including p53-p21 Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
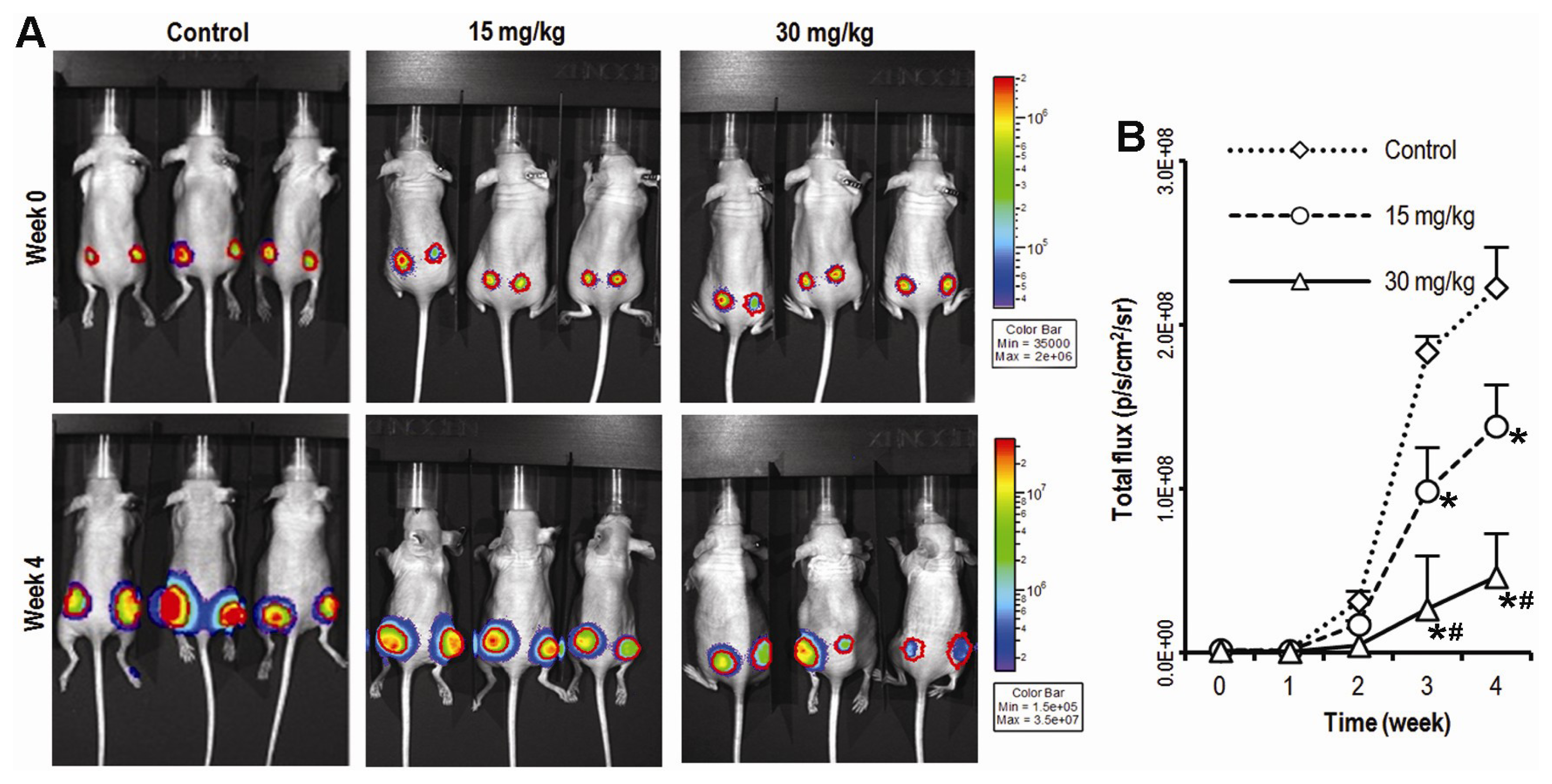
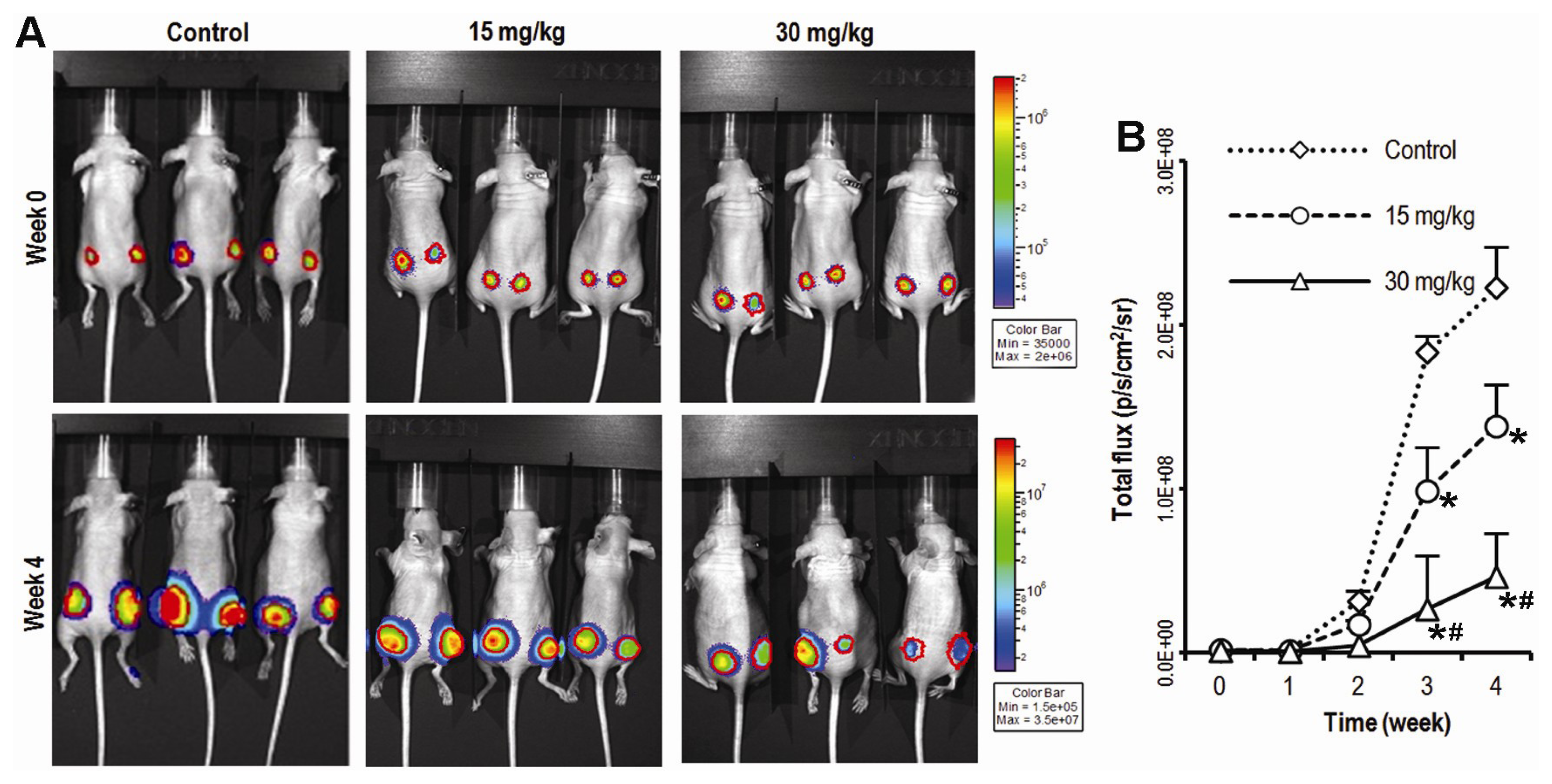
2.1. Compound K Inhibits Tumor Growth in a Xenograft Model of Human Colorectal Cancer Cells in Vivo
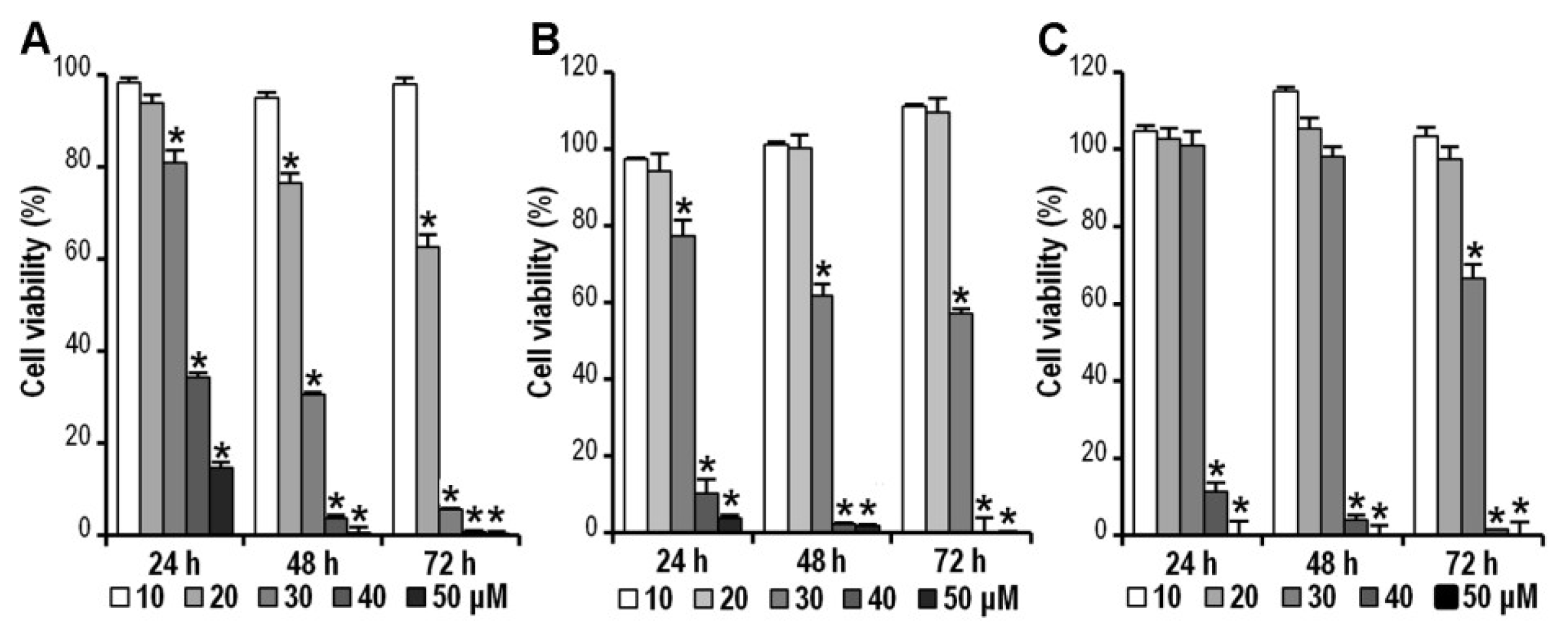
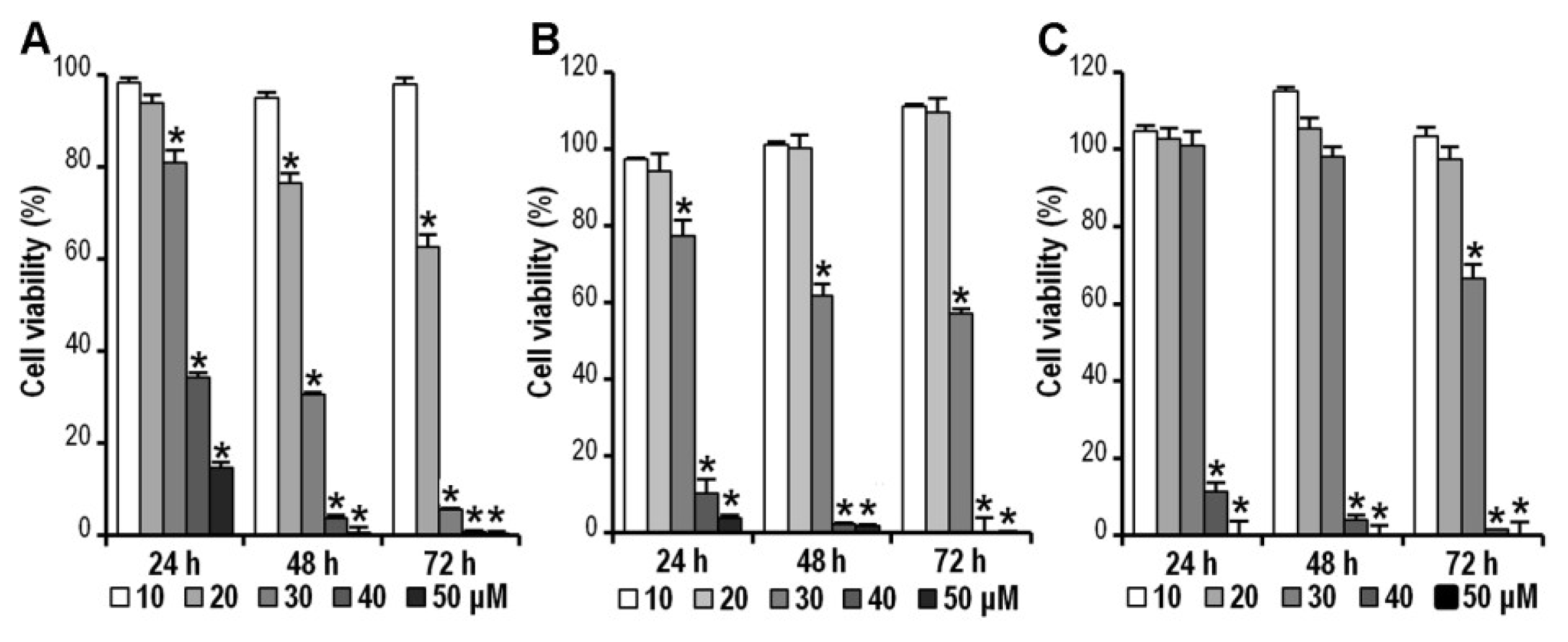
2.2. CK Inhibits HCT-116, SW-480 and HT-29 Cell Viability
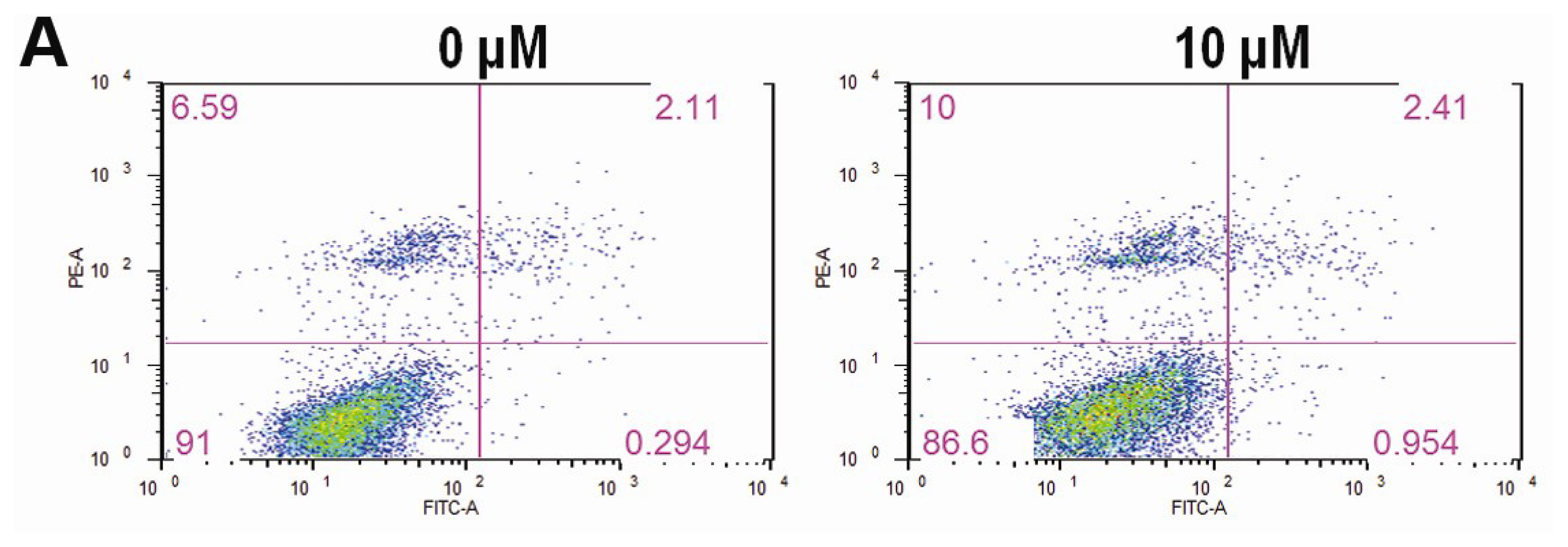
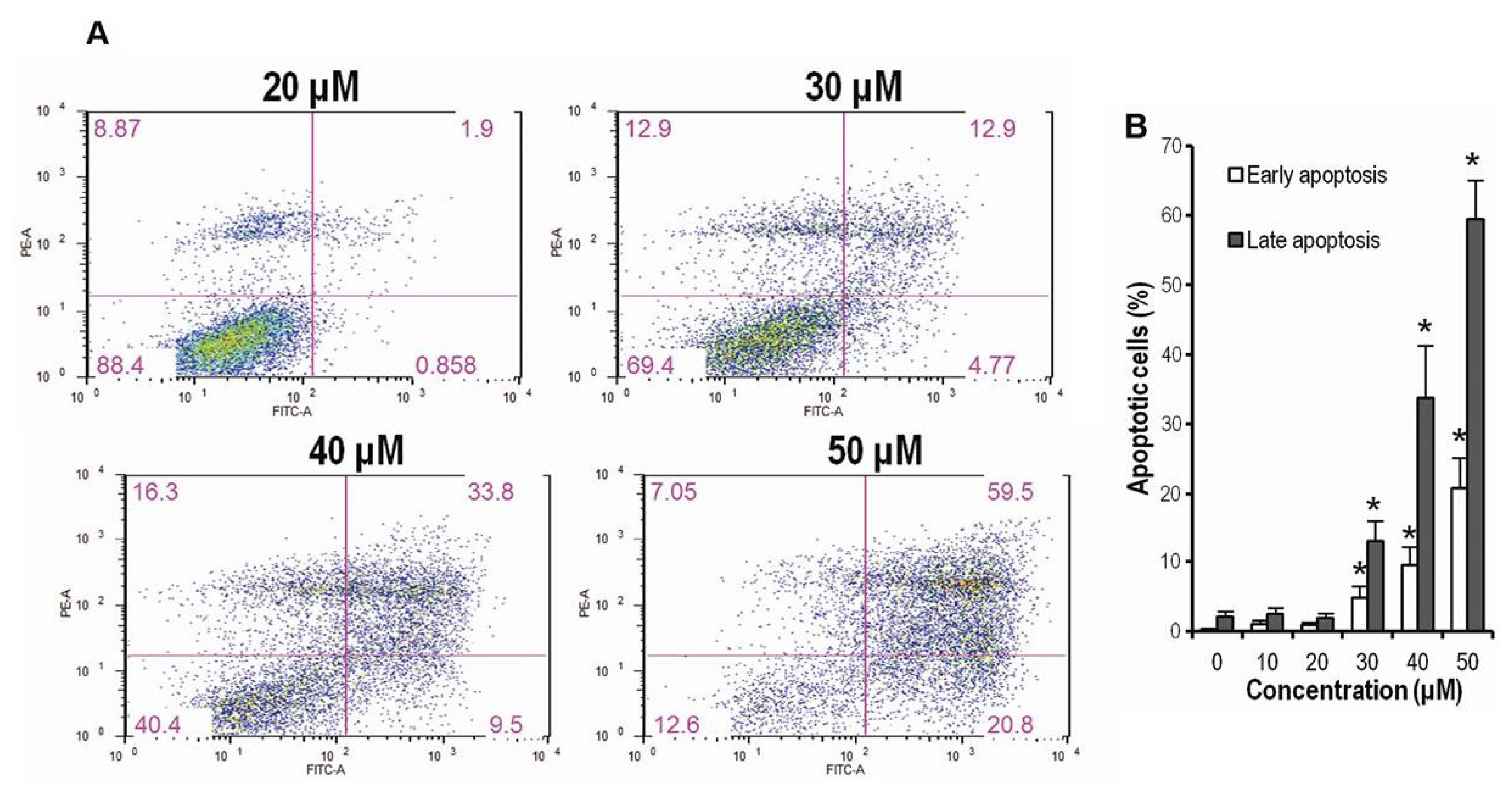
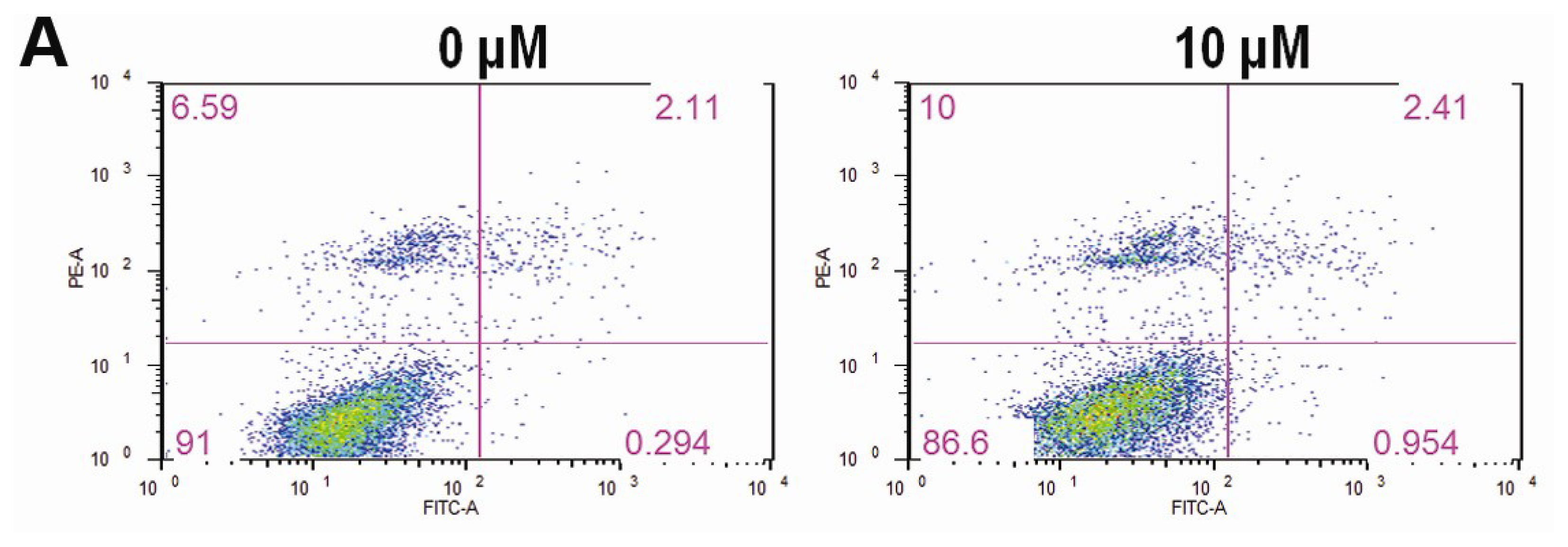
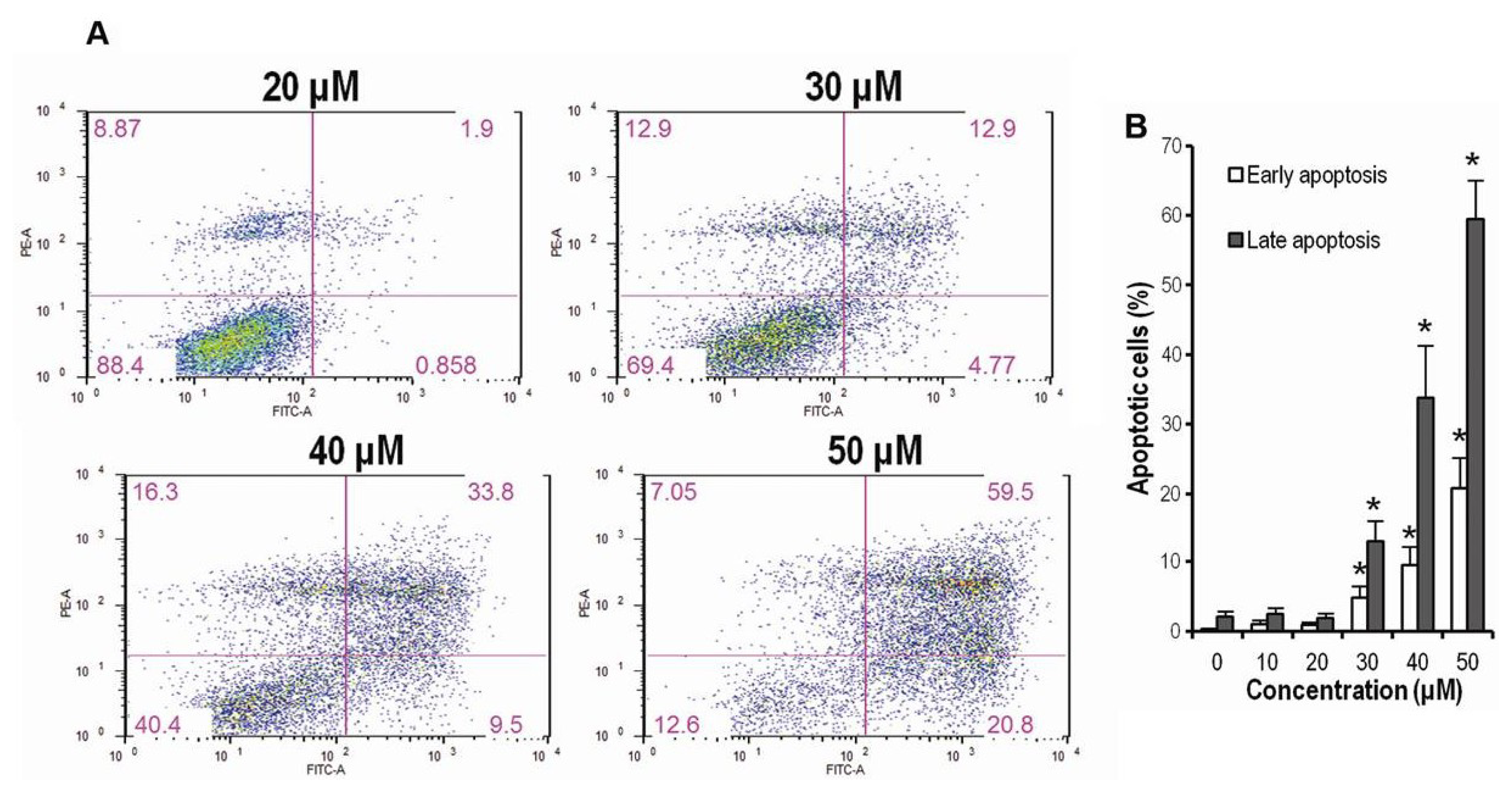
2.3. CK Promotes Apoptosis in HCT-116 Cells
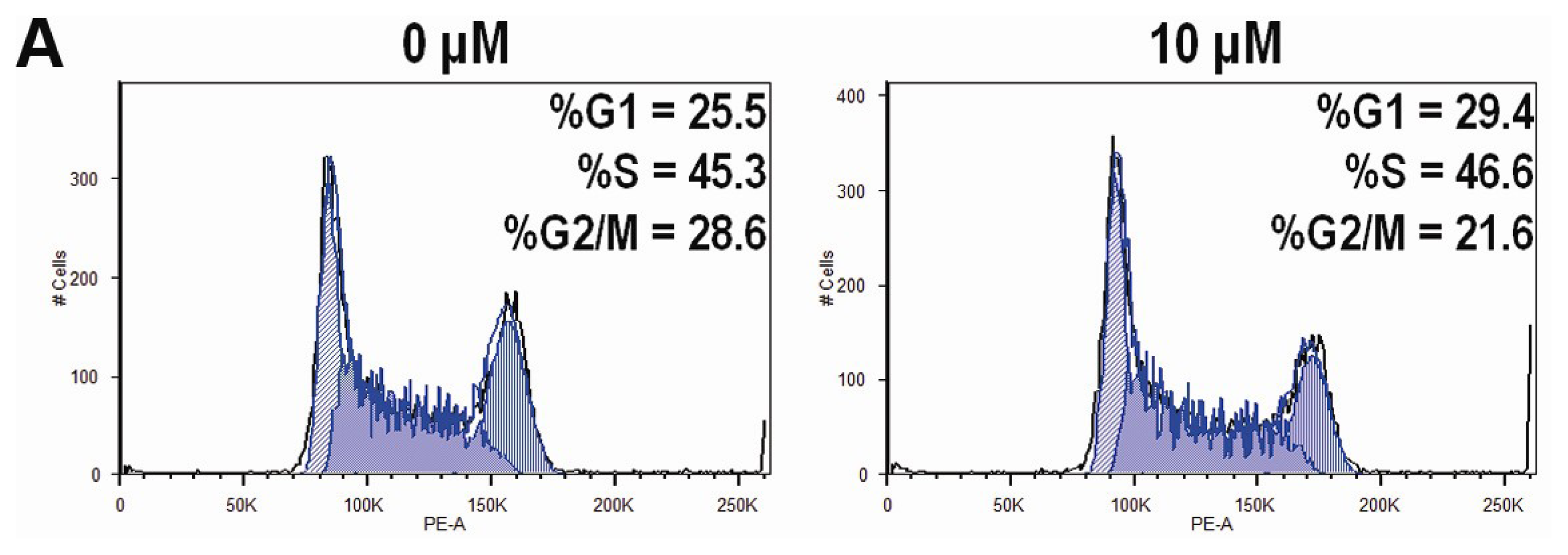
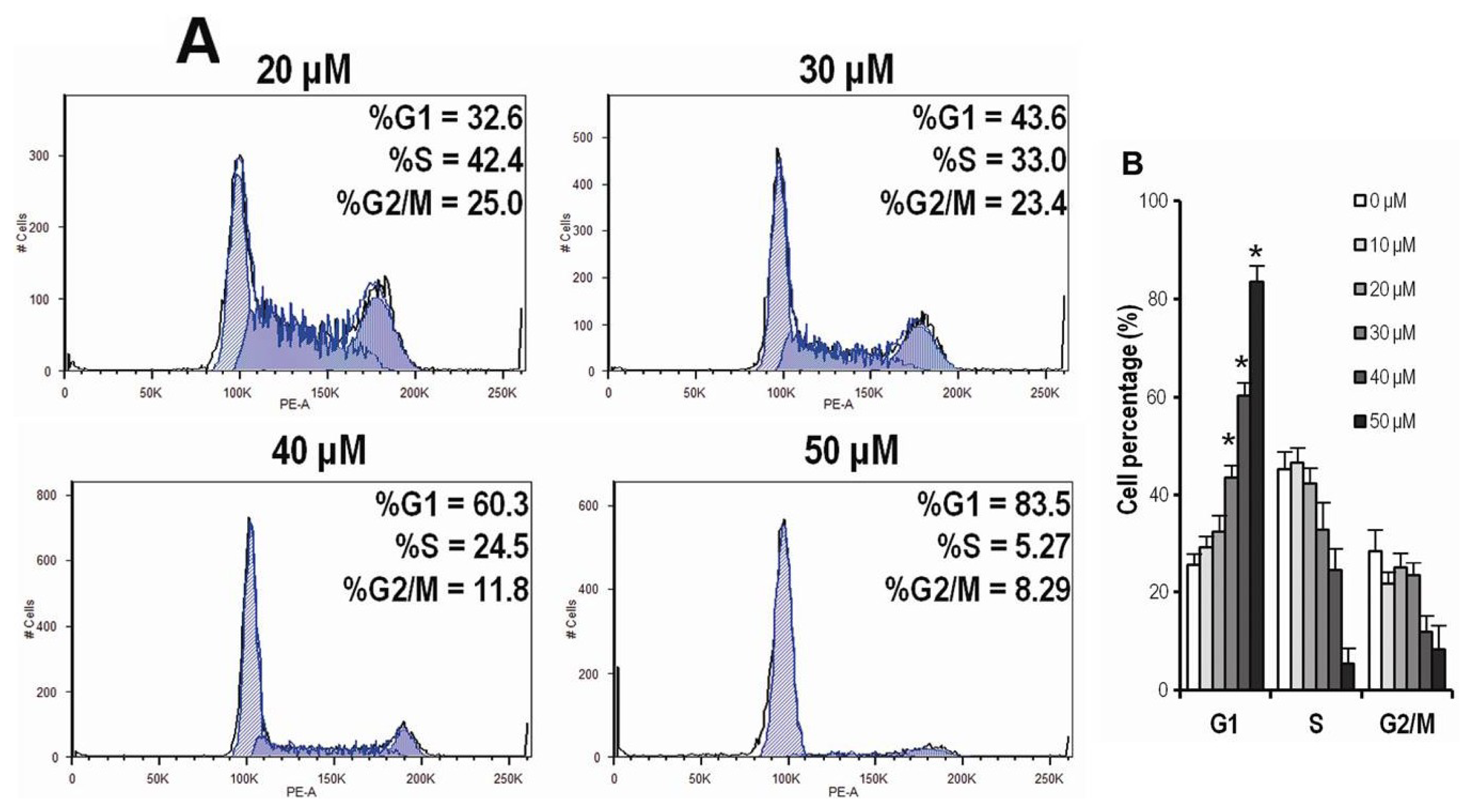
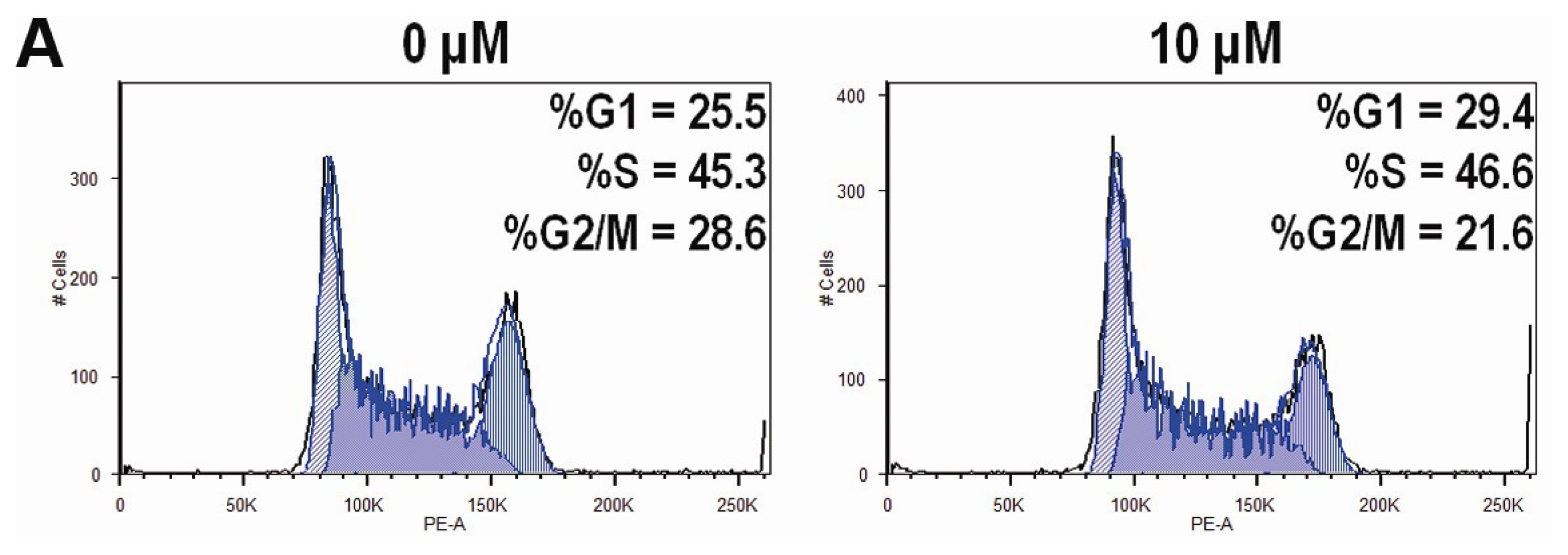
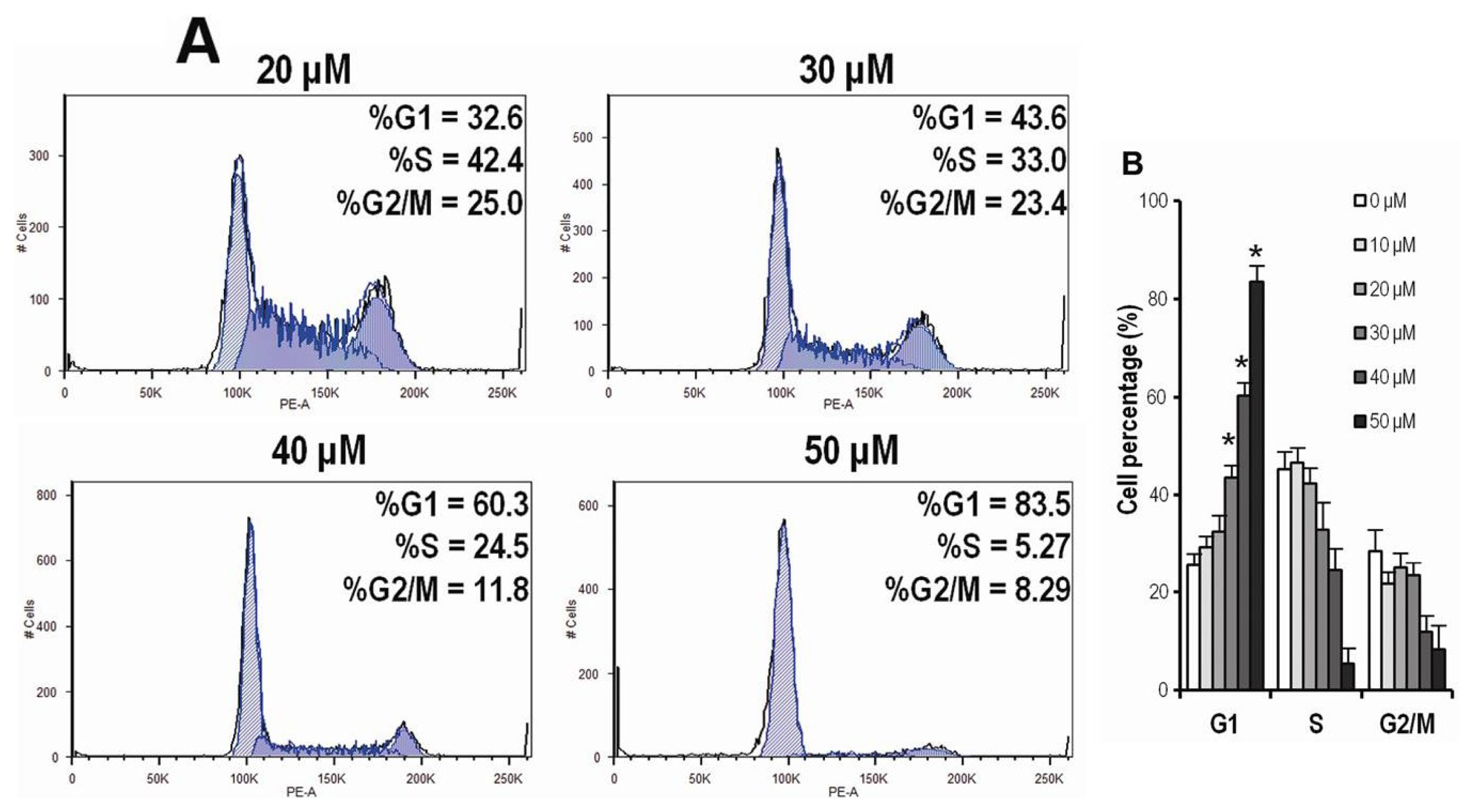
2.4. CK Arrests Cell Cycle at the G1 Phase in HCT-116 Cells
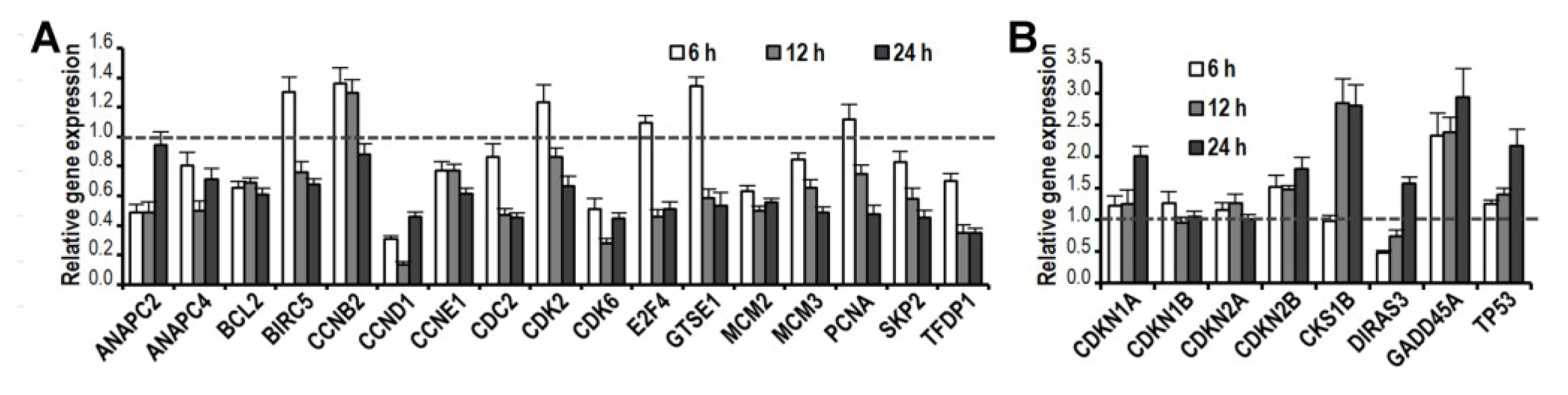
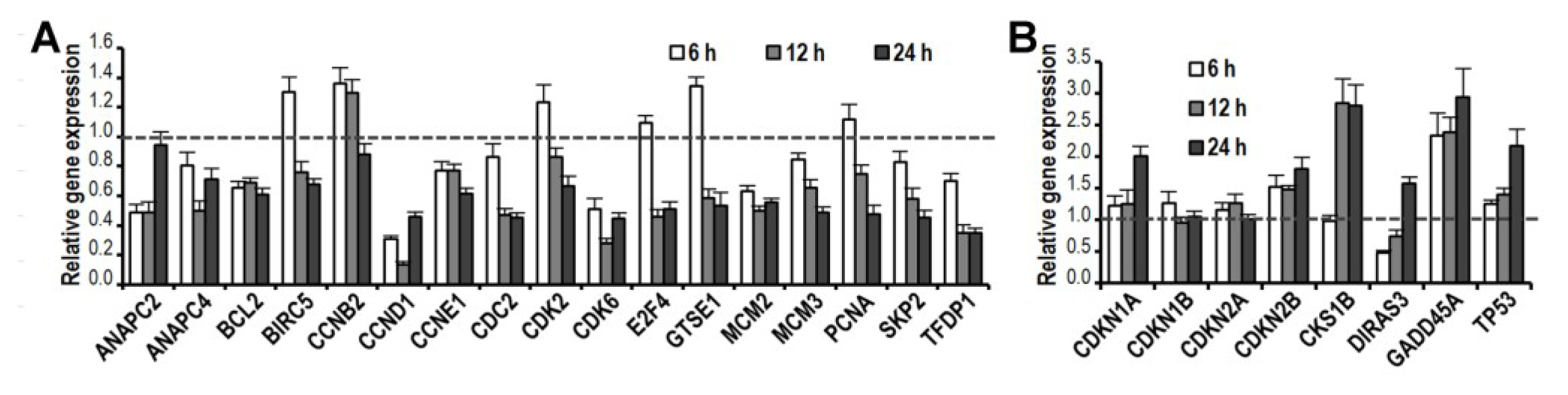
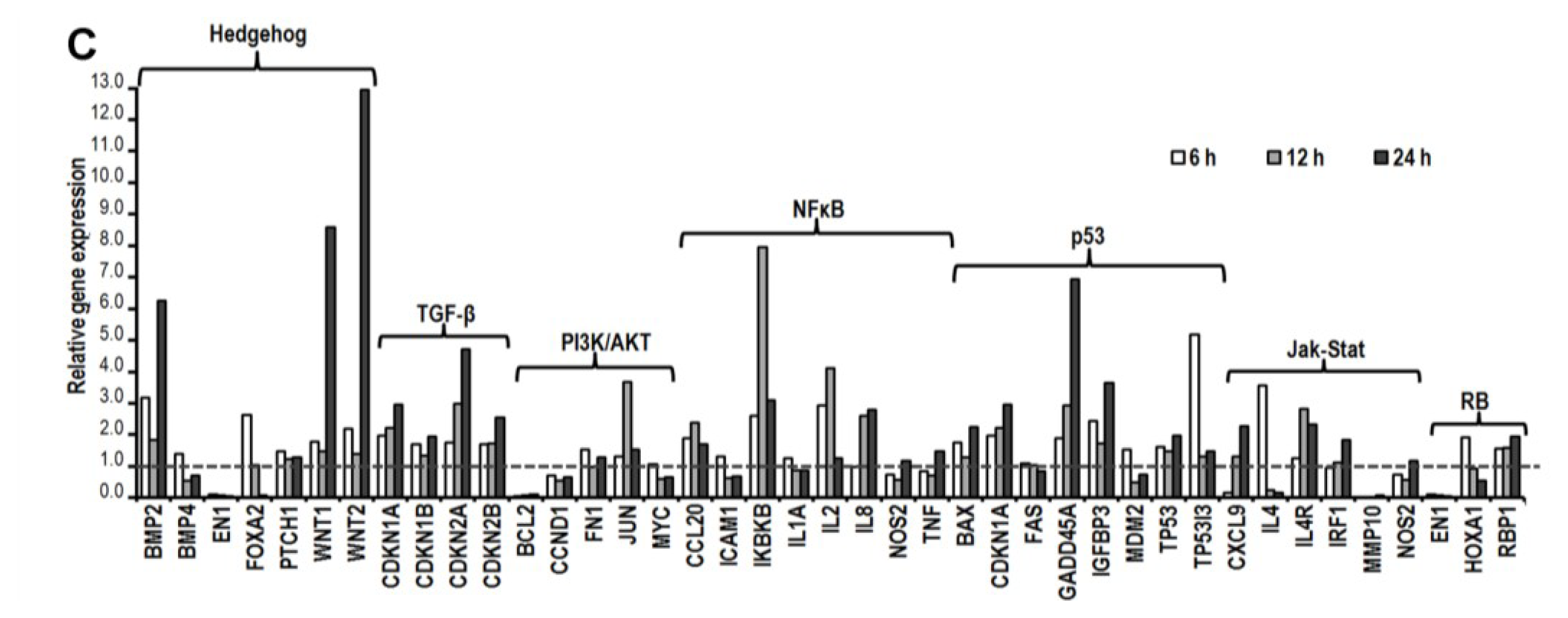
2.5. CK Induces Gene Expression Changes and Pathways Activation Involved in G1 Cell Cycle Arrest
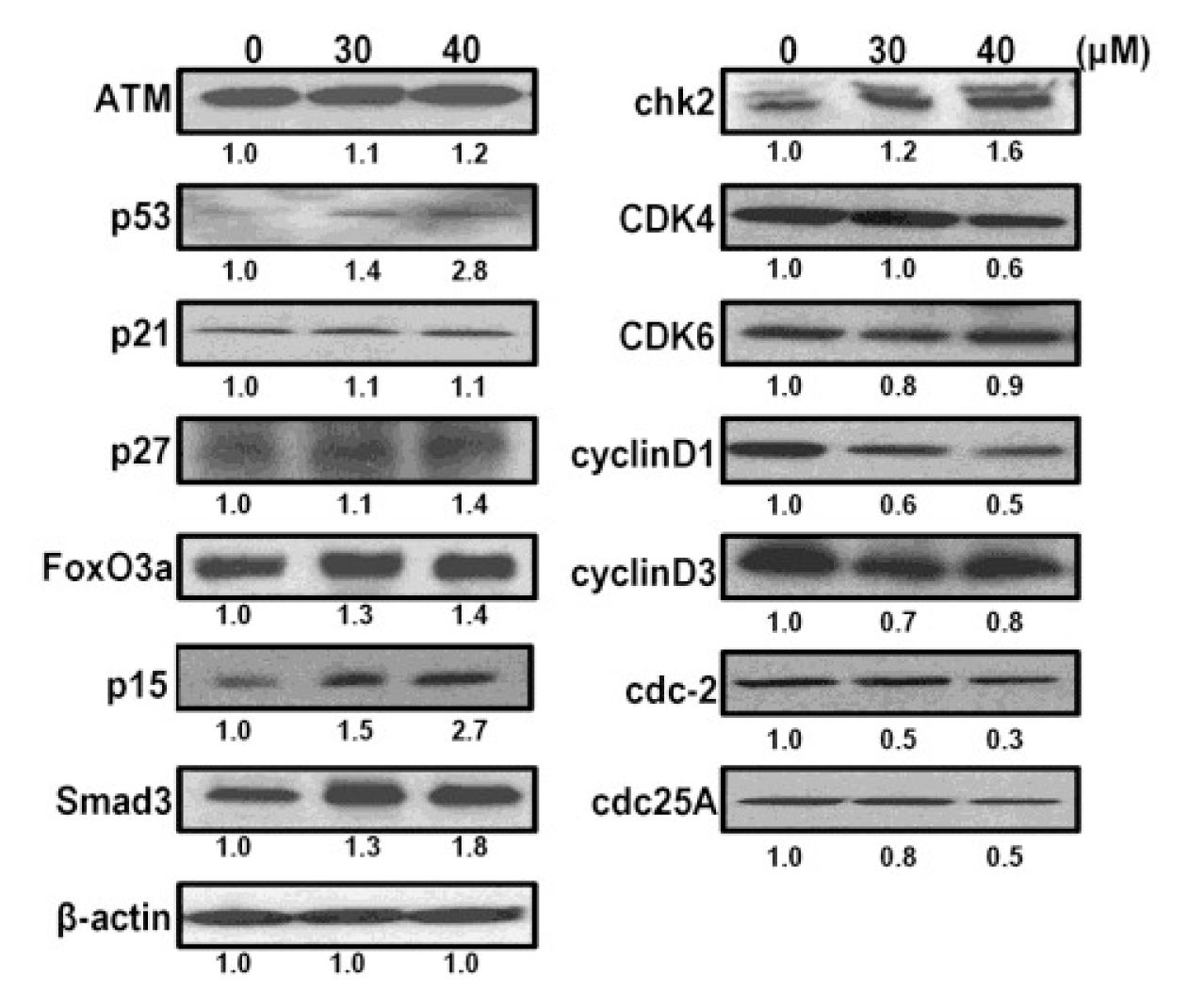
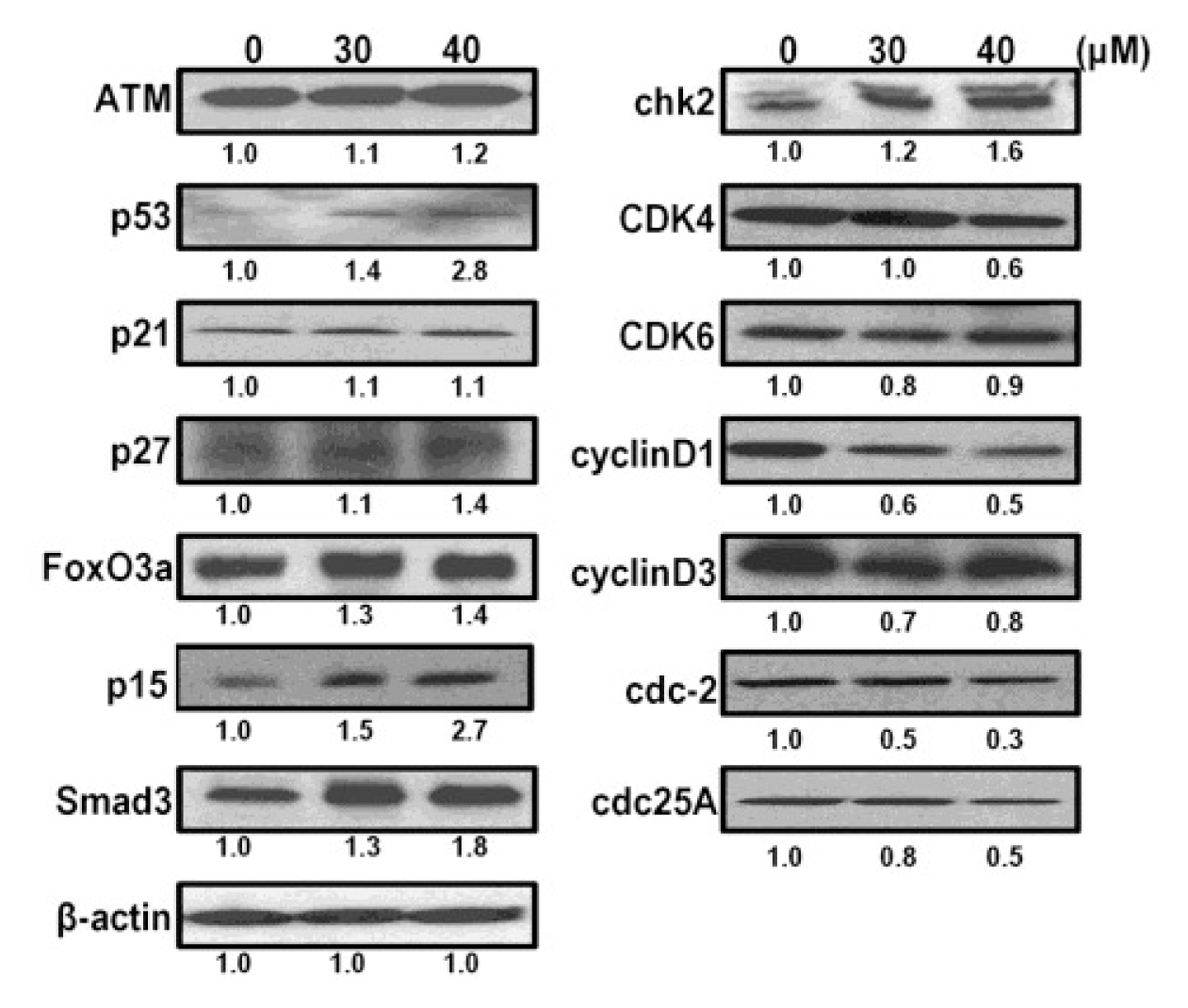
2.6. CK Induces G1 Cell Cycle Arrest in HCT-116 Cells by Activation of Multiple Pathways, Including ATM/p53-p21 and FoxO3a-p27/p15
2.7. Discussion
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture Conditions
3.3. Xenograft Tumor Model of Human Colon Cancer, Xenogen Bioluminescence Imaging
3.4. MTS Assays
3.5. Apoptosis Assay
3.6. Cell Cycle Assay
