Thermal Stability Threshold for Amyloid Formation in Light Chain Amyloidosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
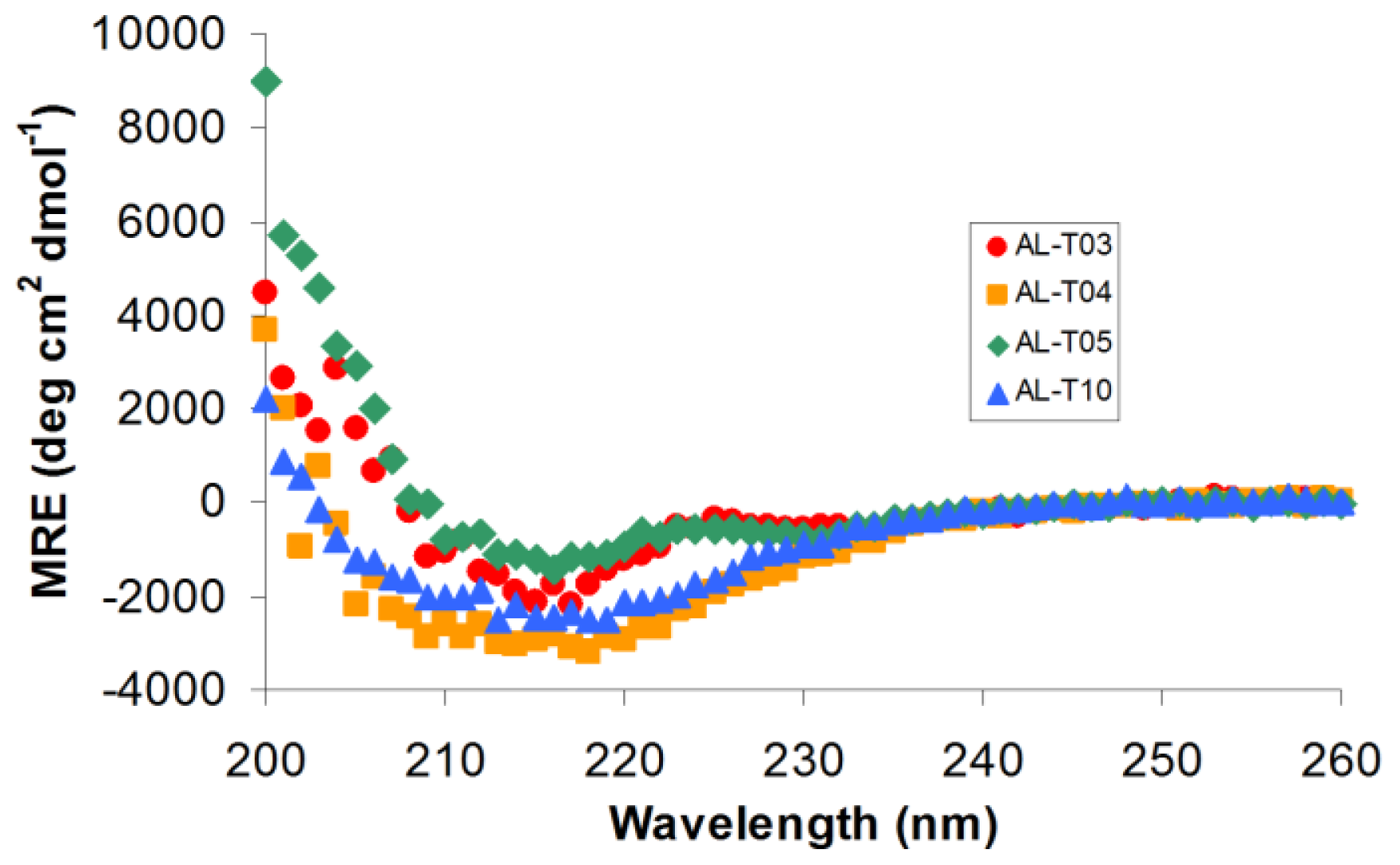
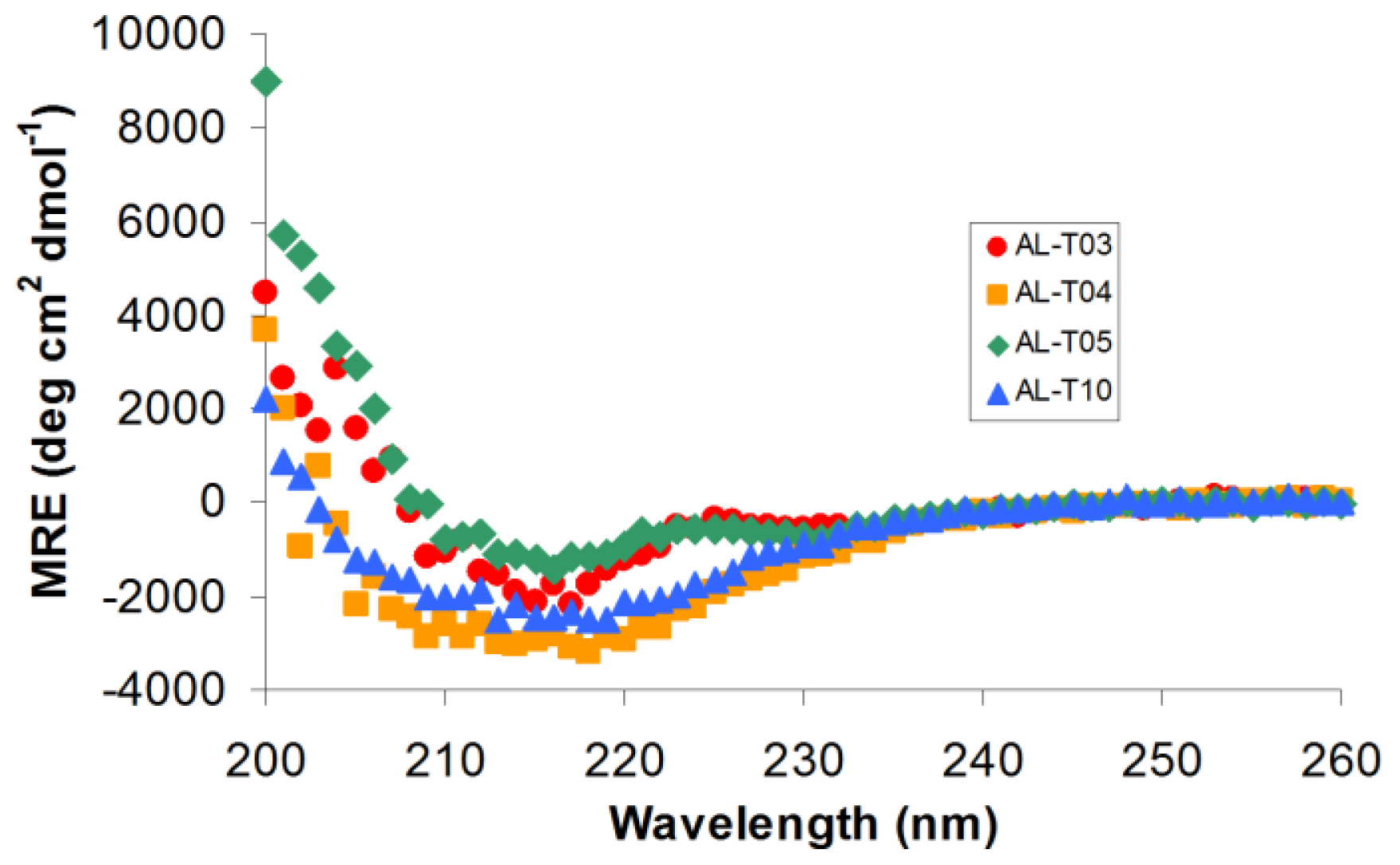
2.1. AL Proteins from Different Patients Adopt an Overall β-Sheet Structure
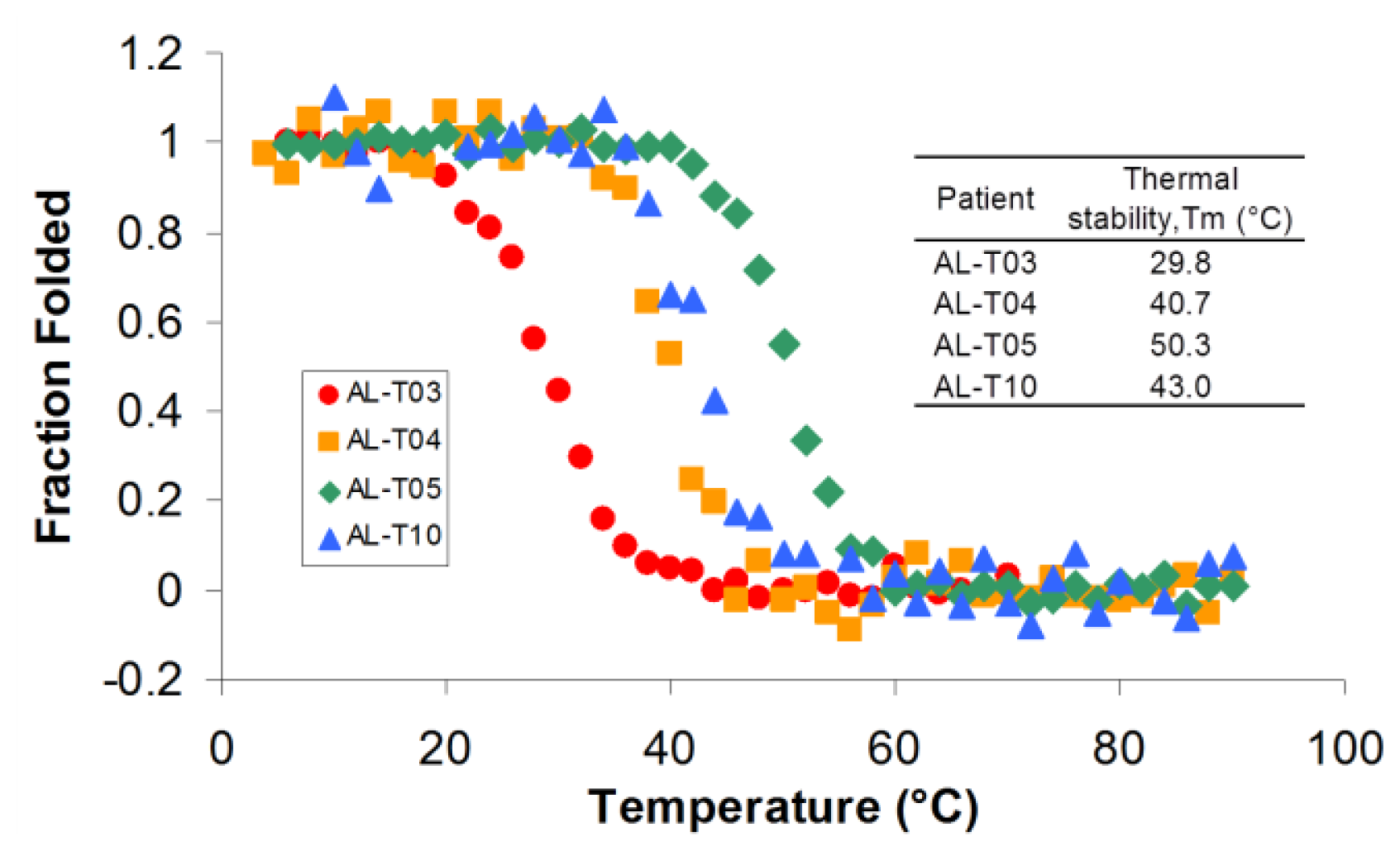
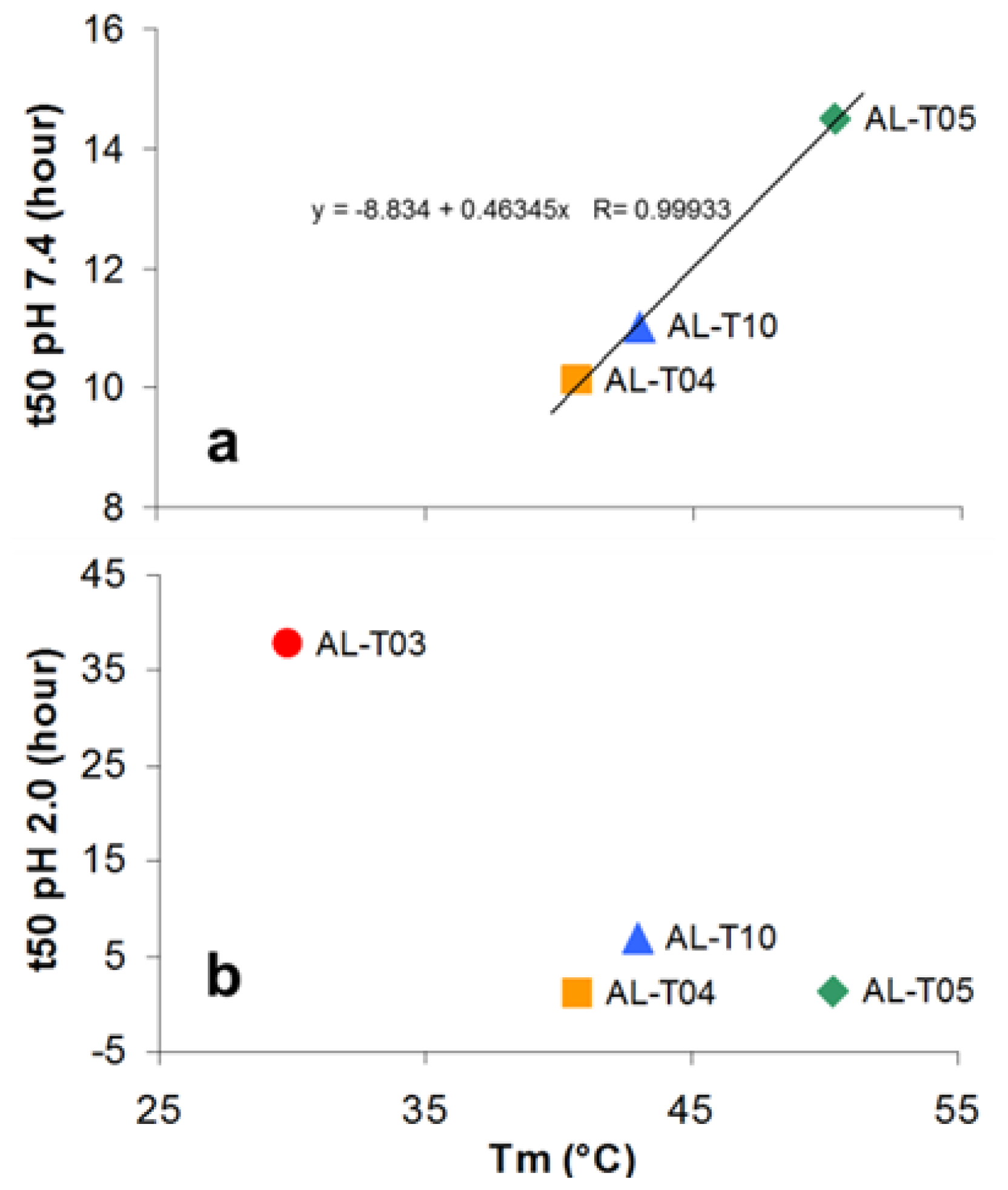
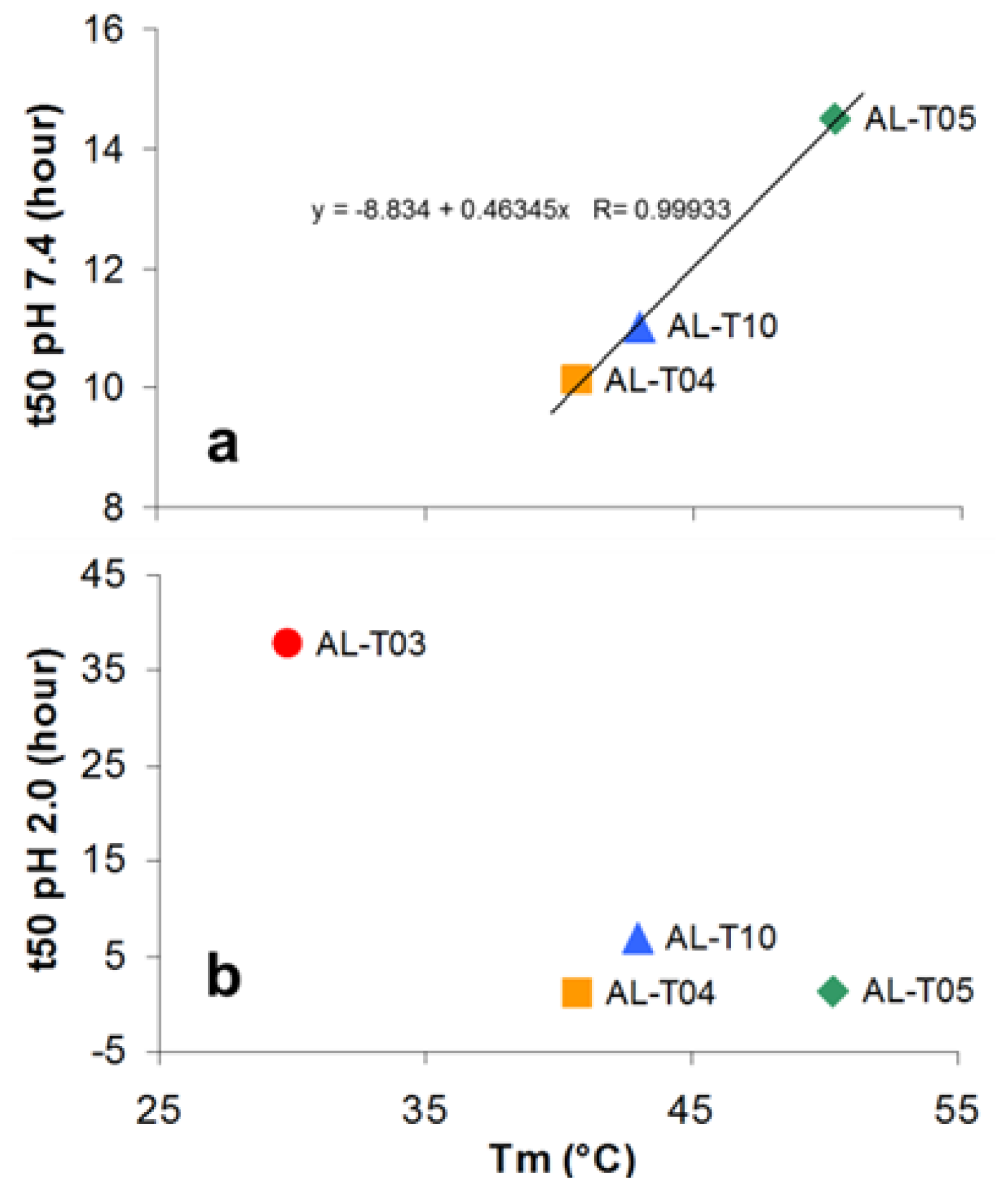
2.2. AL Proteins from Different Patients Present Different Thermal Stability
