Structural Variation of Bamboo Lignin before and after Ethanol Organosolv Pretreatment




Abstract
:1. Introduction
2. Results and Discussion
2.1. Sugar Analysis
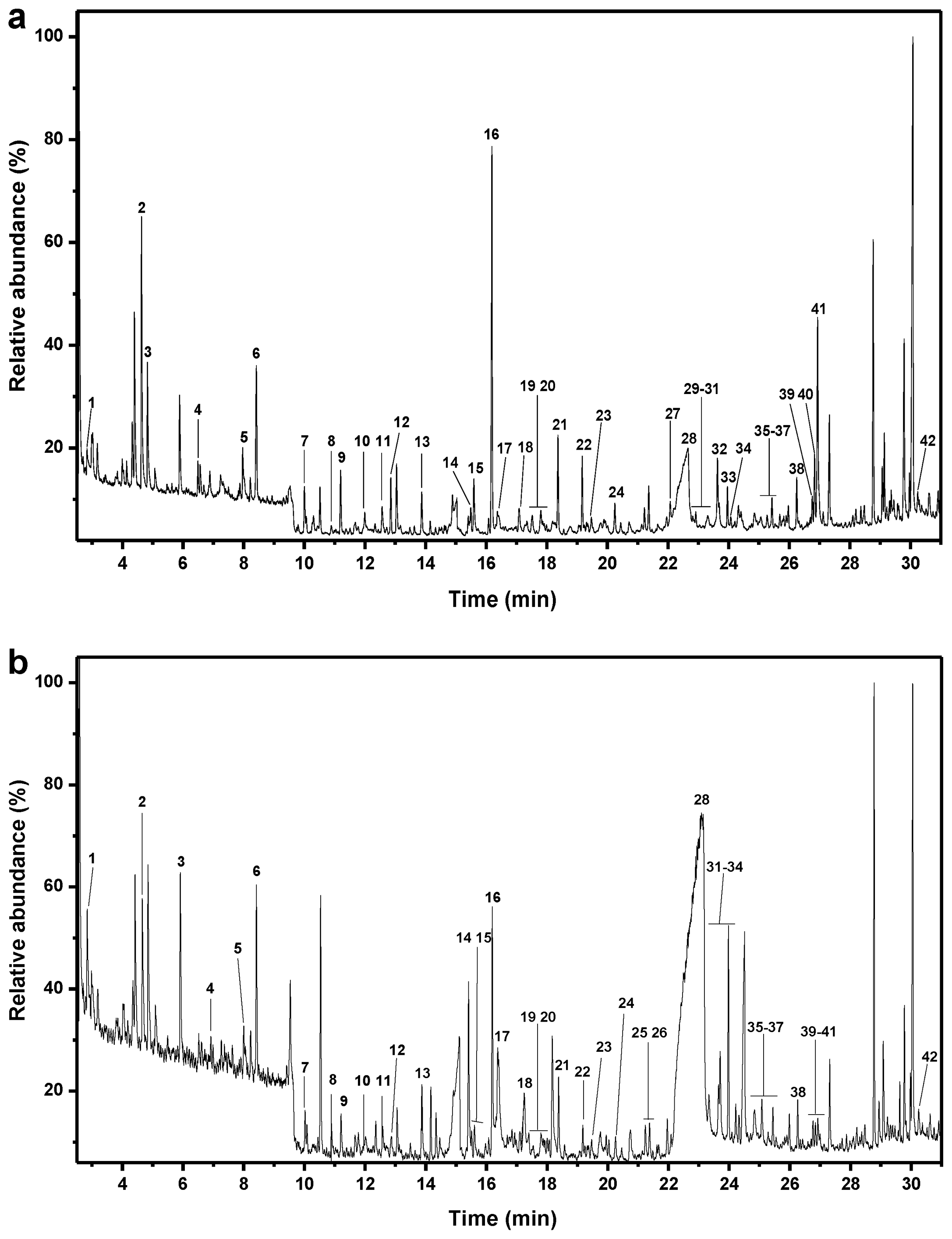
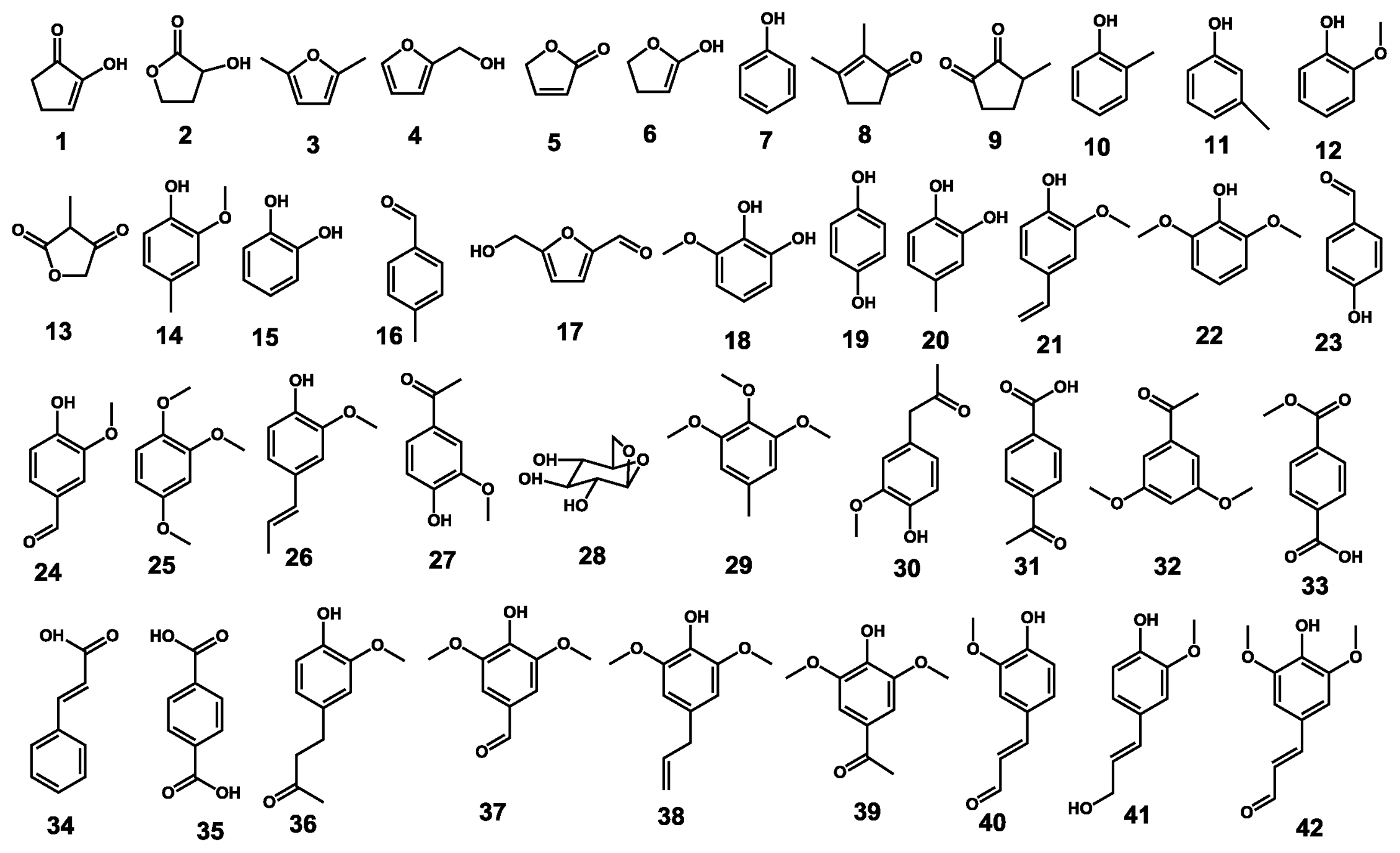
2.2. Pyrolysis-Gas Chromatography/Mass Spectrometry
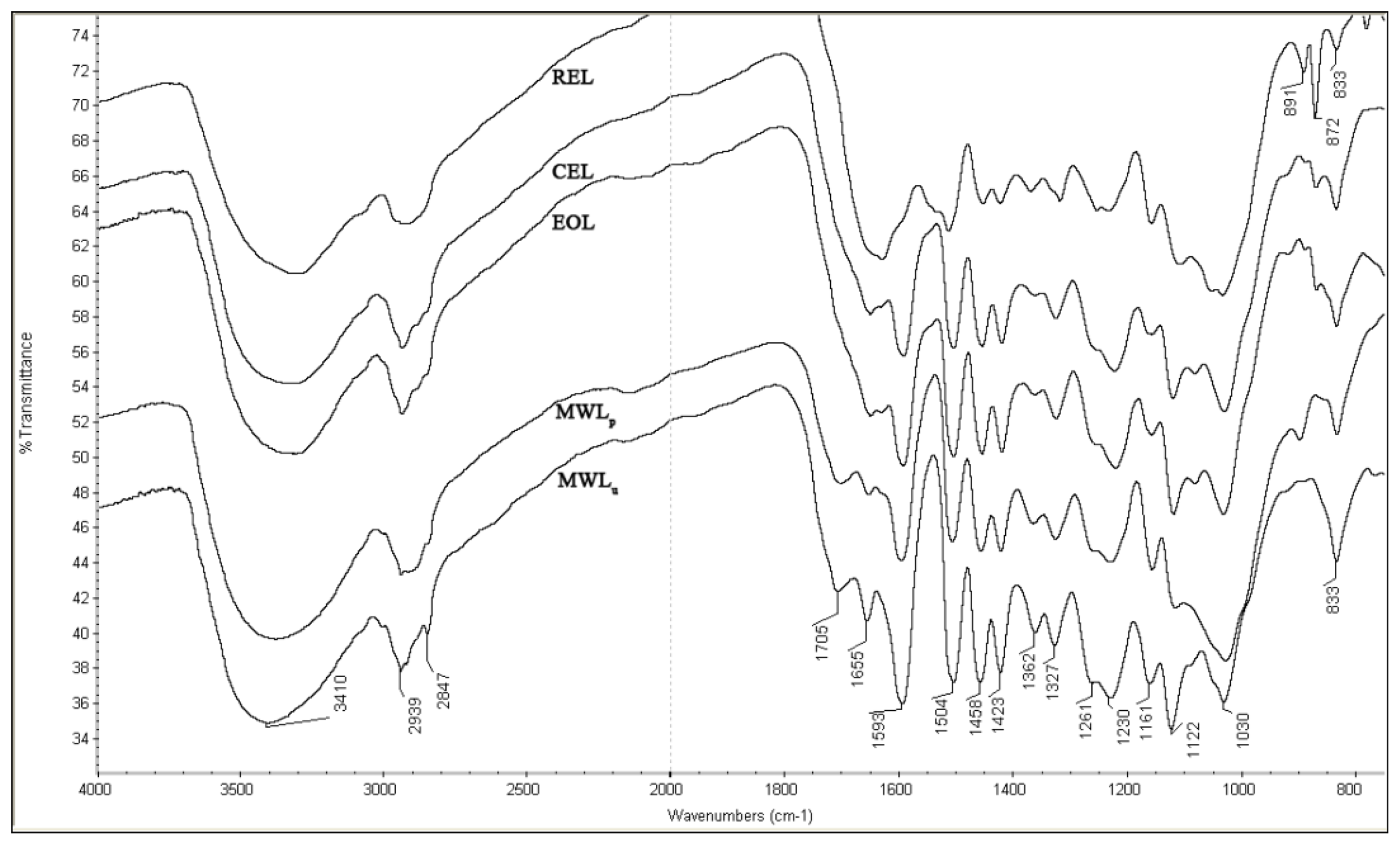
2.3. FT-IR Spectra
2.4. Molecular Weight Distribution
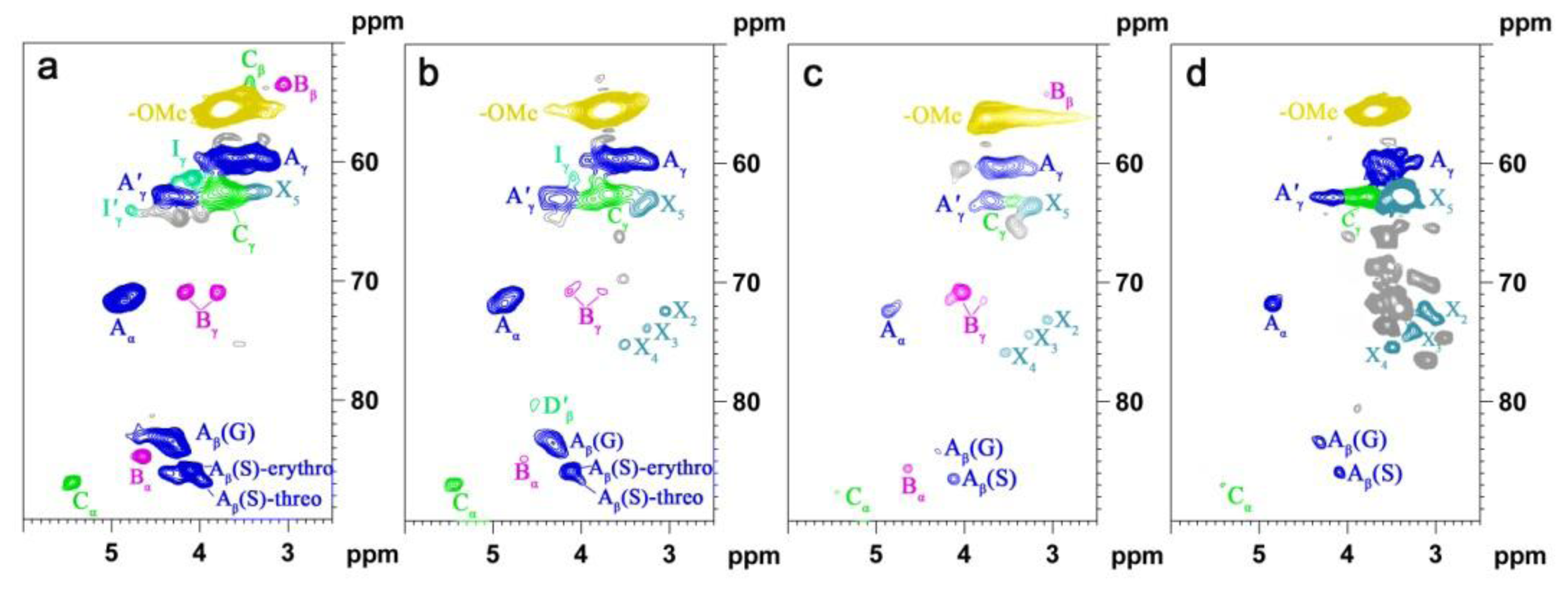
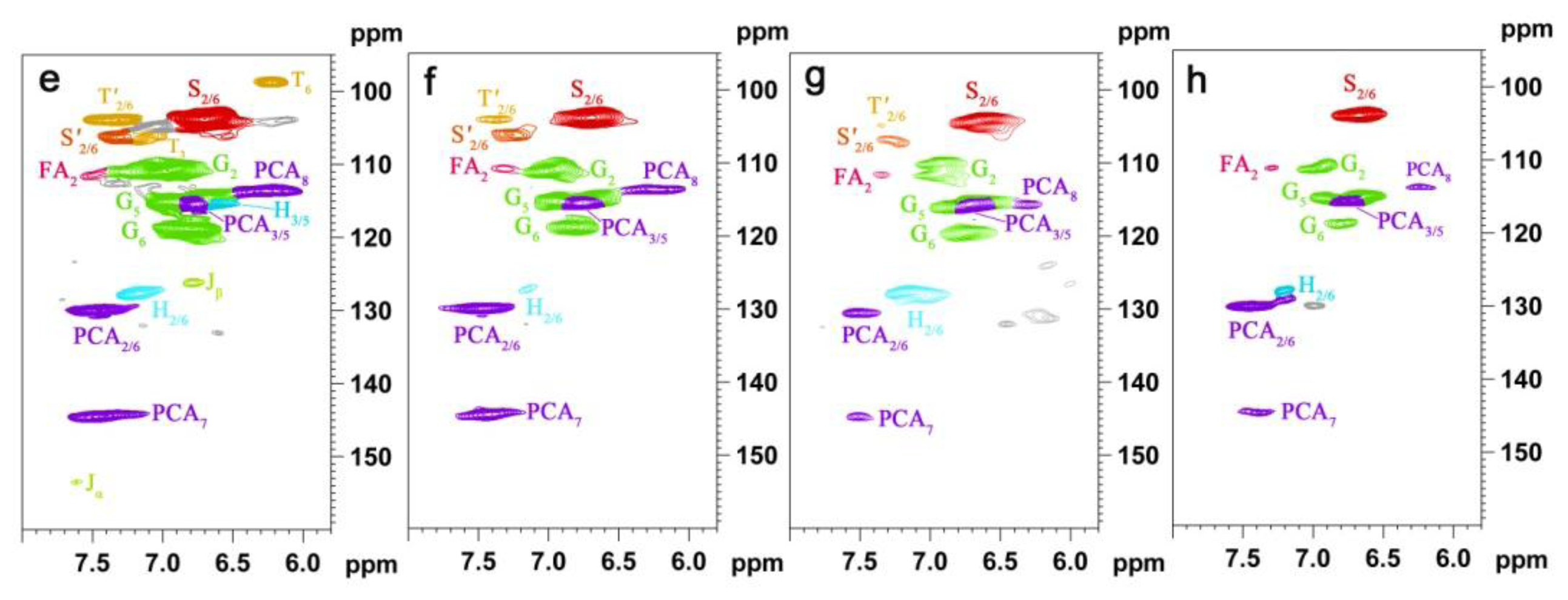
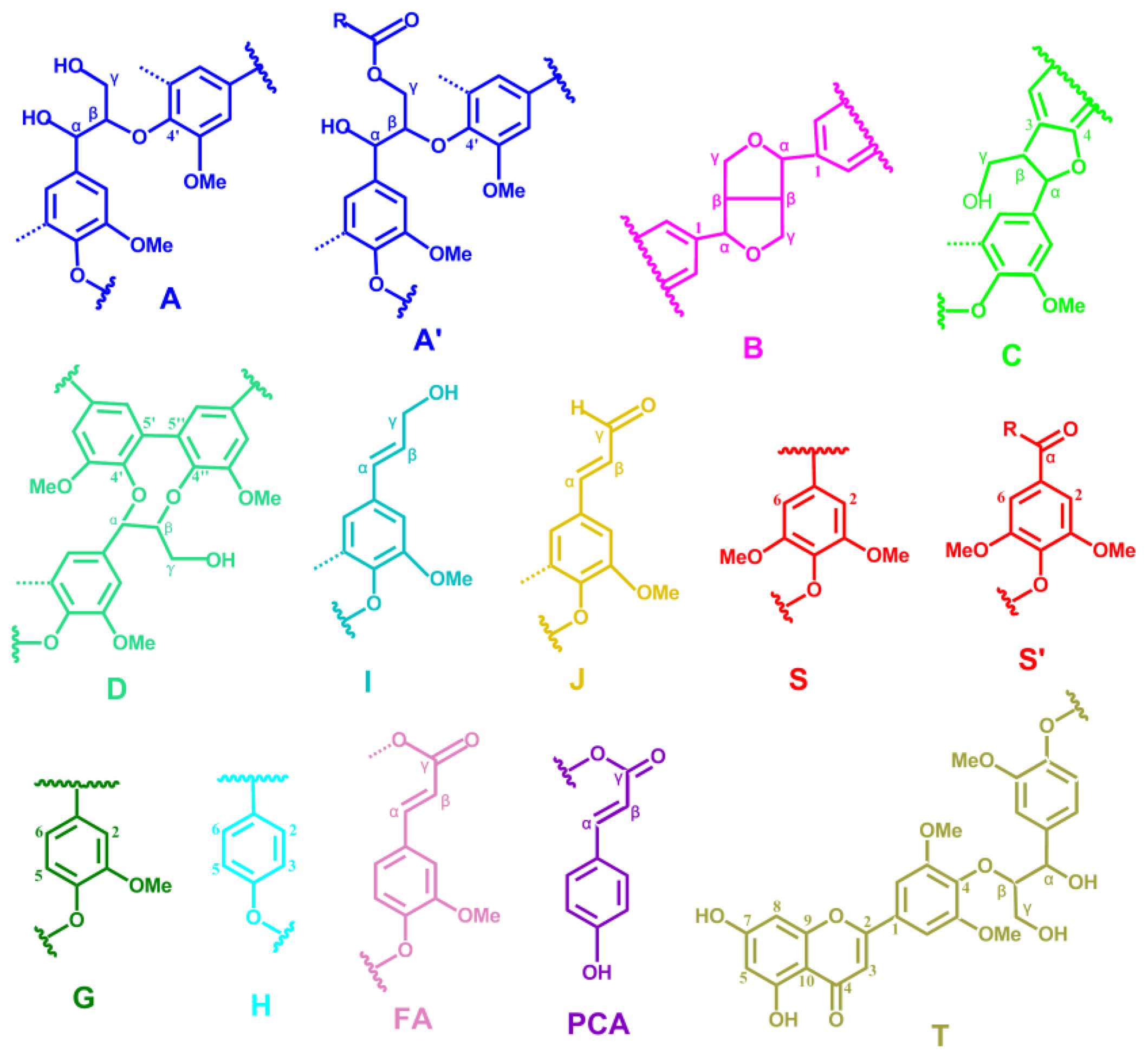
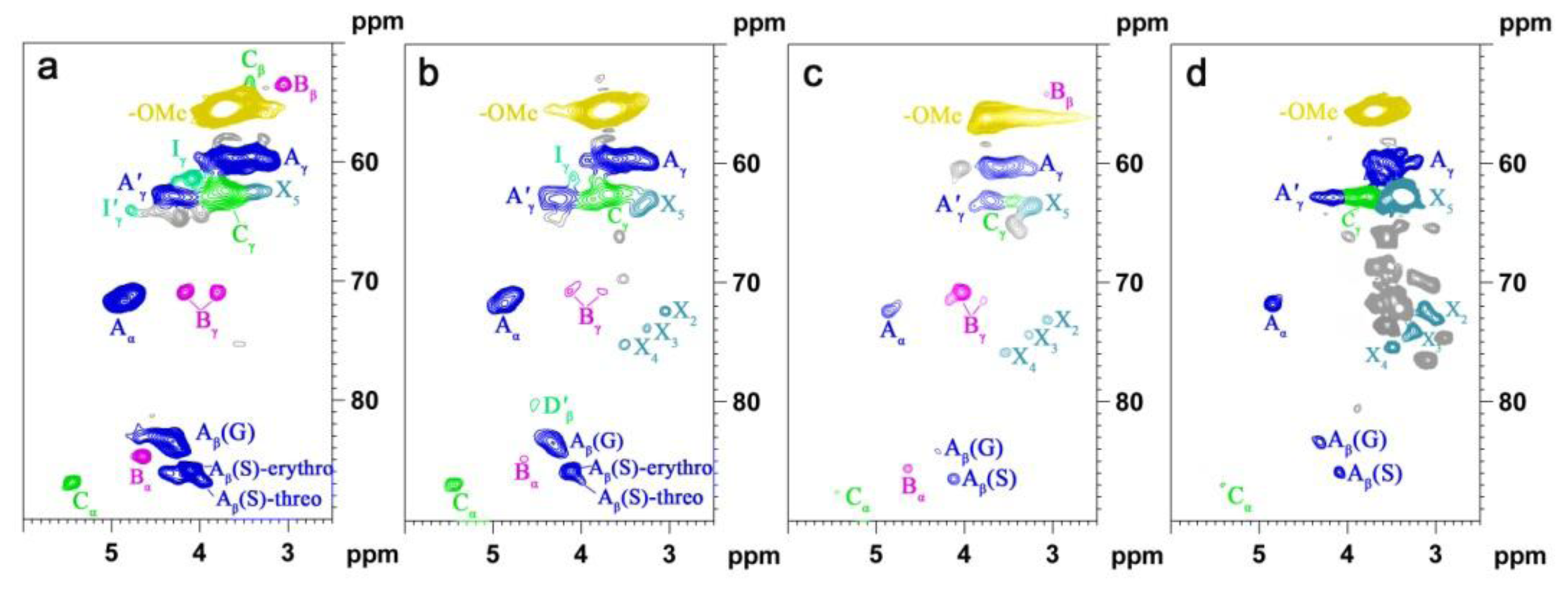
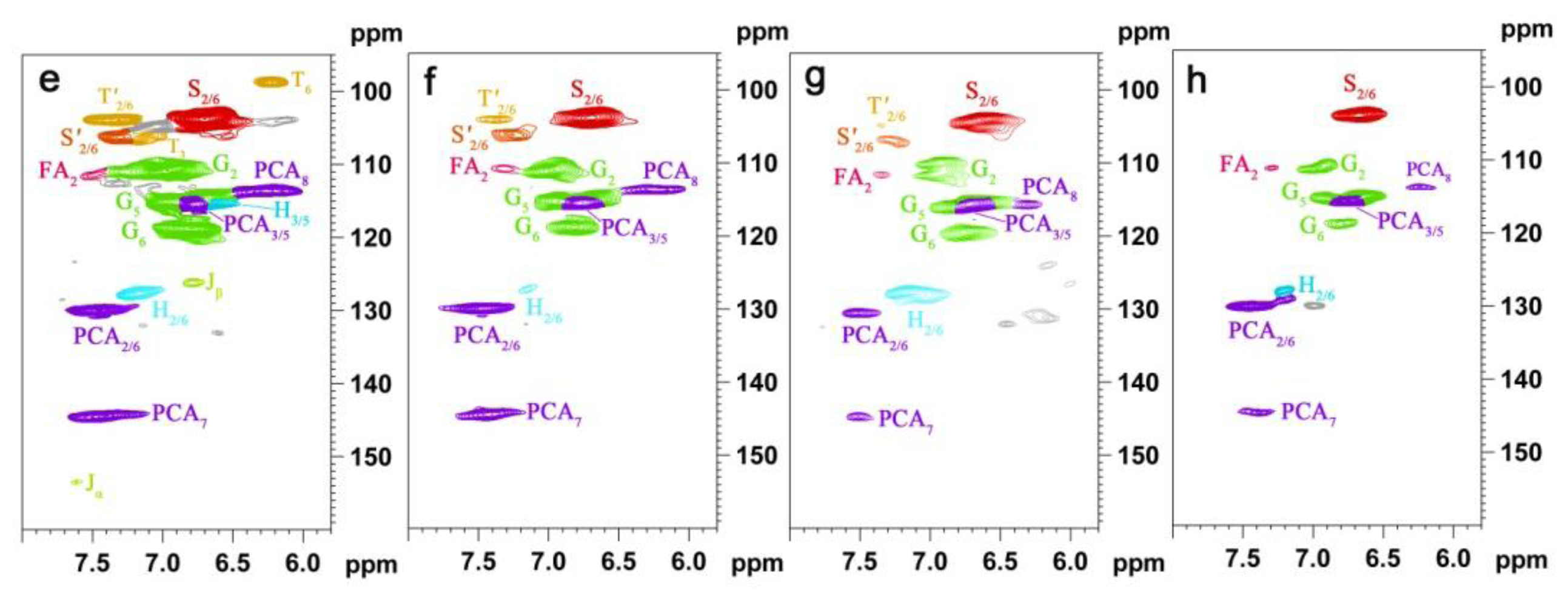
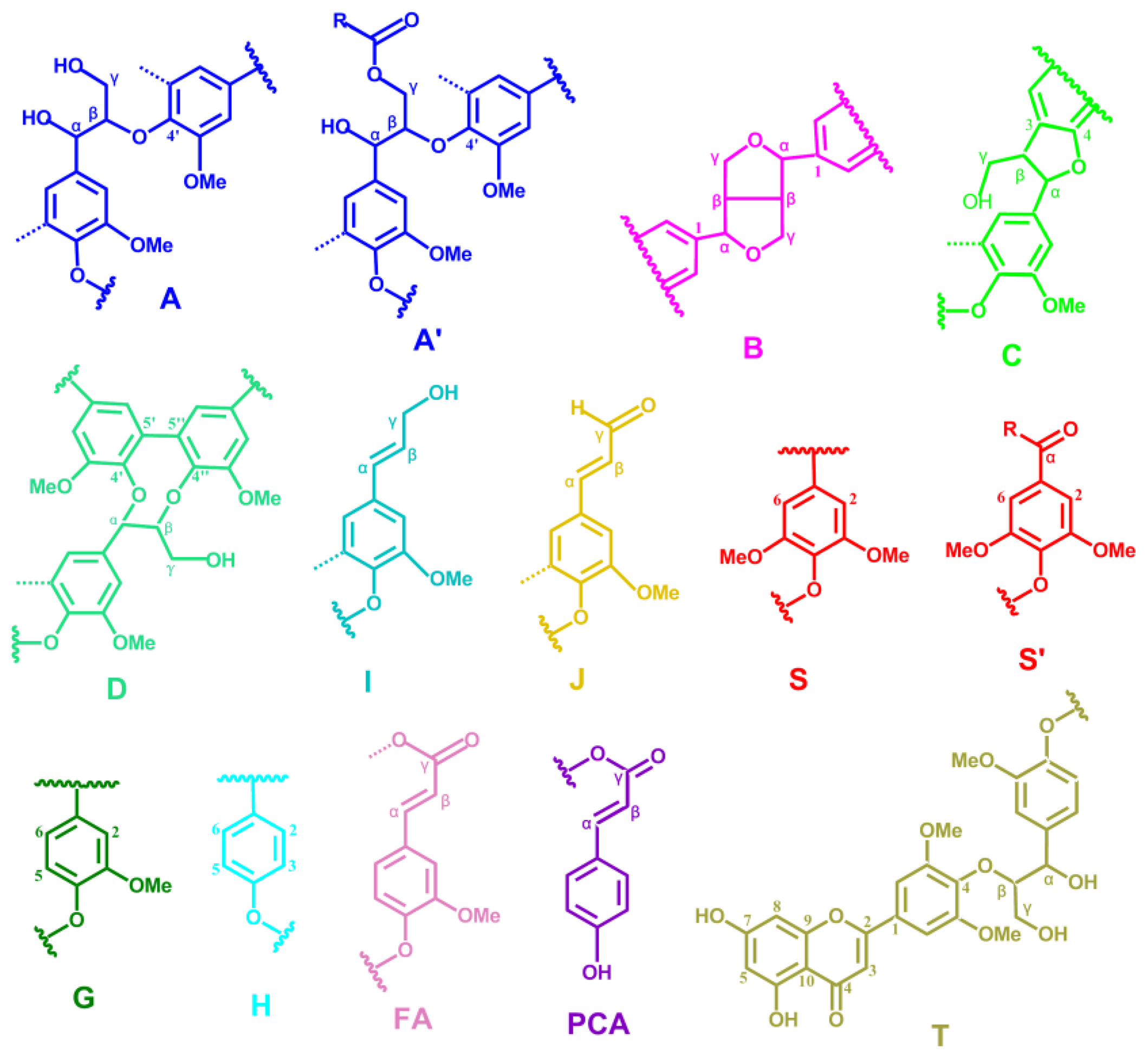
2.5. HSQC NMR Spectra
3. Experimental Section
3.1. Materials
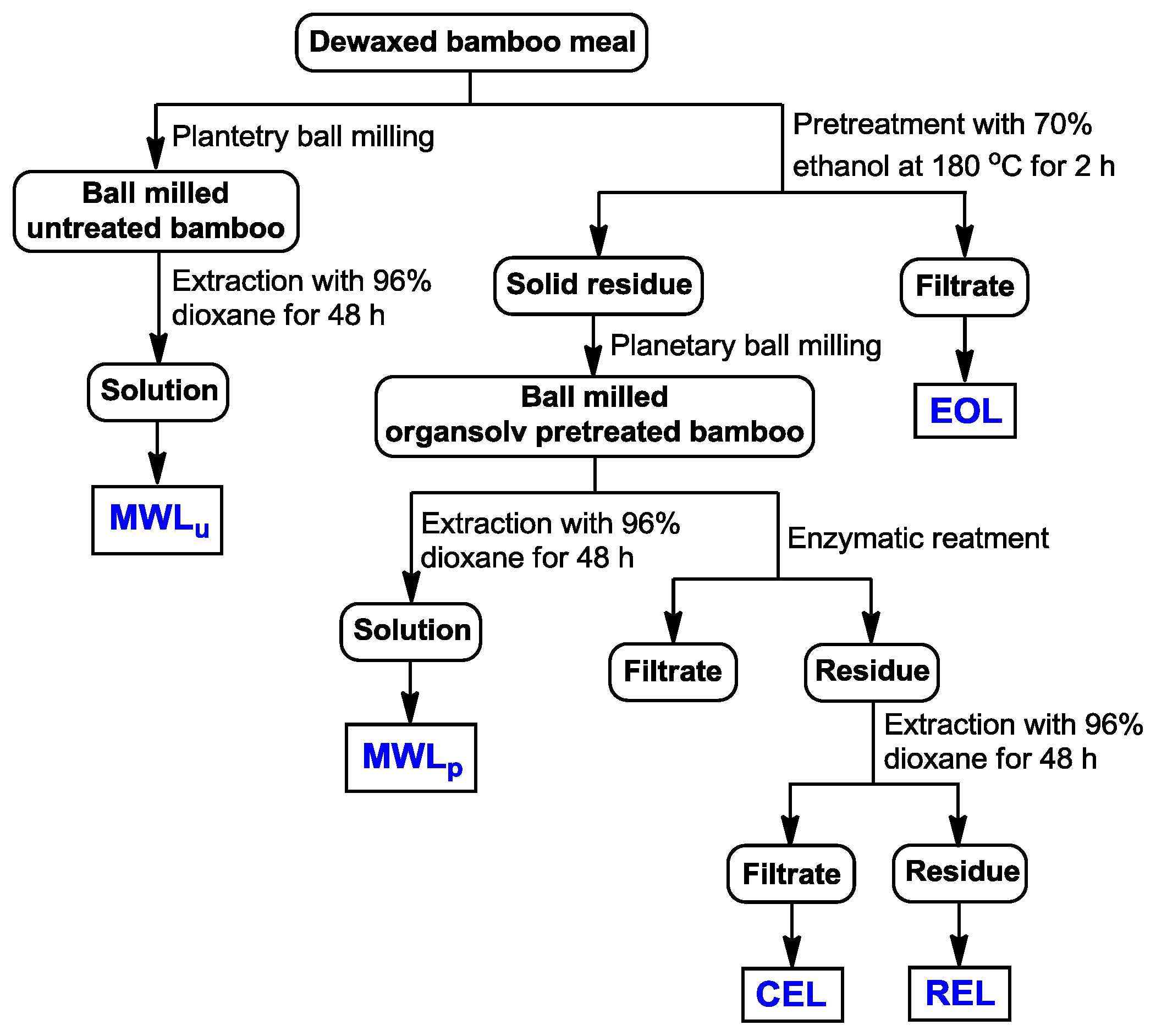
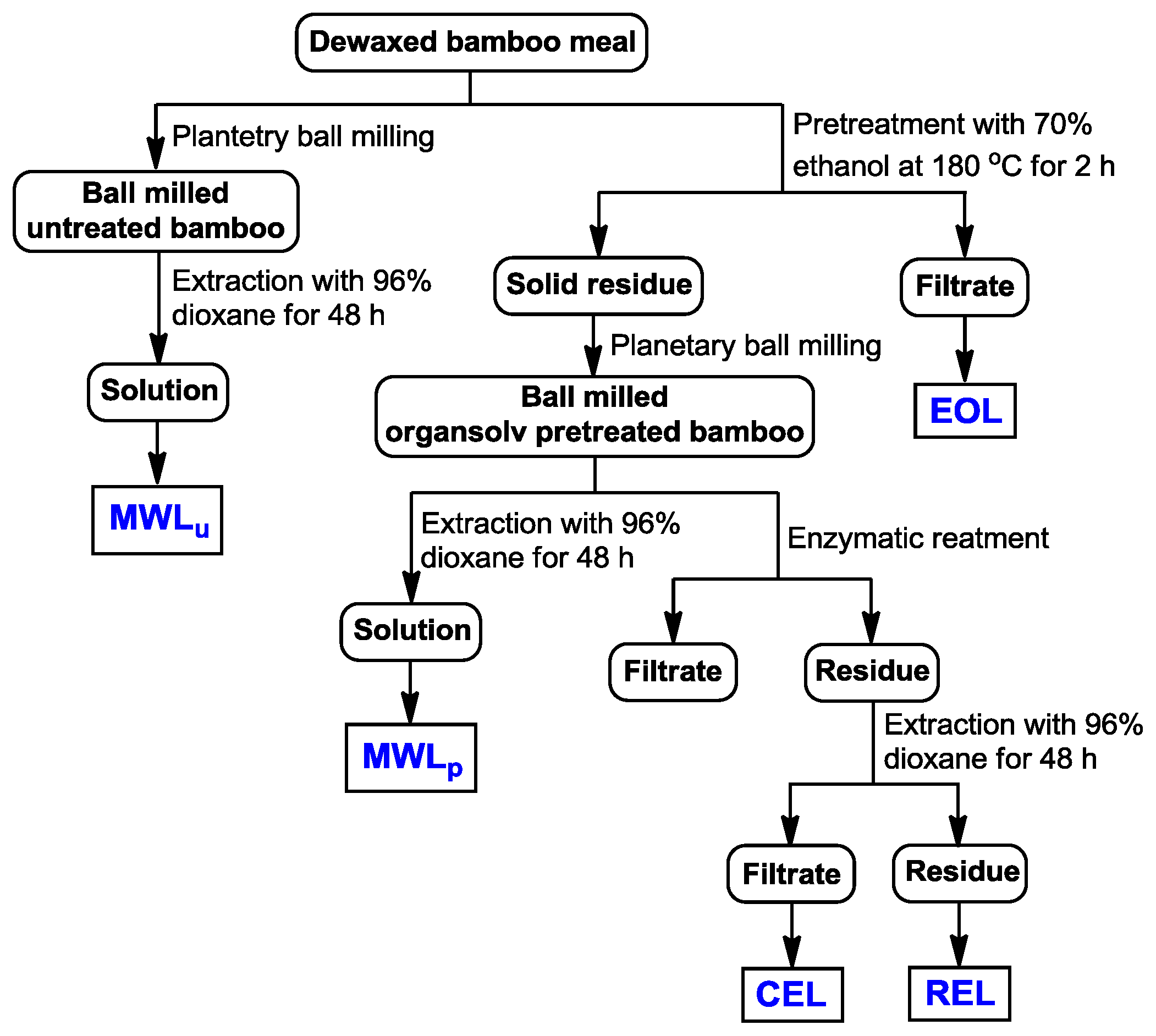
3.2. Isolation of Lignins
3.3. Chemical Composition Analysis
3.4. Analytical Pyrolysis
3.5. FT-IR Analysis
3.6. Molecular Weight Analysis
3.7. NMR Spectra
4. Conclusions
Supplementary Information
ijms-14-21394-s001.pdf



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical composition | Untreated bamboo | Pretreated bamboo | ||||
|---|---|---|---|---|---|---|
| Cellulose (as glucan) | 47.24 ± 1.15 | 52.34 ± 0.32 | ||||
| Hemicellulose sugars | 23.85 ± 1.79 | 23.41 ± 0.03 | ||||
| Xylan | 22.12 ± 1.53 | 22.53 ± 0.03 | ||||
| Arabinan | 1.24 ± 0.23 | 0.68 ± 0.00 | ||||
| Galactan | 0.47 ± 0.05 | 0.20 ± 0.00 | ||||
| Mannan | 0.07 ± 0.00 | ND | ||||
| Galacturonic acid | 0.03 ± 0.01 | ND | ||||
| Klason lignin | 23.84 ± 1.09 | 17.27 ± 0.11 | ||||
| Acid-soluble lignin | 1.51 ± 0.06 | 1.06 ± 0.02 | ||||
| Ash | 1.37 ± 0.03 | 1.16 ± 0.06 | ||||
| Samples | Carb a | Ara a | Gal a | Glc a | Xly a | Man a |
| MWLu | 10.51 ± 0.25 | 0.22 ± 0.05 | 0.10 ± 0.01 | 6.68 ± 0.05 | 3.49 ± 0.07 | Trace |
| MWLp | 8.11 ± 0.87 | 0.04 ± 0.00 | 0.02 ± 0.00 | 6.63 ± 0.71 | 1.43 ± 0.15 | Trace |
| EOL | 5.26 ± 0.42 | 0.58 ± 0.05 | 0.32 ± 0.02 | 1.01 ± 0.11 | 3.35 ± 0.22 | Trace |
| CEL | 12.24 ± 1.24 | 0.83 ± 0.01 | 0.49 ± 0.02 | 5.17 ± 0.12 | 4.79 ± 0.92 | 0.95 ± 0.22 |
| REL | 24.96 ± 1.38 | 0.46 ± 0.03 | 0.92 ± 0.05 | 17.32 ± 0.91 | 4.47 ± 0.30 | 1.47 ± 0.07 |
| Label | R.T. (min) | Compound name | Formula | Mw | Untreated | Pretreated |
|---|---|---|---|---|---|---|
| Carbohydrate derived compounds | ||||||
| 1 | 2.82 | 2-Cyclopenten-1-one, 2-hydroxy- | C5H6O2 | 98 | 0.9 | 1.6 |