3.7. Real-Time PCR Array for Cell Cycle and Signal Pathway Analysis
3.8. Immunoblot Assay
3.9. Statistical Analysis
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Arber, N.; Levin, B. Chemoprevention of colorectal cancer: Ready for routine use? Recent Results Cancer Res 2005, 166, 213–230. [Google Scholar]
- Rodriguez, M.; Du, G.J.; Wang, C.Z.; Yuan, C.S. Letter to the editor: Panaxadiol’s anticancer activity is enhanced by epicatechin. Am. J. Chin. Med 2010, 38, 1233–1235. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin 2009, 59, 225–249. [Google Scholar]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin 2012, 62, 10–29. [Google Scholar]
- Wils, J.; O’Dwyer, P.; Labianca, R. Adjuvant treatment of colorectal cancer at the turn of the century: European and US perspectives. Ann. Oncol 2001, 12, 13–22. [Google Scholar]
- Segal, N.H.; Saltz, L.B. Evolving treatment of advanced colon cancer. Annu. Rev. Med 2009, 60, 207–219. [Google Scholar]
- Thomasset, S.C.; Berry, D.P.; Garcea, G.; Marczylo, T.; Steward, W.P.; Gescher, A.J. Dietary polyphenolic phytochemicals—Promising cancer chemopreventive agents in humans? A review of their clinical properties. Int. J. Cancer 2007, 120, 451–458. [Google Scholar]
- Xu, Z.; Chen, X.; Zhong, Z.; Chen, L.; Wang, Y. Ganoderma lucidum polysaccharides: Immunomodulation and potential anti-tumor activities. Am. J. Chin. Med 2011, 39, 15–27. [Google Scholar]
- Qi, L.W.; Wang, C.Z.; Yuan, C.S. Isolation and analysis of ginseng: Advances and challenges. Nat. Prod. Rep 2011, 28, 467–495. [Google Scholar]