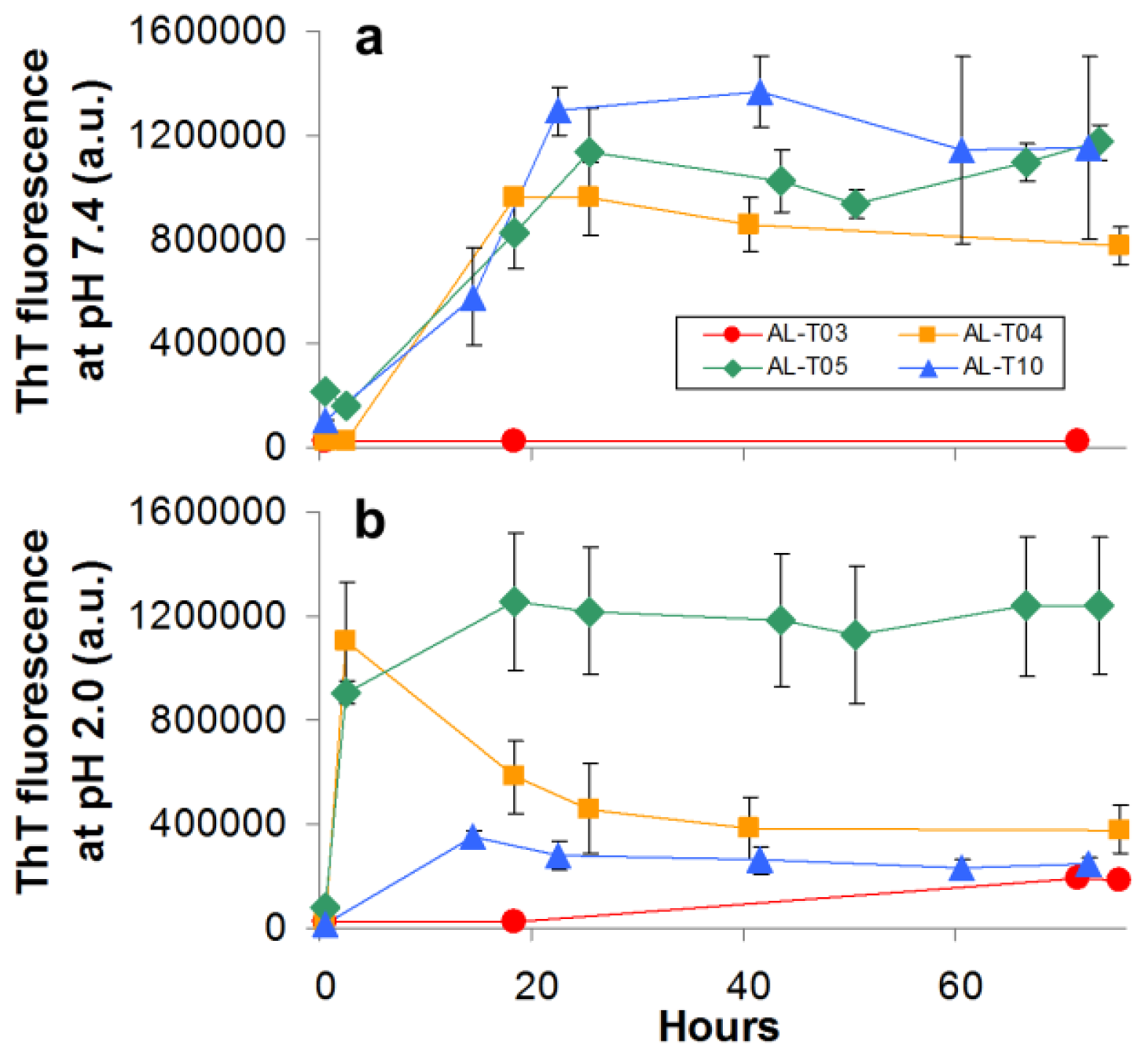
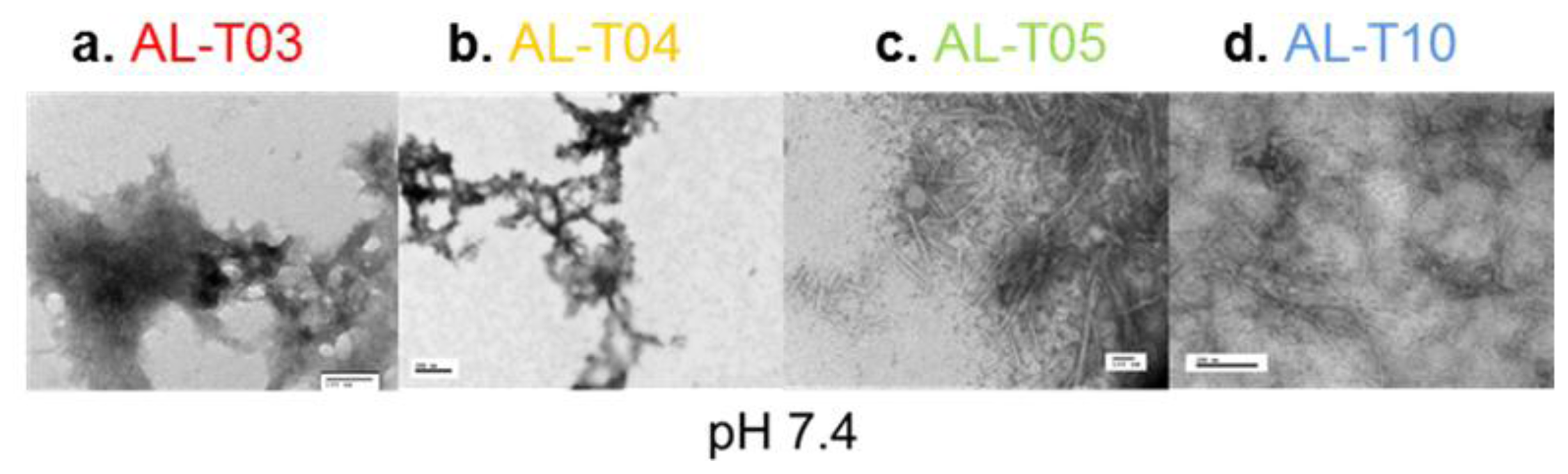
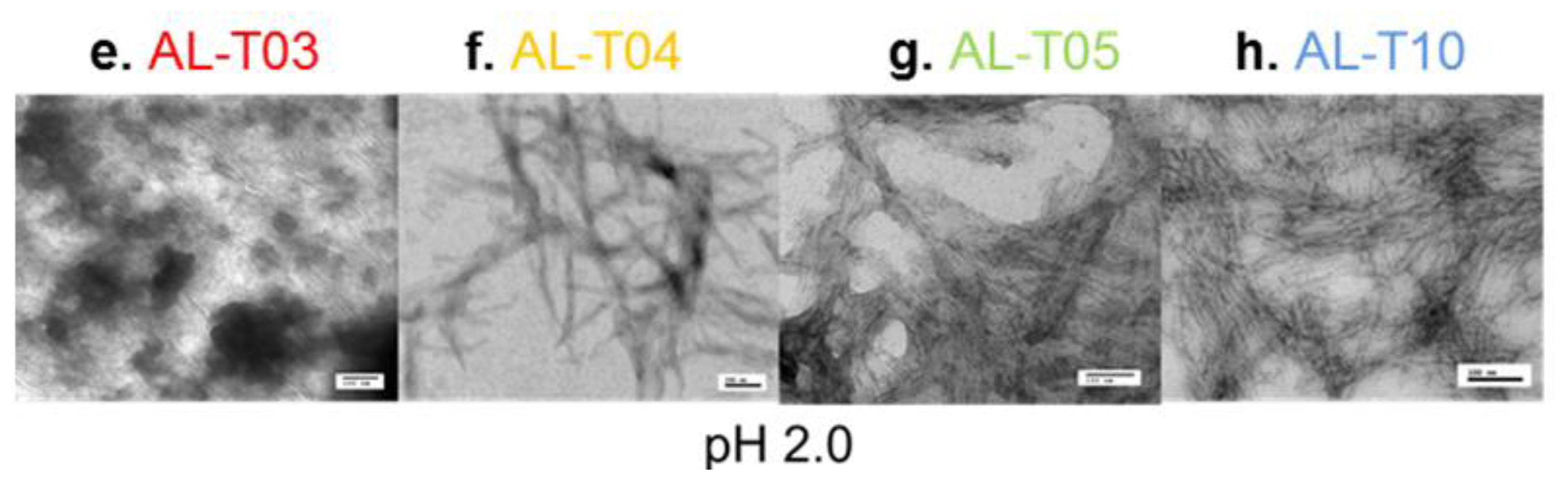
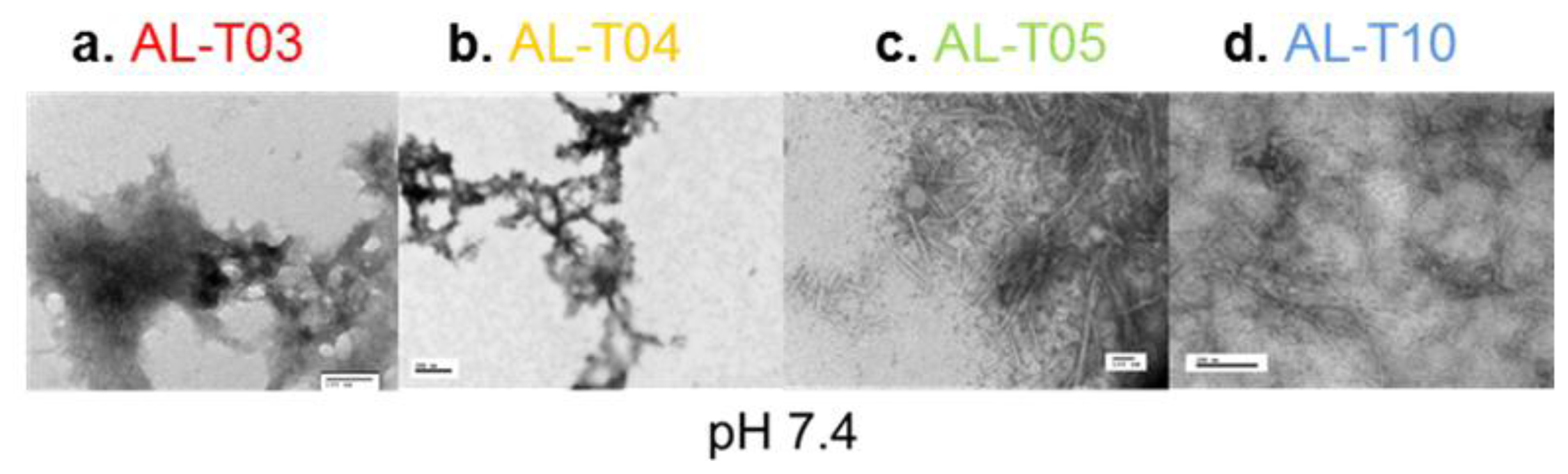
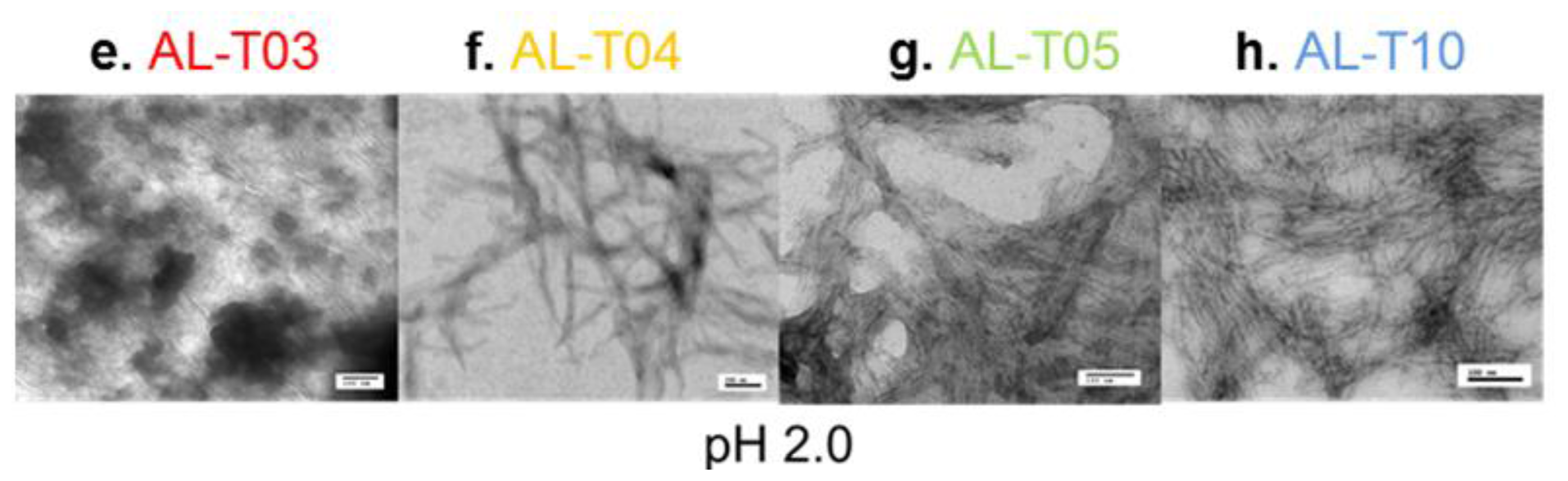
2.3. Amyloid Fibril Formation Kinetics Correlates with Thermal Stability at pH 7.4
- AL-T03 ------ Renal response after one year post transplant
- AL-T04 ------ No renal response after one year post-transplant
- AL-T05 ------ No renal response after one year post-transplant