| 2 | 4.63 | 2-Hydroxy-gamma-butyrolactone | C4H6O3 | 102 | 6.5 | 2.2 |
| 3 | 5.88 | Furan, 2,5-dimethyl- | C6H8O | 96 | 3.3 | 2.2 |
| 4 | 6.49 | 2-Furanmethanol | C5H6O2 | 98 | 0.8 | 0.5 |
| 5 | 7.97 | 2(5H)-furanone | C4H4O2 | 84 | 2.3 | 0.6 |
| 6 | 8.42 | 2-Cyclopenten-1-one, 2-hydroxy- | C5H6O2 | 98 | 4.5 | 2.3 |
| 8 | 10.89 | 2-Cyclopenten-1-one, 2-hydroxy-3-methyl- | C6H8O2 | 112 | 0.3 | 0.6 |
| 9 | 11.20 | 1,2-Cyclopentanedione, 3-methyl- | C6H8O2 | 112 | 1.8 | 0.6 |
| 13 | 13.87 | 2,4(3H,5H)-furandione, 3-methyl- | C5H6O3 | 114 | 1.4 | 1.1 |
| 17 | 16.38 | 2-Furancarboxaldehyde, 5-(hydroxymethyl)- | C6H6O3 | 126 | 1.1 | 2.3 |
| 28 | 22.67 | β-D-Glucopyranose, 1,6-anhydro- | C6H10O5 | 162 | 21.0 | 65.8 |
| Sum | 43.9 | 79.9 | ||||
| Lignin guaiacyl-type | ||||||
| 10 | 12.00 | Phenol, 2-methyl- | C7H8O | 108 | 1.2 | 0.4 |
| 12 | 12.85 | Phenol, 2-methoxy- | C7H8O2 | 124 | 1.5 | 0.4 |
| 14 | 15.49 | Phenol, 2-methoxy-4-methyl- | C8H10O2 | 138 | 0.9 | 0.3 |
| 15 | 15.59 | 1,2-Benzenediol | C6H6O2 | 110 | 1.8 | 0.5 |
| 20 | 17.80 | 1,2-Benzenediol, 4-methyl- | C7H8O2 | 124 | 0.9 | 0.5 |
| 21 | 18.37 | 2-Methoxy-4-vinylphenol | C9H10O2 | 150 | 2.8 | 0.9 |
| 24 | 20.25 | Vanillin | C8H8O3 | 152 | 1.1 | 0.4 |
| 25 | 21.23 | 1,2,4-Trimethoxybenzene | C9H12O3 | 168 | 0.7 | 0.4 |
| 26 | 21.37 | Phenol, 2-methoxy-4-(1-propenyl)- | C10H12O2 | 164 | 1.2 | 0.4 |
| 27 | 22.07 | Ethanone, 1-(4-hydroxy-3-methoxyphenyl)- | C9H10O3 | 166 | 1.6 | 0.3 |
| 30 | 22.92 | 2-Propanone, 1-(4-hydroxy-3-methoxyphenyl)- | C10H12O3 | 180 | 0.4 | Trace |
| 36 | 25.27 | 2-Butanone, 4-(4-hydroxy-3-methoxyphenyl)- | C11H14O3 | 194 | 0.5 | 0.2 |
| 40 | 26.84 | 2-Propenal, 3-(4-hydroxy-3-methoxyphenyl)- | C10H10O3 | 178 | 1.6 | 0.2 |
| 41 | 26.94 | 4-((1E)-3-Hydroxy-1-propenyl)-2-methoxyphenol | C10H12O3 | 180 | 6.4 | 0.4 |
| Sum | 22.7 | 5.3 | ||||
| Lignin syringyl-type | ||||||
| 18 | 17.08 | 1,2-Benzenediol, 3-methoxy- | C7H8O3 | 140 | 1.1 | 0.3 |
| 22 | 19.17 | Phenol, 2,6-dimethoxy- | C8H10O3 | 154 | 1.9 | 0.4 |
| 29 | 22.82 | Benzene, 1,2,3-trimethoxy-5-methyl- | C10H14O3 | 182 | 0.5 | Trace |
| 32 | 23.63 | 3′,5′-Dimethoxyacetophenone | C10H12O3 | 180 | 2.4 | 0.7 |
| 37 | 25.43 | Benzaldehyde, 4-hydroxy-3,5-dimethoxy- | C9H10O4 | 182 | 0.9 | 0.4 |
| 38 | 26.25 | Phenol, 2,6-dimethoxy-4-(2-propenyl)- | C11H14O3 | 194 | 1.6 | 0.5 |
| 39 | 26.76 | Ethanone, 1-(4-hydroxy-3,5-dimethoxyphenyl)- | C10H12O4 | 196 | 0.7 | 0.3 |