- Qi, L.W.; Wang, C.Z.; Du, G.J.; Zhang, Z.Y.; Calway, T.; Yuan, C.S. Metabolism of ginseng and its interactions with drugs. Curr. Drug. Metab 2011, 12, 818–822. [Google Scholar]
- Wang, H.Y.; Qi, L.W.; Wang, C.Z.; Li, P. Bioactivity enhancement of herbal supplements by intestinal microbiota focusing on ginsenosides. Am. J. Chin. Med 2011, 39, 1103–1115. [Google Scholar]
- Wang, C.Z.; Kim, K.E.; Du, G.J.; Qi, L.W.; Wen, X.D.; Li, P.; Bauer, B.A.; Bissonnette, M.B.; Musch, M.W.; Chang, E.B.; Yuan, C.S. Ultra-performance liquid chromatography and time-of-flight mass spectrometry analysis of ginsenoside metabolites in human plasma. Am. J. Chin. Med 2011, 39, 1161–1171. [Google Scholar]
- Akao, T.; Kanaoka, M.; Kobashi, K. Appearance of compound K, a major metabolite of ginsenoside Rb1 by intestinal bacteria, in rat plasma after oral administration—Measurement of compound K by enzyme immunoassay. Biol. Pharm. Bull 1998, 21, 245–249. [Google Scholar]
- Hasegawa, H.; Sung, J.H.; Matsumiya, S.; Uchiyama, M. Main ginseng saponin metabolites formed by intestinal bacteria. Plant. Med 1996, 62, 453–457. [Google Scholar]
- Oh, S.H.; Lee, B.H. A ginseng saponin metabolite-induced apoptosis in HepG2 cells involves a mitochondria-mediated pathway and its downstream caspase-8 activation and Bid cleavage. Toxicol. Appl. Pharmacol 2004, 194, 221–229. [Google Scholar]
