Early Amyloidogenic Oligomerization Studied through Fluorescence Lifetime Correlation Spectroscopy




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
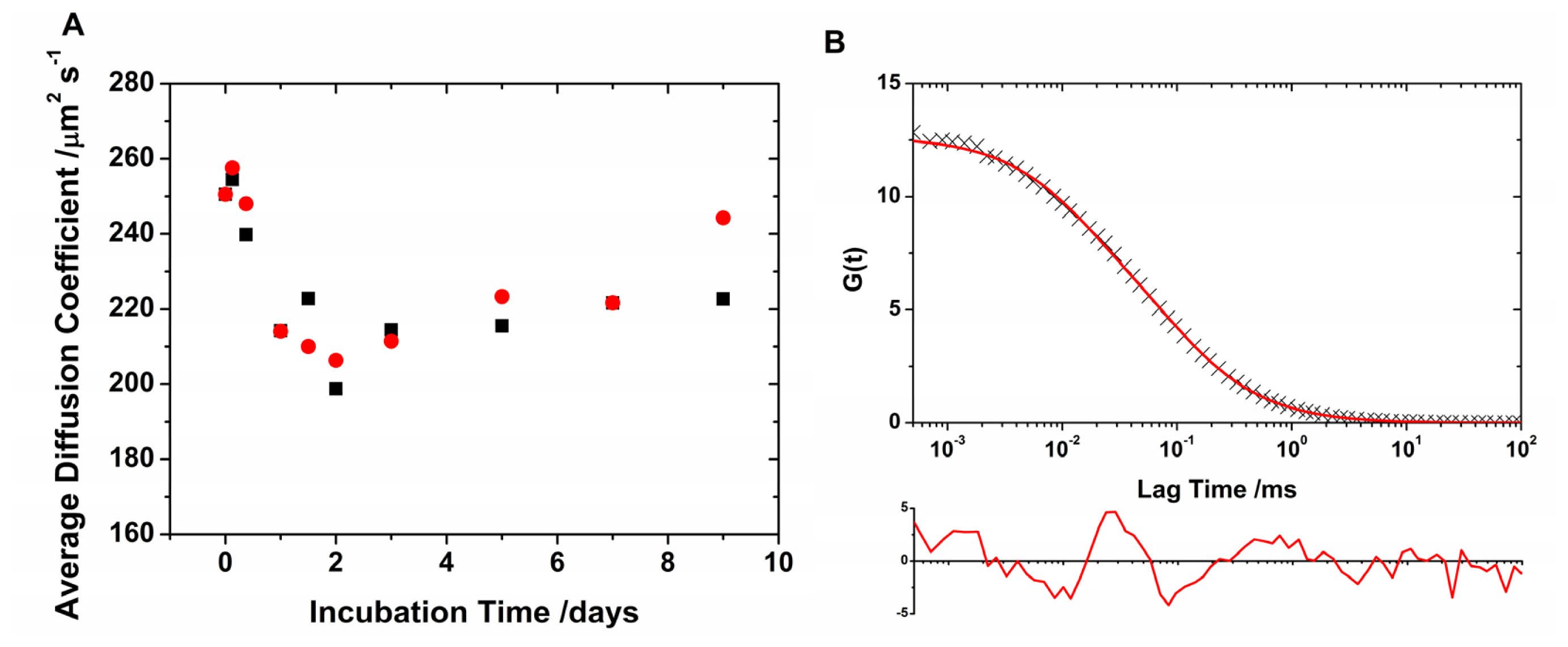
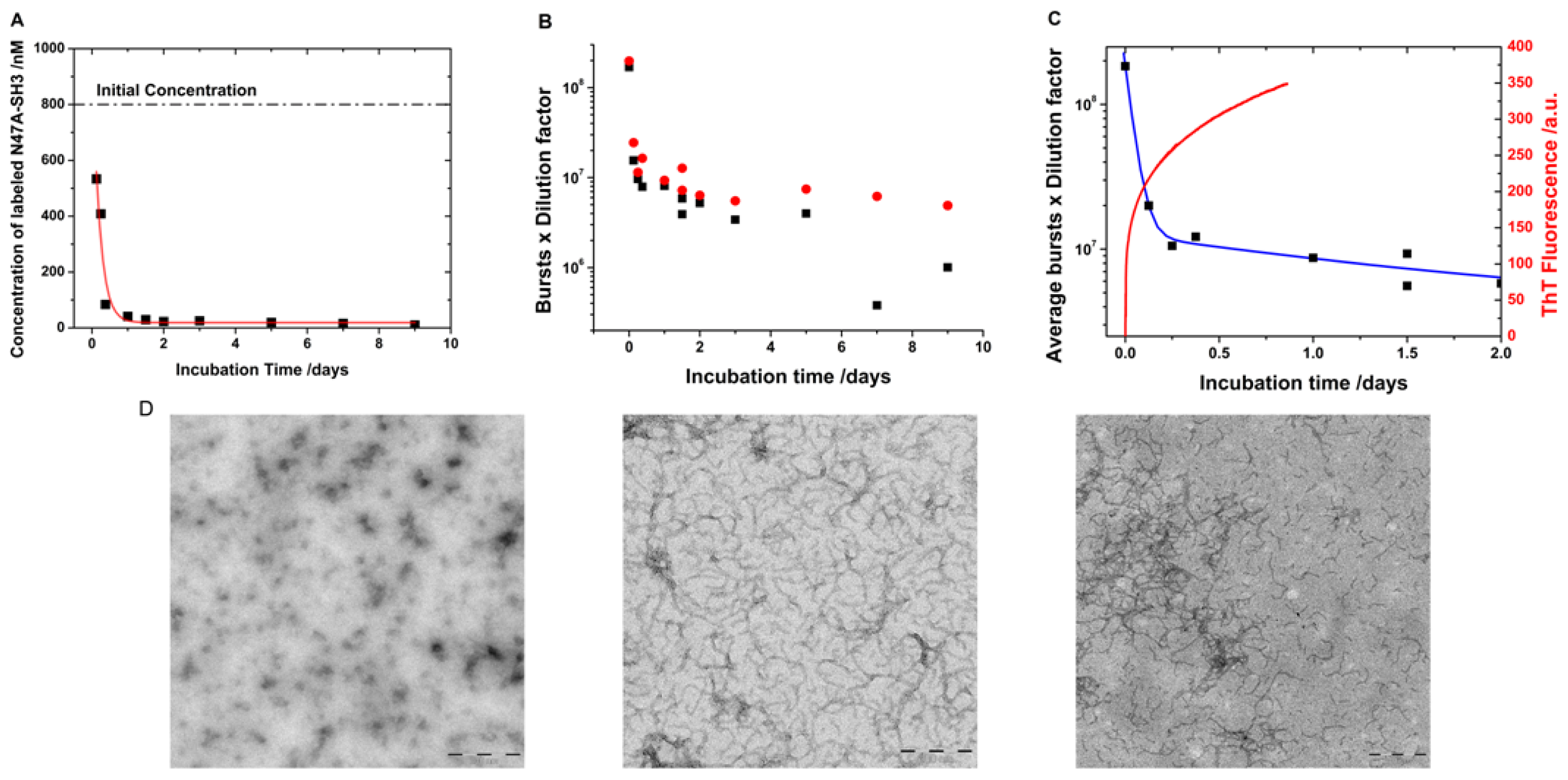
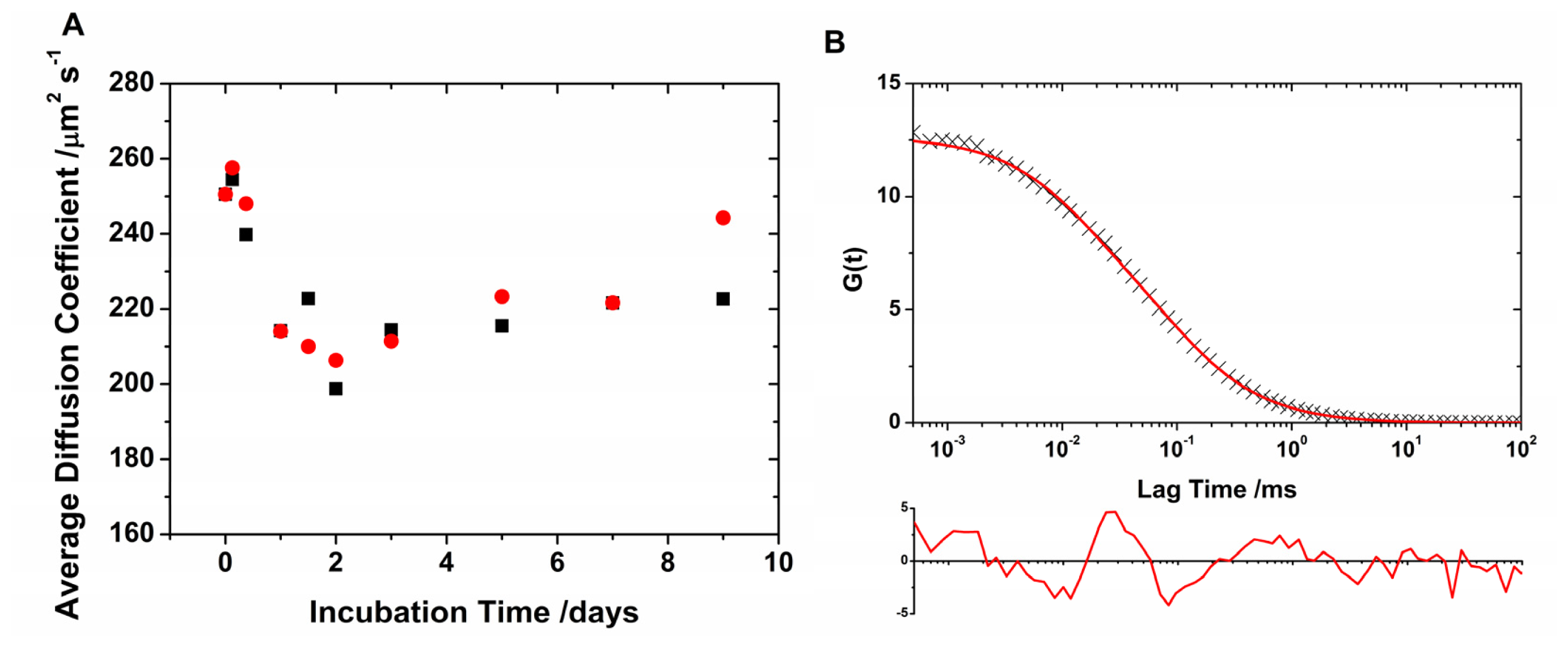
2.1. Translational Diffusion Coefficients Show Oligomer Growth Kinetics
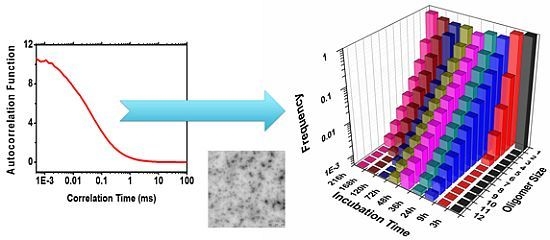
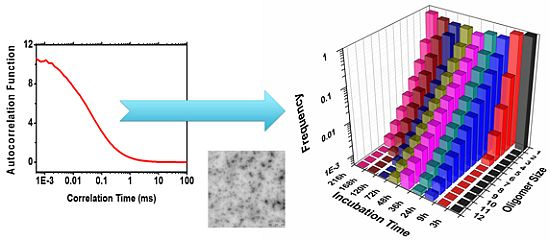
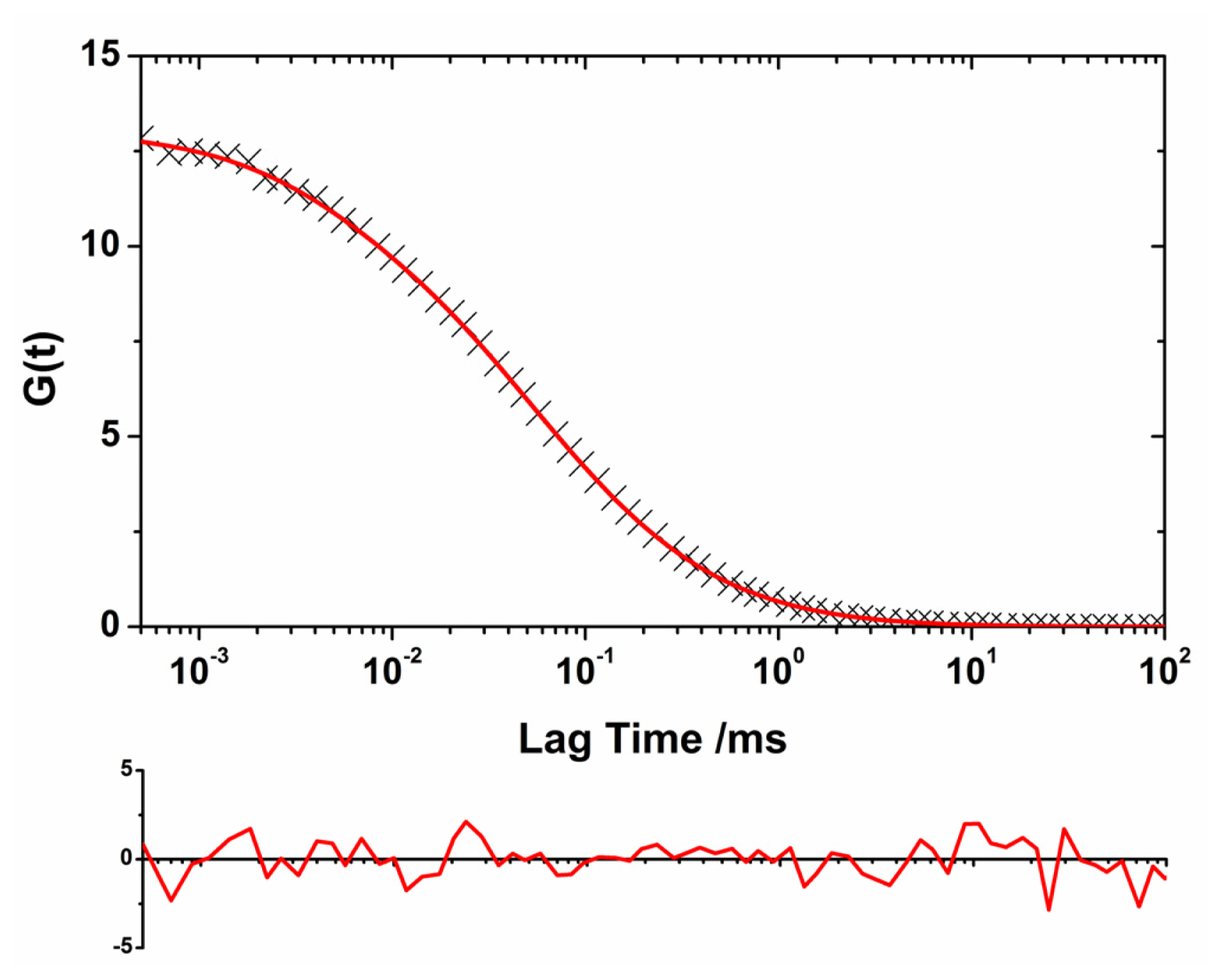
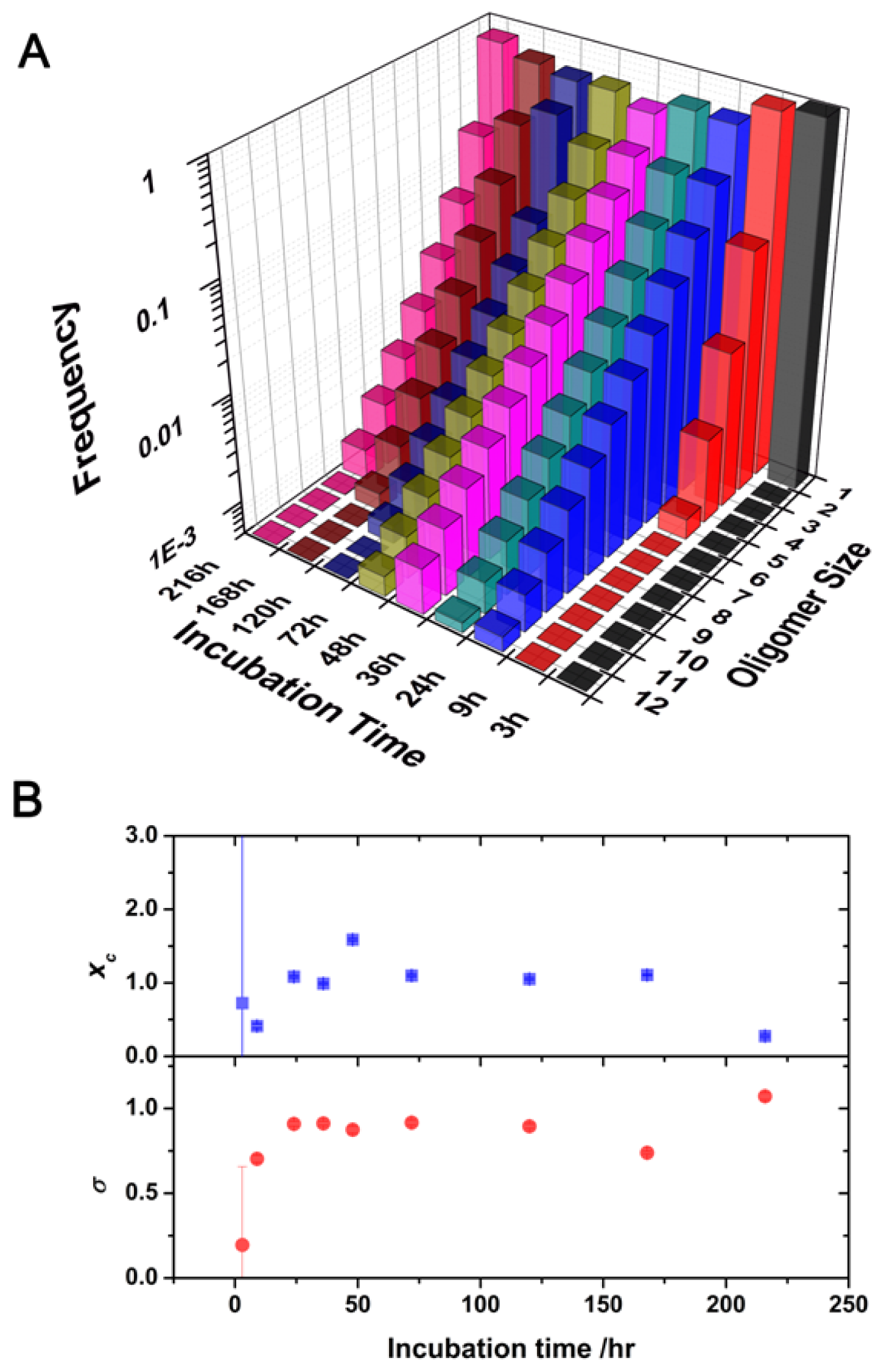
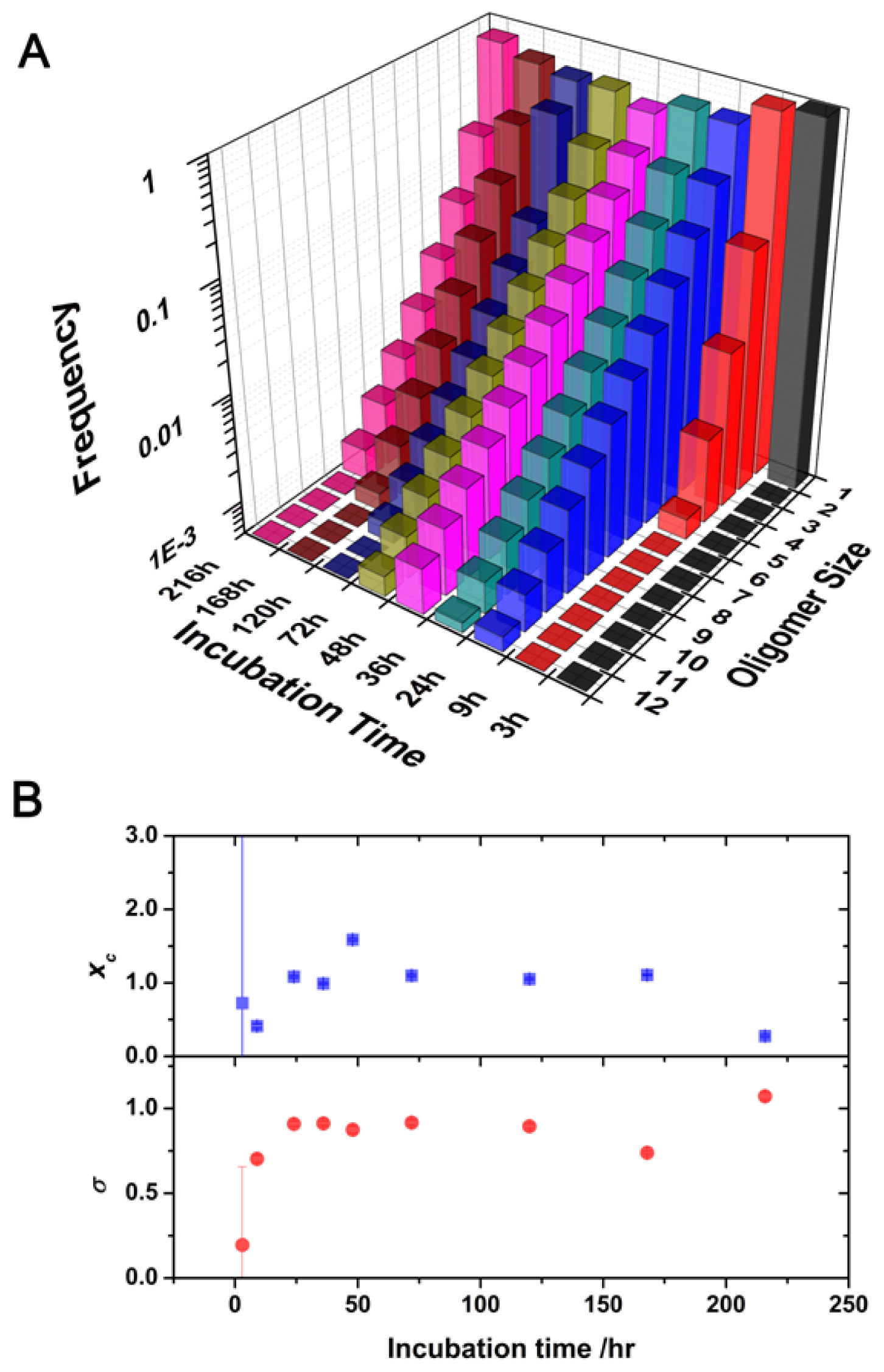
2.2. Oligomer Size Distributions from the FLCS Curves
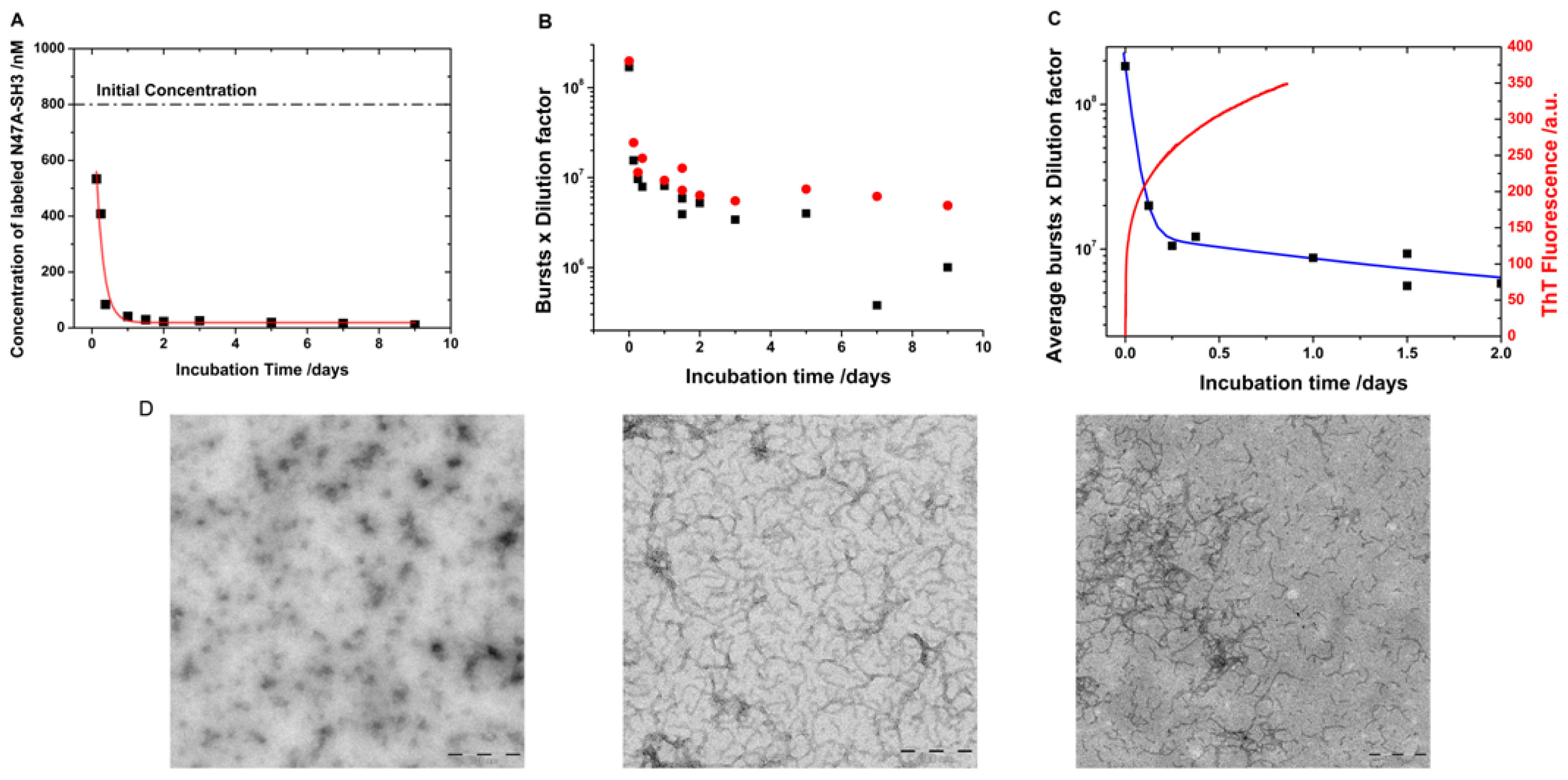
2.3. Kinetics of Monomer Incorporation
3. Experimental Section
3.1. Expression of the N47A Spc-SH3 Domain
3.2. Labeling and Purification of the Protein
3.3. Incubation and Formation of Amyloid Fibrils
3.4. Thioflavin T Binding Assay