| 43 | 30.25 | 3,5-Dimethoxy-4-hydroxycinnamaldehyde | C11H12O4 | 208 | 0.6 | 0.4 |
| Sum | 9.7 | 3.1 | ||||
| Other lignin derived products | ||||||
| 7 | 10.01 | Phenol | C6H6O | 94 | 1.4 | 0.5 |
| 11 | 12.57 | Phenol, 3-methyl- | C7H8O2 | 108 | 1.0 | 0.5 |
| 16 | 16.19 | 4-Methyl-benzaldehyde | C8H8O | 120 | 10.0 | 2.6 |
| 19 | 17.52 | Hydroquinone | C6H6O2 | 110 | 0.9 | 0.2 |
| 23 | 19.47 | Benzaldehyde, 4-hydroxy- | C7H6O2 | 122 | 0.7 | 0.3 |
| 31 | 23.31 | 4-Acetylbenzoic acid | C9H8O3 | 164 | 0.9 | 1.2 |
| 33 | 23.69 | 1,4-Benzenedicarboxylic acid, methyl ester | C9H8O4 | 180 | 0.9 | 1.3 |
| 34 | 24.07 | trans-Cinnamic acid | C9H8O2 | 148 | 0.3 | 0.1 |
| 35 | 25.07 | 1,4-Benzenedicarboxylic acid | C8H6O4 | 166 | 0.9 | 0.6 |
| Sum | 16.8 | 7.4 | ||||
| S/G | 0.4 | 0.6 | ||||
| Heading | MWLu | MWLp | EOL | CEL |
|---|---|---|---|---|
| Mw (g/mol) | 7692 | 10657 | 5873 | 15307 |
| Mn (g/mol) | 4406 | 5997 | 3072 | 9721 |
| Mw/Mn | 1.75 | 1.78 | 1.91 | 1.57 |
| Labels | δC/δH (ppm) | Assignment |
|---|---|---|
| Lignin cross-signals | ||
| Cβ | 53.2/3.43 | Cβ–Hβ in β-5′ (phenylcoumaran) substructures (C) |
| Bβ | 53.5/3.05 | Cβ–Hβ in β-β′ (resinol) substructures (B) |
| –OMe | 55.9/3.72 | C–H in methoxyls (MeO) |
| Aγ | 60.1/3.22 and 59.67/3.59 | Cγ–Hγ in β-O-4′ substructures (A) and others |
| Iγ | 61.4/4.09 | Cγ–Hγ in cinnamyl alcohol end-groups (I) |
| A′γ | 62.8/4.28 | Cγ–Hγ in γ-acylated β-O-4′ substructures (A′) |
| Cγ | 62.4/3.71 | Cγ–Hγ in β-5′ (phenylcoumaran) substructures (C) |
| I′γ | 64.1/4.77 | Cγ–Hγ in γ-acylated cinnamyl alcohol end-groups (I′) |
| Aα | 72.4/4.85 | Cα–Hα in β-O-4′ substructures (A) |
| Bγ | 71.0/4.17 and 70.9/3.80 | Cγ–Hγ in β-β′ (resinol) substructures (B) |
| Aβ(G) | 83.6/4.30 | Cβ–Hβ in β-O-4′ substructures linked to a guaiacyl unit (A) |
| Cα | 87.1/5.45 | Cα–Hα in β-5′ (phenylcoumaran) substructures (C) |
| Aβ(S) | 85.8/4.10 | Cβ–Hβ in β-O-4′ substructures linked to a syringyl unit (A, erythro) |
| Aβ(S) | 86.2/3.99 | Cβ–Hβ in β-O-4′ substructures linked to a syringyl unit (A, threo) |
| Bα | 84.7/4.65 | Cα–Hα in β-β′ (resinol) substructures (B) |
| T′2/6 | 103.9/7.32 | C′2,6–H′2,6 in tricin (T) |
| T3 | 106.1/7.04 | C3–H3 in tricin (T) |
| T6 | 98.8/6.22 | C2,6–H2,6 in tricin (T) |
| S2/6 | 104.3/6.70 | C2,6–H2,6 in syringyl units (S) |