- Ming, Y.L.; Song, G.; Chen, L.H.; Zheng, Z.Z.; Chen, Z.Y.; Ouyang, G.L.; Tong, Q.X. Anti-proliferation and apoptosis induced by a novel intestinal metabolite of ginseng saponin in human hepatocellular carcinoma cells. Cell Biol. Int 2007, 31, 1265–1273. [Google Scholar]
- Chae, S.; Kang, K.A.; Chang, W.Y.; Kim, M.J.; Lee, S.J.; Lee, Y.S.; Kim, H.S.; Kim, D.H.; Hyun, J.W. Effect of compound K, a metabolite of ginseng saponin, combined with γ-ray radiation in human lung cancer cells in vitro and in vivo. J. Agric. Food Chem 2009, 57, 5777–5782. [Google Scholar]
- Kim do, Y.; Park, M.W.; Yuan, H.D.; Lee, H.J.; Kim, S.H.; Chung, S.H. Compound K induces apoptosis via CAMK-IV/AMPK pathways in HT-29 colon cancer cells. J. Agric. Food Chem 2009, 57, 10573–10578. [Google Scholar]
- Yu, H.M.; Wang, T.C. Mechanism of cisplatin resistance in human urothelial carcinoma cells. Food Chem. Toxicol 2012, 50, 1226–1237. [Google Scholar]
- Jung, S.H.; Woo, M.S.; Kim, S.Y.; Kim, W.K.; Hyun, J.W.; Kim, E.J.; Kim, D.H.; Kim, H.S. Ginseng saponin metabolite suppresses phorbol ester-induced matrix metalloproteinase-9 expression through inhibition of activator protein-1 and mitogen-activated protein kinase signaling pathways in human astroglioma cells. Int. J. Cancer 2006, 118, 490–497. [Google Scholar]
- Lee, S.J.; Sung, J.H.; Moon, C.K.; Lee, B.H. Antitumor activity of a novel ginseng saponin metabolite in human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer Lett 1999, 144, 39–43. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar]