3.5. Transmission Electron Micrographs
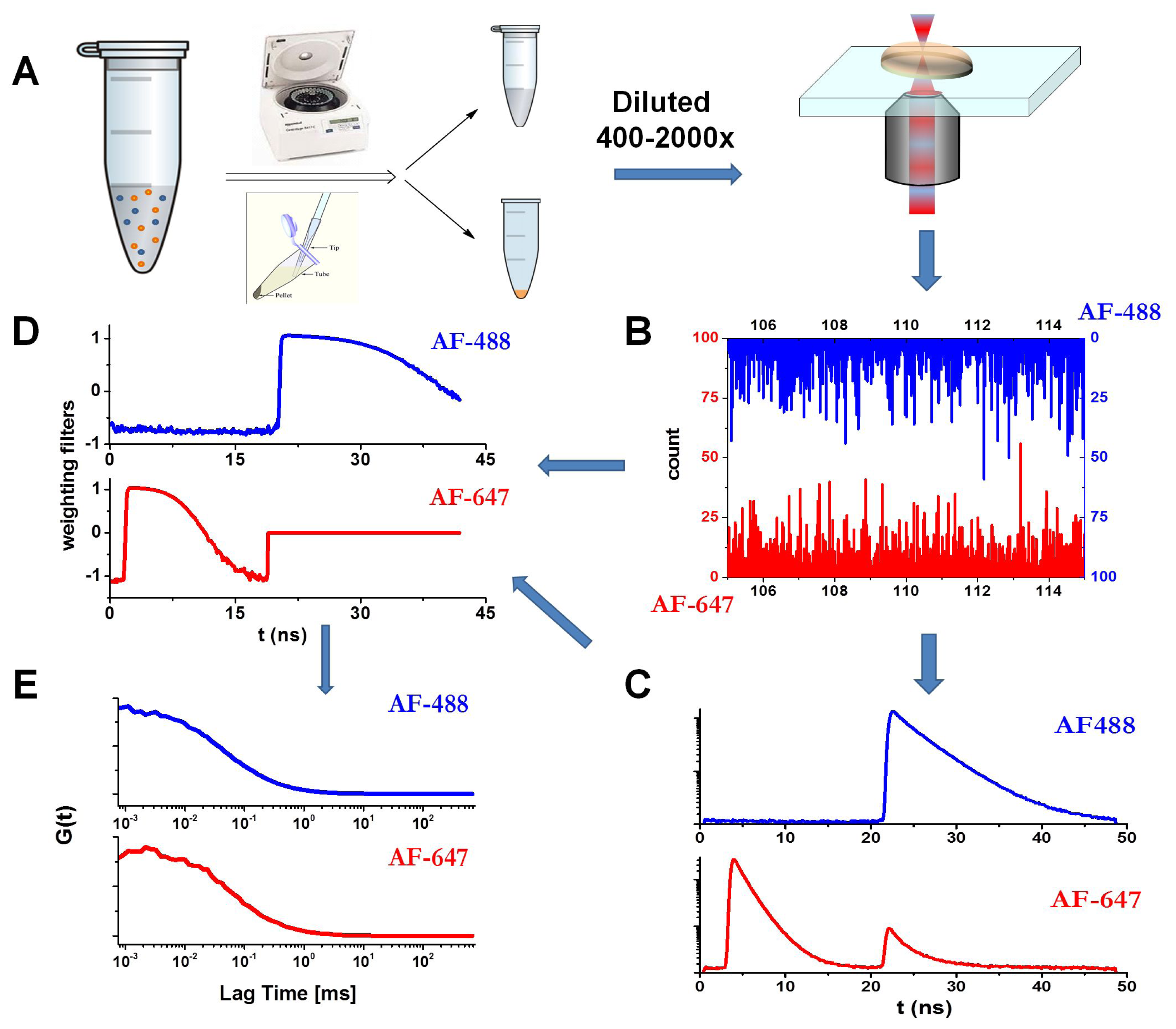
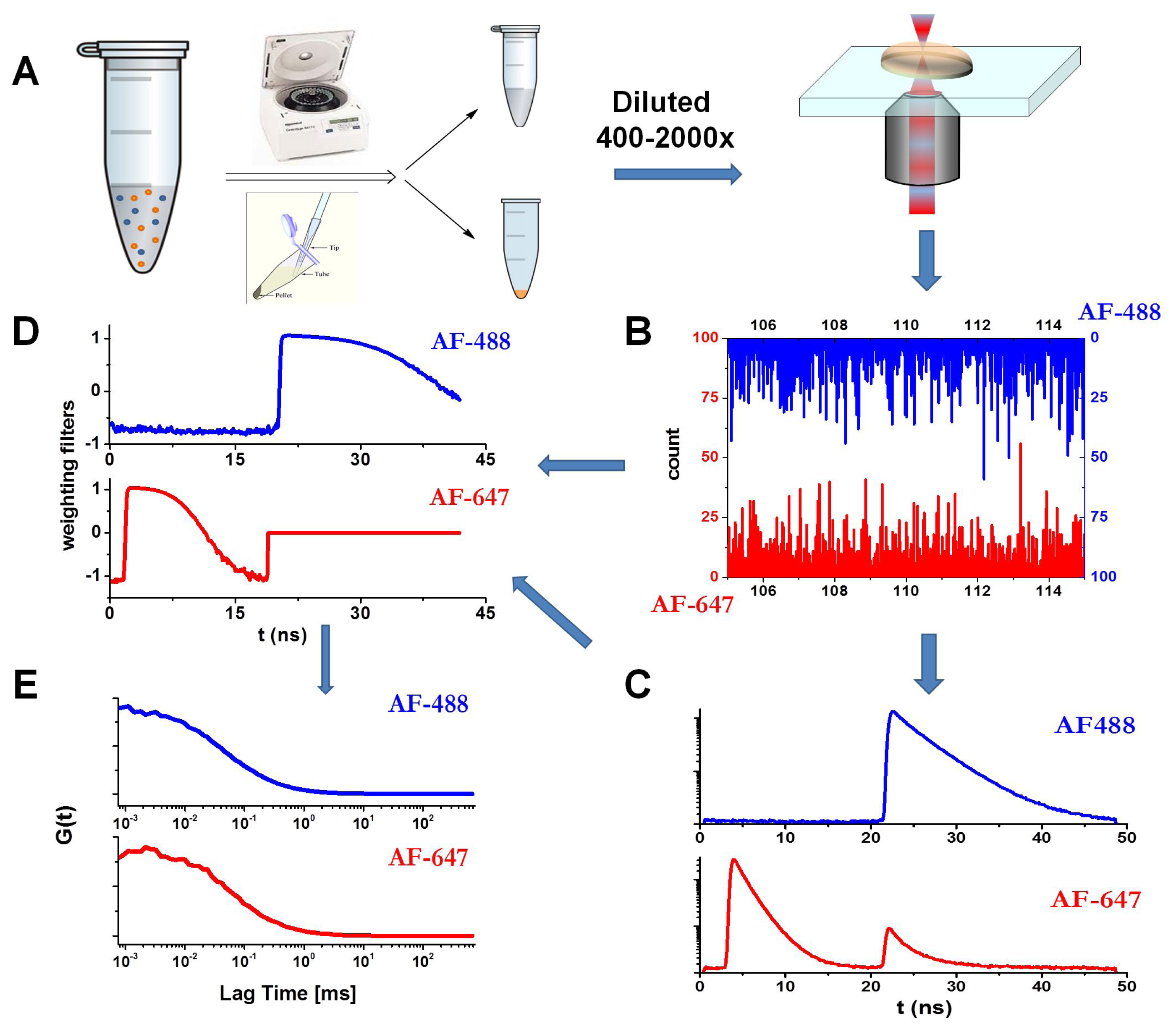
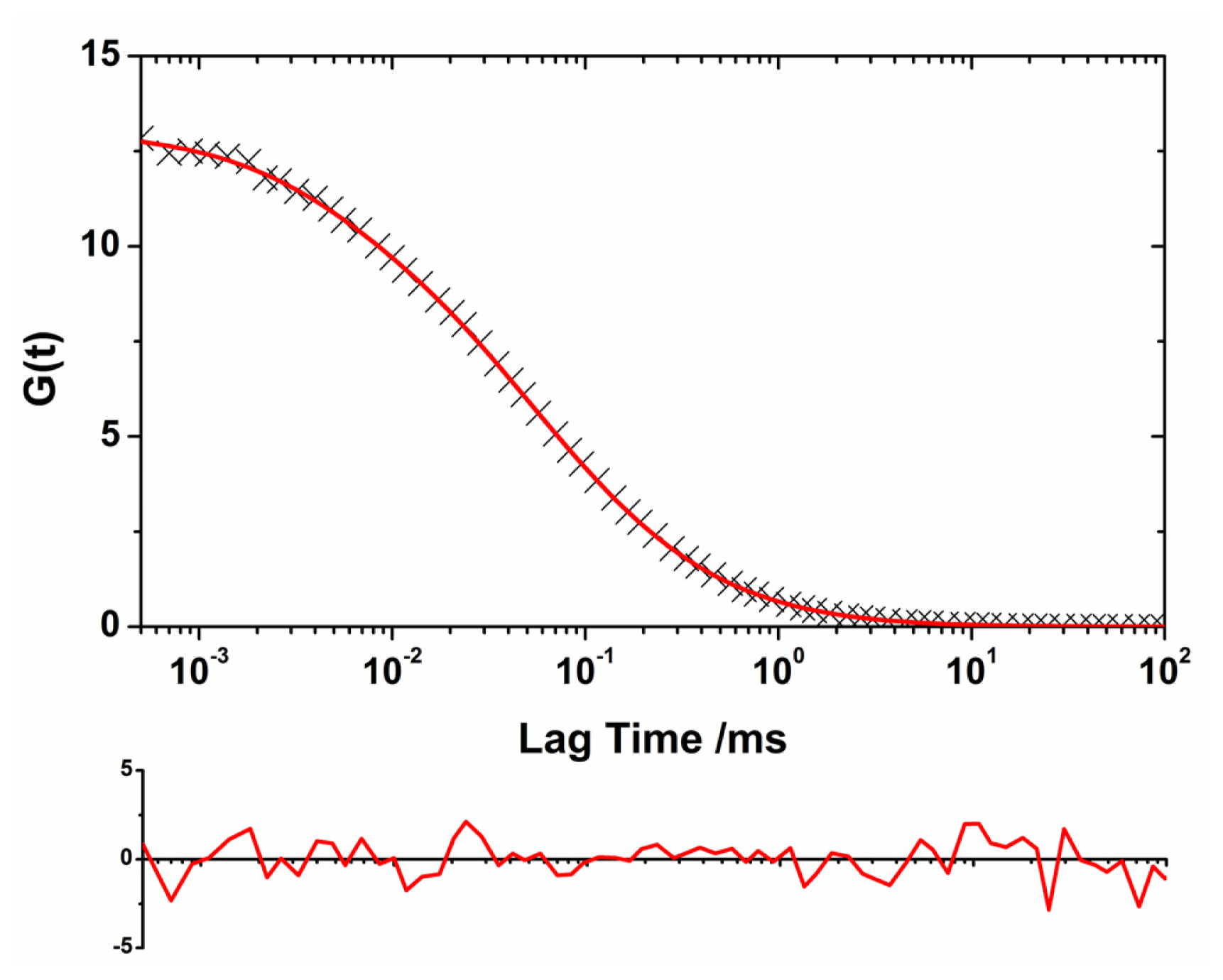
3.6. Fluorescence Lifetime Correlation Spectroscopy (FLCS) with Pulsed Interleaved Excitation (PIE)
3.7. Convolution of the Autocorrelation Function with Oligomer Size Distributions
4. Conclusions
Acknowledgments
References
- Rochet, J.-C.; Lansbury, P.T., Jr. Amyloid fibrillogenesis: Themes and variations. Curr. Opin. Struct. Biol 2000, 10, 60–68. [Google Scholar]
- Ferrone, F. Analysis of protein aggregation kinetics. Methods Enzymol 1999, 309, 256–274. [Google Scholar]
- Morris, A.M.; Watzky, M.A.; Finke, R.G. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim. Biophys. Acta 2009, 1794, 375–397. [Google Scholar]