| S′2/6 | 106.3/7.30 | C2,6–H2,6 in oxidized (CαOOH) syringyl units (S′) |
| G2 | 111.1/6.97 | C2–H2 in guaiacyl units (G) |
| G5 | 115.8/6.69 | C5–H5 and C6–H6 in guaiacyl units (G) |
| G6 | 119.1/6.79 | C6–H6 in guaiacyl units (G) |
| PCA7 | 144.5/7.43 | C7–H7 in p-coumaroylated substructures (PCA) |
| PCA2/6 | 130.2/7.46 | C2.6–H2.6 in p-coumaroylated substructures (PCA) |
| PCA3/5 | 115.4/6.76 | C3–H3 and C5–H5 in p-coumaroylated substructures (PCA) |
| PCA8 | 113.6/6.26 | C8–H8 in p-coumaroylated substructures (PCA) |
| FA2 | 111.5/7.49 | C2–H2 in ferulate (FA) |
| H2/6 | 128.0/7.17 | C2.6–C2.6 in p-hydroxyphenyl units (H) |
| H3/5 | 115.2/6.57 | C3.5–C3.5 in p-hydroxyphenyl units (H) |
| Jα | 153.5/7.61 | Cα–Hα in cinnamyl aldehyde end-groups (J) |
| Jβ | 126.2/6.79 | Cβ–Hβ in cinnamyl aldehydes end-groups (J) |
| D′β | 80.3/4.54 | C′β–H′β in spirodienone substructure (D) |
| Polysaccharide cross-signals | ||
| X2 | 70.1/3.33 | C2–H2 in β-d-xylopyranoside |
| X3 | 72.0/3.42 | C3–H3 in β-d-xylopyranoside |
| X4 | 75.3/3.54 | C4–H4 in β-d-xylopyranoside |
| X5 | 62.8/3.40 | C5–H5 in β-d-xylopyranoside |
| MWLu (%) | MWLp (%) | EOL (%) | CEL (%) | |
|---|---|---|---|---|
| Lignin interunit linkages | ||||
| β-O-4′ substructure (A) | 89.4 | 82.1 | 72.3 | 94.5 |
| β-β′ resinol substructures (B) | 5.5 | 2.6 | 20.0 | 0 |
| β-5′ phenylcoumaran substructures (C) | 5.1 | 15.3 | 7.7 | 5.5 |
| Lignin aromatic units | ||||
| H | 3.5 | – | 19.6 | 8.0 |
| G | 49.5 | 48.5 | 42.4 | 47.5 |
| S | 47.0 | 51.5 | 38.0 | 44.5 |
| S/G ratio | 0.95 | 1.06 | 0.90 | 0.94 |
| p-Hydroxycinnamates | ||||
| p-Coumarates | 97.5 | 84.9 | 82.1 | 76.6 |
| Ferulates | 9.3 | 15.1 | 17.9 | 23.4 |
| p-Coumarates/ferulates ratio | 9.75 | 5.62 | 4.59 | 3.27 |
Acknowledgments
Conflicts and Interest
References
- Samuel, R.; Pu, Y.; Raman, B.; Ragauskas, A.J. Structural characterization and comparison of switchgrass ball-milled lignin before and after dilute acid pretreatment. Appl. Biochem. Biotechnol 2010, 162, 62–74. [Google Scholar]
- del Río, J.C.; Prinsen, P.; Rencoret, J.; Nieto, L.; Jiménez-Barbero, J.; Ralph, J.; Martínez, Á.T.; Gutiérrez, A. Structural characterization of the lignin in the cortex and pith of elephant grass (Pennisetum purpureum) stems. J. Agric. Food. Chem 2012, 60, 3619–3634. [Google Scholar]