- Agarwal, M.K.; Hastak, K.; Jackson, M.W.; Breit, S.N.; Stark, G.R.; Agarwal, M.L. Macrophage inhibitory cytokine 1 mediates a p53-dependent protective arrest in S phase in response to starvation for DNA precursors. Proc. Natl. Acad. Sci. USA 2006, 103, 16278–16283. [Google Scholar]
- Dougherty, U.; Mustafi, R.; Wang, Y.; Musch, M.W.; Wang, C.Z.; Konda, V.J.; Kulkarni, A.; Hart, J.; Dawson, G.; Kim, K.E.; et al. American ginseng suppresses Western diet-promoted tumorigenesis in model of inflammation-associated colon cancer: Role of EGFR. BMC Complement Altern. Med 2011, 11, 111. [Google Scholar]
- Park, J.A.; Lee, K.Y.; Oh, Y.J.; Kim, K.W.; Lee, S.K. Activation of caspase-3 protease via a Bcl-2-insensitive pathway during the process of ginsenoside Rh2-induced apoptosis. Cancer Lett 1997, 121, 73–81. [Google Scholar]
- Shinkai, K.; Akedo, H.; Mukai, M.; Imamura, F.; Isoai, A.; Kobayashi, M.; Kitagawa, I. Inhibition of in vitro tumor cell invasion by ginsenoside Rg3. Jpn. J. Cancer Res 1996, 87, 357–362. [Google Scholar]
- Hwang, J.W.; Oh, J.H.; Yoo, H.S.; Lee, Y.W.; Cho, C.K.; Kwon, K.R.; Yoon, J.H.; Park, J.; Her, S.; Lee, Z.W.; et al. Mountain ginseng extract exhibits anti-lung cancer activity by inhibiting the nuclear translocation of NF-κB. Am. J. Chin. Med 2012, 40, 187–202. [Google Scholar]