- Knowles, T.P.J.; Waudby, C.A.; Devlin, G.L.; Cohen, S.I.A.; Aguzzi, A.; Vendruscolo, M.; Terentjev, E.M.; Welland, M.E.; Dobson, C.M. An analytical solution to the kinetics of breakable filament assembly. Science 2009, 326, 1533–1537. [Google Scholar]
- Guijarro, J.L.; Sunde, M.; Jones, J.A.; Campbell, I.D.; Dobson, C.M. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA 1998, 95, 4224–4228. [Google Scholar]
- Bemporad, F.; Chiti, F. Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol 2012, 19, 315–327. [Google Scholar]
- Glabe, C.G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 2006, 27, 570–575. [Google Scholar]
- Morel, B.; Casares, S.; Conejero-Lara, F. A single mutation induces amyloid aggregation in the alpha-spectrin SH3 domain: Analysis of the early stages of fibril formation. J. Mol. Biol 2006, 356, 453–468. [Google Scholar]
- Varela, L.; Morel, B.; Azuaga, A.I.; Conejero-Lara, F. A single mutation in an SH3 domain increases amyloid aggregation by accelerating nucleation, but not by destabilizing thermodynamically the native state. FEBS Lett 2009, 583, 801–806. [Google Scholar]
- Morel, B.; Varela, L.; Azuaga, A.I.; Conejero-Lara, F. Environmental conditions affect the kinetics of nucleation of amyloid fibrils and determine their morphology. Biophys. J 2010, 99, 3801–3810. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar]
- Glabe, C.G. amyloid oligomer structures and toxicity. Open Biol. J 2009, 2, 222–227. [Google Scholar]
- Tomic, J.L.; Pensalfini, A.; Head, E.; Glabe, C.G. Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol. Dis 2009, 35, 352–358. [Google Scholar]
- Gurlo, T.; Ryazantsev, S.; Huang, C.-J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for Proteotoxicity in β Cells in Type 2 Diabetes: Toxic Islet Amyloid Polypeptide Oligomers Form Intracellularly in the Secretory Pathway. Am. J. Pathol 2010, 176, 861–869. [Google Scholar]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol 2007, 8, 101–112. [Google Scholar]