- Xu, F.; Yu, J.M.; Tesso, T.; Dowell, F.; Wang, D.H. Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: A mini-review. Appl. Energy 2013, 104, 801–809. [Google Scholar]
- Gao, A.H.; Bule, M.V.; Laskar, D.D.; Chen, S. Structural and thermal characterization of wheat straw pretreated with aqueous ammonia soaking. J. Agric. Food. Chem 2012, 60, 8632–8639. [Google Scholar]
- Guerra, A.; Filpponen, I.; Lucia, L.A.; Argyropoulos, D.S. Comparative evaluation of three lignin isolation protocols for various wood species. J. Agric. Food Chem 2006, 54, 9696–9705. [Google Scholar]
- Sasaki, C.; Wanaka, M.; Takagi, H.; Tamura, S.; Asada, C.; Nakamura, Y. Evaluation of epoxy resins synthesized from steam-exploded bamboo lignin. Ind. Crop. Prod 2013, 43, 757–761. [Google Scholar]
- Shi, Z.J.; Xiao, L.P.; Xu, F.; Sun, R.C. Physicochemical characterization of lignin fractions sequentially isolated from bamboo (Dendrocalamus brandisii) with hot water and alkaline ethanol solution. J. Appl. Polym. Sci 2012, 125, 3290–3301. [Google Scholar]
- Obama, P.; Ricochon, G.; Muniglia, L.; Brosse, N. Combination of enzymatic hydrolysis and ethanol organosolv pretreatments: Effect on lignin structures, delignification yields and cellulose-to-glucose conversion. Bioresour. Technol 2012, 112, 156–163. [Google Scholar]
- Romaní, A.; Garrote, G.; López, F.; Parajó, J.C. Eucalyptus globulus wood fractionation by autohydrolysis and organosolv delignification. Bioresour. Technol 2011, 102, 5896–5904. [Google Scholar]
- El Hage, R.; Perrin, D.; Brosse, N. Effect of the pre-treatment severity on the antioxidant properties of ethanol organosolv Miscanthus x giganteus lignin. Nature Resour 2012, 3, 29–34. [Google Scholar]
- Hu, G.; Cateto, C.; Pu, Y.; Samuel, R.; Ragauskas, A.J. Structural characterization of switchgrass lignin after ethanol organosolv pretreatment. Energy Fuels 2011, 26, 740–745. [Google Scholar]
- Bauer, S.; Sorek, H.; Mitchell, V.D.; Ibáñez, A.B.; Wemmer, D.E. Characterization of Miscanthus giganteus lignin isolated by ethanol organosolv process under reflux condition. J. Agric. Food Chem 2012, 60, 8203–8212. [Google Scholar]
- Chang, H.; Cowling, E.B.; Brown, W. Comparative studies on cellulolytic enzyme lignin and milled wood lignin of sweetgum and spruce. Holzforschung 1975, 29, 153–159. [Google Scholar]
- Shi, Z.J.; Xiao, L.P.; Deng, J.; Xu, F.; Sun, R.C. Isolation and characterization of soluble polysaccharides ofDendrocalamus brandisii. BioResources 2011, 6, 5151–5166. [Google Scholar]