- AL-T10 ------ No renal response after one year post-transplant
3. Experimental Section
3.1. Patient Eligibility for the Clinical Trial
- i)
- Required Characteristics
- (a)
- Histologic proof of amyloidosis.
- (b)
- The amyloidosis must be of AL type. The patients must have a monoclonal protein by immunoelectrophoresis or immunofixation of the serum or urine or have an elevation in their free light chain ratio [20]. Secondary and familial amyloidosis must be excluded, and available tissues stained with appropriate anti-sera to confirm the type.
- (c)
- Typical amyloid syndromes considered eligible include amyloid hepatomegaly, cardiomyopathy, proteinuria, peripheral or autonomic neuropathy, or soft tissue involvement including the tongue, submandibular tissues, and vascular claudication. Occasional patients with diffuse interstitial pulmonary AL would be eligible if their pulmonary function is adequate to allow safe transplantation.
- (d)
- ≥18 years of age.
- (e)
- Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0, 1, or 2.
- (f)
- The following laboratory values must be obtained ≤8 weeks of registration:
- Platelets (PLT) ≥ 100,000/μL
- Direct bilirubin ≤2.0 × ULN
- Alkaline phosphatase ≤6 × ULN
- Serum creatinine ≤3.0 mg/dL.
- (g)
- Must be previously untreated.
- (h)
- Must have a compensated cardiac status.
- (i)
- New York Heart Association classification I, II, or III.
- ii)
- Contraindications
- (a)
- Multiple myeloma with lytic bone lesions or in excess of 30% plasma cells in the bone marrow.
- (b)
- Must not have amyloidosis that is manifest only by carpal tunnel syndrome or purpura. The presence of amyloid deposits in a plasmacytoma or in bone marrow vessels in an asymptomatic individual does not constitute an amyloid syndrome. Patients with overt multiple myeloma with lytic or destructive bone lesions or myeloma cast nephropathy.
- (c)
- Previous exposure to alkylating agents, immunosuppressive drugs, or steroids.
- (d)
- Infection at the time of registration.
- (e)
- HIV positive.
- (f)
- Any of the following:
- Pregnant women
- Nursing women
- Men or women of childbearing potential who are unwilling to employ adequate contraception (condoms, diaphragm, birth control pills, injections, under-the-skin implants, intrauterine device [IUD], surgical sterilization, abstinence, etc.) This study involves an agent that has known genotoxic, mutagenic, and teratogenic effects.