- Joo, C.; Balci, H.; Ishitsuka, Y.; Buranachai, C.; Ha, T. Advances in Single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem 2008, 77, 51–76. [Google Scholar]
- Kapanidis, A.N.; Strick, T. Biology, one molecule at a time. Trends Biochem. Sci 2009, 34, 234–243. [Google Scholar]
- Orte, A.; Clarke, R.; Klenerman, D. Single-molecule two-colour coincidence detection to probe biomolecular associations. Biochem. Soc. Trans 2010, 038, 914–918. [Google Scholar]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem 2008, 283, 10992–11003. [Google Scholar]
- Gerard, M.; Debyser, Z.; Desender, L.; Kahle, P.J.; Baert, J.; Baekelandt, V.; Engelborghs, Y. The aggregation of alpha-synuclein is stimulated by FK506 binding proteins as shown by fluorescence correlation spectroscopy. FASEB J 2006, 20, 524–526. [Google Scholar]
- Nath, S.; Meuvis, J.; Hendrix, J.; Carl, S.A.; Engelborghs, Y. Early aggregation steps in α-synuclein as measured by fcs and fret: evidence for a contagious conformational change. Biophys. J 2010, 98, 1302–1311. [Google Scholar]
- Orte, A.; Clarke, R.; Balasubramanian, S.; Klenerman, D. Determination of the fraction and stoichiometry of femtomolar levels of biomolecular complexes in an excess of monomer using single-molecule, two-color coincidence detection. Anal. Chem 2006, 78, 7707–7715. [Google Scholar]
- Orte, A.; Birkett, N.R.; Clarke, R.W.; Devlin, G.L.; Dobson, C.M.; Klenerman, D. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2008, 105, 14424–14429. [Google Scholar]
- Chiou, A.; Hägglöf, P.; Orte, A.; Chen, A.Y.; Dunne, P.D.; Belorgey, D.; Karlsson-Li, S.; Lomas, D.A.; Klenerman, D. Probing neuroserpin polymerization