- Zhang, A.; Lu, F.; Sun, R.C.; Ralph, J. Isolation of cellulolytic enzyme lignin from wood preswollen/dissolved in dimethyl sulfoxide/N-methylimidazole. J. Agric. Food Chem 2010, 58, 3446–3450. [Google Scholar]
- Yuan, T.Q.; Sun, S.N.; Xu, F.; Sun, R.C. Structural characterization of lignin from triploid of Populus tomentosa Carr. J. Agric. Food Chem 2011, 59, 6605–6615. [Google Scholar]
- Mihalcik, D.J.; Mullen, C.A.; Boateng, A.A. Screening acidic zeolites for catalytic fast pyrolysis of biomass and its components. J. Anal. Appl. Pyrolysis 2011, 92, 224–232. [Google Scholar]
- Del Río, J.C.; Gutiérrez, A.; Rodríguez, I.M.; Ibarra, D.; Martinez, A.T. Composition of non-woody plant lignins and cinnamic acids by Py-GC/MS, Py/TMAH and FT-IR. J. Anal. Appl. Pyrolysis 2007, 79, 39–46. [Google Scholar]
- Lima, M.A.; Lavorente, G.B.; da Silva, H.K.P.; Bragatto, J.; Rezende, C.A.; Bernardinelli, O.D.; de Azevedo, E.R.; Gomez, L.D.; McQueen-Mason, S.J.; Labate, C.A.; et al. Effects of pretreatment on morphology, chemical composition and enzymatic digestibility of eucalyptus bark: A potentially valuable source of fermentable sugars for biofuel production—Part 1. Biotechnol. Biofuels 2013, 6, 1–17. [Google Scholar]
- Peng, P.; Peng, F.; Bian, J.; Xu, F.; Sun, R.C. A comparative study of bamboo (Phyllostachys incarnata Wen) milled wood lignin and the successively alkali-fractionated lignins. Wood Sci. Technol 2012, 46, 871–885. [Google Scholar]
- Faix, O. Classification of lignins from different botanical origins by FT-IR spectroscopy. Holzforschung 2009, 45, 21–22. [Google Scholar]
- Xiao, L.P.; Shi, Z.J.; Xu, F.; Sun, R.C. Characterization of MWLs from Tamarix ramosissima isolated before and after hydrothermal treatment by spectroscopical and wet chemical methods. Holzforschung 2012, 66, 295–302. [Google Scholar]
- Kim, H.; Ralph, J. Solution-state 2D NMR of ball-milled plant cell wall gels in DMSO-d6/pyridine-d5. Org. Biomol. Chem 2010, 8, 576–591. [Google Scholar]
- Samuel, R.; Foston, M.; Jiang, N.; Allison, L.; Ragauskas, A.J. Structural changes in switchgrass lignin and hemicelluloses during pretreatments by NMR analysis. Polym. Degrad. Stab 2011, 96, 2002–2009. [Google Scholar]