- Lee, I.K.; Kang, K.A.; Lim, C.M.; Kim, K.C.; Kim, H.S.; Kim, D.H.; Kim, B.J.; Chang, W.Y.; Choi, J.H.; Hyun, J.W. Compound K, a metabolite of ginseng saponin, induces mitochondria-dependent and caspase-dependent apoptosis via the generation of reactive oxygen species in human colon cancer cells. Int. J. Mol. Sci 2010, 11, 4916–4931. [Google Scholar]
- Hu, C.; Song, G.; Zhang, B.; Liu, Z.; Chen, R.; Zhang, H.; Hu, T. Intestinal metabolite compound K of panaxoside inhibits the growth of gastric carcinoma by augmenting apoptosis via Bid-mediated mitochondrial pathway. J. Cell Mol. Med 2012, 16, 96–106. [Google Scholar]
- Kang, K.A.; Kim, Y.W.; Kim, S.U.; Chae, S.; Koh, Y.S.; Kim, H.S.; Choo, M.K.; Kim, D.H.; Hyun, J.W. G1 phase arrest of the cell cycle by a ginseng metabolite, compound K, in U937 human monocytic leukamia cells. Arch. Pharm. Res 2005, 28, 685–690. [Google Scholar]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar]
- Sherr, C.J. Cell cycle control and cancer. Harvey Lect 2000, 96, 73–92. [Google Scholar]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998, 12, 3499–3511. [Google Scholar]
- Carlson, B.; Lahusen, T.; Singh, S.; Loaiza-Perez, A.; Worland, P.J.; Pestell, R.; Albanese, C.; Sausville, E.A.; Senderowicz, A.M. Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res 1999, 59, 4634–4641. [Google Scholar]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar]
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv. Cancer Res 1996, 68, 67–108. [Google Scholar]
- Levesque, A.A.; Eastman, A. p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis 2007, 28, 13–20. [Google Scholar]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar]