3.2. Sample Collection
3.3. Identification of the Clonal Ig Light Chain Variable Gene (VL)
3.4. Protein Expression
3.5. Circular Dichroism Spectroscopy
3.6. Amyloid Fibril Formation In Vitro
3.7. Electron Microscopy
4. Conclusions



Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Gertz, M.A. Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin. Hematol 1995, 32, 45–59. [Google Scholar]
- Dispenzieri, A.; Gertz, M.A.; Buadi, F. What do I need to know about immunoglobulin light chain (AL) amyloidosis? Blood Rev 2012, 26, 137–154. [Google Scholar]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2013 update on diagnosis, prognosis, and treatment. Am. J. Hematol 2013, 88, 416–425. [Google Scholar]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J. Clin. Oncol 2012, 30, 989–995. [Google Scholar]
- Gatt, M.E.; Palladini, G. Light chain amyloidosis 2012: A new era. Br. J. Haematol 2013. [Google Scholar] [CrossRef]
- Stevens, F.J. Four structural risk factors identify most fibril-forming kappa light chains. Amyloid 2000, 7, 200–211. [Google Scholar]
- Poshusta, T.L.; Sikkink, L.A.; Leung, N.; Clark, R.J.; Dispenzieri, A.; Ramirez-Alvarado, M. Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One 2009, 4, e5169. [Google Scholar]
- Blancas-Mejia, L.M.; Tellez, L.A.; del Pozo-Yauner, L.; Becerril, B.; Sanchez-Ruiz, J.M.; Fernandez-Velasco, D.A. Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: The relation between unfolding and fibrillogenesis. J. Mol. Biol 2009, 386, 1153–1166. [Google Scholar]
- DiCostanzo, A.C.; Thompson, J.R.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M. Tyrosine residues mediate fibril formation in a dynamic light chain dimer interface. J. Biol. Chem 2012, 287, 27997–28006. [Google Scholar]
- Khurana, R.; Gillespie, J.R.; Talapatra, A.; Minert, L.J.; Ionescu-Zanetti, C.; Millett, I.; Fink, A.L. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 2001, 40, 3525–3535. [Google Scholar]
- McLaughlin, R.W.; De Stigter, J.K.; Sikkink, L.A.; Baden, E.M.; Ramirez-Alvarado, M. The effects of sodium sulfate, glycosaminoglycans, and Congo red on the structure, stability, and amyloid formation of an immunoglobulin light-chain protein. Protein Sci 2006, 15, 1710–1722. [Google Scholar]
- Sikkink, L.A.; Ramirez-Alvarado, M. Salts enhance both protein stability and amyloid formation of an immunoglobulin light chain. Biophys. Chem 2008, 135, 25–31. [Google Scholar]
- Blancas-Mejía, L.M.; Tischer, A.; Thompson, J.R.; Tai, J.; Wang, L.; Auton, M.; Ramirez-Alvarado, M. Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations. J. Mol. Biol 2013. [Google Scholar] [CrossRef]
- Blancas-Mejia, L.M.; Ramirez-Alvarado, M. Systemic amyloidoses. Annu. Rev. Biochem 2013, 82, 745–774. [Google Scholar]
- Martin, D.J.; Ramirez-Alvarado, M. Comparison of amyloid fibril formation by two closely related immunoglobulin light chain variable domains. Amyloid 2010, 17, 129–136. [Google Scholar]
- Wilson, M.R.; Yerbury, J.J.; Poon, S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mole. BioSyst 2008, 4, 42–52. [Google Scholar]
- Ionescu-Zanetti, C.; Khurana, R.; Gillespie, J.R.; Petrick, J.S.; Trabachino, L.C.; Minert, L.J.; Carter, S.A.; Fink, A.L. Monitoring the assembly of Ig light-chain amyloid fibrils by atomic force microscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 13175–13179. [Google Scholar]
- Fujita, H.; Hishizawa, M.; Sakamoto, S.; Kondo, T.; Kadowaki, N.; Ishikawa, T.; Itoh, J.; Fukatsu, A.; Uchiyama, T.; Takaori-Kondo, A. Durable hematological response and improvement of nephrotic syndrome on thalidomide therapy in a patient with refractory light chain deposition disease. Int. J. Hematol 2011, 93, 673–676. [Google Scholar]
- Jimenez-Zepeda, V.H. Light chain deposition disease: Novel biological insights and treatment advances. Int. J. Lab. Hematol 2012, 34, 347–355. [Google Scholar]
- Abraham, R.S.; Katzmann, J.A.; Clark, R.J.; Bradwell, A.R.; Kyle, R.A.; Gertz, M.A. Quantitative analysis of serum free light chains. A new marker for the diagnostic evaluation of primary systemic amyloidosis. Am. J. Clin. Pathol 2003, 119, 274–278. [Google Scholar]
- Baden, E.M.; Owen, B.A.; Peterson, F.C.; Volkman, B.F.; Ramirez-Alvarado, M.; Thompson, J.R. Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem 2008, 283, 15853–15860. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Poshusta, T.L.; Katoh, N.; Gertz, M.A.; Dispenzieri, A.; Ramirez-Alvarado, M. Thermal Stability Threshold for Amyloid Formation in Light Chain Amyloidosis. Int. J. Mol. Sci. 2013, 14, 22604-22617. https://doi.org/10.3390/ijms141122604
Poshusta TL, Katoh N, Gertz MA, Dispenzieri A, Ramirez-Alvarado M. Thermal Stability Threshold for Amyloid Formation in Light Chain Amyloidosis. International Journal of Molecular Sciences. 2013; 14(11):22604-22617. https://doi.org/10.3390/ijms141122604
Chicago/Turabian StylePoshusta, Tanya L., Nagaaki Katoh, Morie A. Gertz, Angela Dispenzieri, and Marina Ramirez-Alvarado. 2013. "Thermal Stability Threshold for Amyloid Formation in Light Chain Amyloidosis" International Journal of Molecular Sciences 14, no. 11: 22604-22617. https://doi.org/10.3390/ijms141122604