- Vanderghem, C.; Richel, A.; Jacquet, N.; Blecker, C.; Paquot, M. Impact of formic/acetic acid and ammonia pre-treatments on chemical structure and physico-chemical properties of Miscanthus x giganteus lignins. Polym. Degrad. Stab 2011, 96, 1761–1770. [Google Scholar]
- Rencoret, J.; Marques, G.; Gutiérrez, A.; Nieto, L.; Jiménez-Barbero, J.; Martínez, Á.T.; del Río, J.C. Isolation and structural characterization of the milled-wood lignin from Paulownia fortunei wood. Ind. Crop. Prod 2009, 30, 137–143. [Google Scholar]
- El Hage, R.; Brosse, N.; Chrusciel, L.; Sanchez, C.; Sannigrahi, P.; Ragauskas, A. Characterization of milled wood lignin and ethanol organosolv lignin from miscanthus. Polym. Degrad. Stab. 2009, 94, 1632–1638. [Google Scholar]
- Yelle, D.J.; Kaparaju, P.; Hunt, C.G.; Hirth, K.; Kim, H.; Ralph, J.; Felby, C. Two-imensional NMR evidence for cleavage of lignin and xylan substituents in wheat straw through hydrothermal pretreatment and enzymatic hydrolysis. Bioenerg. Res 2013, 6, 211–221. [Google Scholar]
- Zhou, J.M.; Ibrahim, R.K. Tricin—A potential multifunctional nutraceutical. Phytochem. Rev 2010, 9, 413–424. [Google Scholar]
- Del Río, J.C.; Rencoret Pazo, J.; Prinsen, P.; Martínez Ferrer, Á.T.; Ralph, J.; Gutiérrez Suárez, A. Structural characterization of wheat straw lignin as revealed by analytical pyrolysis, 2D-NMR, and reductive cleavage methods. J. Agric. Food Chem 2012, 60, 5922–5953. [Google Scholar]
- Gutiérrez, A.; del Río, J.C.; Ibarra, D.; Rencoret, J.; Romero, J.; Speranza, M.; Camarero, S.; Martínez, M.J.; Martínez, Á.T. Enzymatic removal of free and conjugated sterols forming pitch deposits in environmentally sound bleaching of eucalypt paper pulp. Environ. Sci. Technol 2006, 40, 3416–3422. [Google Scholar]
- Li, M.; Foster, C.; Kelkar, S.; Pu, Y.; Holmes, D.; Ragauskas, A.; Saffron, C.M.; Hodge, D.B. Structural characterization of alkaline hydrogen peroxide pretreated grasses exhibiting diverse lignin phenotypes. Biotechnol. Biofuels 2012, 5, 1–15. [Google Scholar]
- Björkman, A. Studies on finely divided wood. Part 1. Extraction of lignin with neutral solvents. Svensk. Papperst 1956, 59, 477–485. [Google Scholar]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of Structural Carbohydrates and Lignin in Biomass; Technical Report for Laboratory Analytical Procedure (LAP); NREL: Golden, CO, USA, April 2008. [Google Scholar]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D. Determination of Ash in Biomass; Technical Report for Laboratory Analytical Procedure (LAP); NREL: Golden, CO, USA, January 2008. [Google Scholar]