- Kohn, K.W. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol. Biol. Cell 1999, 10, 2703–2734. [Google Scholar]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994, 54, 1169–1174. [Google Scholar]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995, 55, 5187–5190. [Google Scholar]
- Eastman, A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J. Cell Biochem 2004, 91, 223–231. [Google Scholar]
- Samuel, T.; Weber, H.O.; Funk, J.O. Linking DNA damage to cell cycle checkpoints. Cell Cycle 2002, 1, 162–168. [Google Scholar]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA damage checkpoint control in cells exposed to ionizing radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar]
- He, G.; Kuang, J.; Khokhar, A.R.; Siddik, Z.H. The impact of S- and G2-checkpoint response on the fidelity of G1-arrest by cisplatin and its comparison to a non-cross-resistant platinum(IV) analog. Gynecol. Oncol 2011, 122, 402–409. [Google Scholar]
- Iavarone, A.; Massague, J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature 1997, 387, 417–422. [Google Scholar]
- Luo, X.; Chen, J.; Song, W.X.; Tang, N.; Luo, J.; Deng, Z.L.; Sharff, K.A.; He, G.; Bi, Y.; He, B.C.; et al. Osteogenic BMPs promote tumor growth of human osteosarcomas that harbor differentiation defects. Lab. Invest 2008, 88, 1264–1277. [Google Scholar]