- Xiao, L.P.; Shi, Z.J.; Xu, F.; Sun, R.C. Characterization of lignins isolated with alkaline ethanol from the hydrothermal pretreatedTamarix ramosissima. Bioenerg. Res 2013, 6, 519–532. [Google Scholar]
- Ralph, J.; Hatfield, R.D. Pyrolysis-GC-MS characterization of forage materials. J. Agric. Food Chem 1991, 39, 1426–1437. [Google Scholar]
- Del Río, J.C.; Rencoret, J.; Gutiérrez, A.; Nieto, L.; Jiménez-Barbero, J.; Martínez, Á.T. Structural characterization of guaiacyl-rich lignins in flax (Linum usitatissimum) fibers and shives. J. Agric. Food Chem 2011, 59, 11088–11099. [Google Scholar]
- Nunes, C.A.; Lima, C.F.; Barbosa, L.C.; Colodette, J.L.; Gouveia, A.; Silvério, F.O. Determination of Eucalyptus spp lignin S/G ratio: A comparison between methods. Bioresour. Technol 2010, 101, 4056–4061. [Google Scholar]
- Gellerstedt, G. Gel Permeation Chromatography. In Methods in Lignin Chemistry; Stephen, Y.L., Carlton, W.D., Eds.; Springer-Verlag: Heidelberg, Gernmany, 1992; pp. 487–497. [Google Scholar]
- Kang, S.M.; Xiao, L.P.; Meng, L.Y.; Zhang, X.M.; Sun, R.C. Isolation and structural characterization of ligin from cotton stalk treated in an ammonia hydrothermal system. Int. J. Mol. Sci 2012, 13, 15209–15226. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bai, Y.-Y.; Xiao, L.-P.; Shi, Z.-J.; Sun, R.-C. Structural Variation of Bamboo Lignin before and after Ethanol Organosolv Pretreatment. Int. J. Mol. Sci. 2013, 14, 21394-21413. https://doi.org/10.3390/ijms141121394
Bai Y-Y, Xiao L-P, Shi Z-J, Sun R-C. Structural Variation of Bamboo Lignin before and after Ethanol Organosolv Pretreatment. International Journal of Molecular Sciences. 2013; 14(11):21394-21413. https://doi.org/10.3390/ijms141121394
Chicago/Turabian StyleBai, Yuan-Yuan, Ling-Ping Xiao, Zheng-Jun Shi, and Run-Cang Sun. 2013. "Structural Variation of Bamboo Lignin before and after Ethanol Organosolv Pretreatment" International Journal of Molecular Sciences 14, no. 11: 21394-21413. https://doi.org/10.3390/ijms141121394
APA StyleBai, Y.-Y., Xiao, L.-P., Shi, Z.-J., & Sun, R.-C. (2013). Structural Variation of Bamboo Lignin before and after Ethanol Organosolv Pretreatment. International Journal of Molecular Sciences, 14(11), 21394-21413. https://doi.org/10.3390/ijms141121394