- He, B.C.; Chen, L.; Zuo, G.W.; Zhang, W.; Bi, Y.; Huang, J.; Wang, Y.; Jiang, W.; Luo, Q.; Shi, Q.; et al. Synergistic antitumor effect of the activated PPARγ and retinoid receptors on human osteosarcoma. Clin. Cancer Res 2010, 16, 2235–2245. [Google Scholar]
- He, B.C.; Gao, J.L.; Zhang, B.Q.; Luo, Q.; Shi, Q.; Kim, S.H.; Huang, E.; Gao, Y.; Yang, K.; Wagner, E.R.; et al. Tetrandrine inhibits Wnt/β-catenin signaling and suppresses tumor growth of human colorectal cancer. Mol. Pharmacol 2011, 79, 211–219. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Z.; Du, G.-J.; Wang, C.-Z.; Wen, X.-D.; Calway, T.; Li, Z.; He, T.-C.; Du, W.; Bissonnette, M.; Musch, M.W.; et al. Compound K, a Ginsenoside Metabolite, Inhibits Colon Cancer Growth via Multiple Pathways Including p53-p21 Interactions. Int. J. Mol. Sci. 2013, 14, 2980-2995. https://doi.org/10.3390/ijms14022980
Zhang Z, Du G-J, Wang C-Z, Wen X-D, Calway T, Li Z, He T-C, Du W, Bissonnette M, Musch MW, et al. Compound K, a Ginsenoside Metabolite, Inhibits Colon Cancer Growth via Multiple Pathways Including p53-p21 Interactions. International Journal of Molecular Sciences. 2013; 14(2):2980-2995. https://doi.org/10.3390/ijms14022980
Chicago/Turabian StyleZhang, Zhiyu, Guang-Jian Du, Chong-Zhi Wang, Xiao-Dong Wen, Tyler Calway, Zejuan Li, Tong-Chuan He, Wei Du, Marc Bissonnette, Mark W. Musch, and et al. 2013. "Compound K, a Ginsenoside Metabolite, Inhibits Colon Cancer Growth via Multiple Pathways Including p53-p21 Interactions" International Journal of Molecular Sciences 14, no. 2: 2980-2995. https://doi.org/10.3390/ijms14022980