and interaction with amyloid-β peptides using single molecule fluorescence. Biophys. J 2009, 97, 2306–2315. [Google Scholar]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β1–40 peptide. Nat. Struct. Mol. Biol 2012, 19, 79–83. [Google Scholar]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar]
- Widengren, J.; Kudryavtsev, V.; Antonik, M.; Berger, S.; Gerken, M.; Seidel, C.A.M. Single-molecule detection and identification of multiple species by multiparameter fluorescence detection. Anal. Chem 2006, 78, 2039–2050. [Google Scholar]
- Wahl, M.; Erdmann, R.; Lauritsen, K.; Rahn, H.J. Hardware solution for continuous time-resolved burst detection of single molecules in flow. Proc. SPIE 1998, 3259, 173–178. [Google Scholar]
- Lamb, D.C.; Schenk, A.; Röcker, C.; Scalfi-Happ, C.; Nienhaus, G.U. Sensitivity enhancement in fluorescence correlation spectroscopy of multiple species using time-gated detection. Biophys. J 2000, 79, 1129–1138. [Google Scholar]
- Kapusta, P.; Wahl, M.; Benda, A.; Hof, M.; Enderlein, J. Fluorescence lifetime correlation spectroscopy. J. Fluoresc 2007, 17, 43–48. [Google Scholar]
- Rüttinger, S.; Kapusta, P.; Patting, M.; Wahl, M.; Macdonald, R. On the resolution capabilities and limits of fluorescence lifetime correlation spectroscopy (FLCS) measurements. J. Fluoresc 2010, 20, 105–114. [Google Scholar]
- Gregor, I.; Enderlein, J. Time-resolved methods in biophysics. 3. Fluorescence lifetime correlation spectroscopy. Photochem. Photobiol. Sci 2007, 6, 13–18. [Google Scholar]
- Orte, A.; Ruedas-Rama, M.J.; Paredes, J.M.; Crovetto, L.; Alvarez-Pez, J.M. Dynamics of water-in-oil nanoemulsions revealed by fluorescence lifetime correlation spectroscopy. Langmuir 2011, 27, 12792–12799. [Google Scholar]
- Benda, A.; Hof, M.; Wahl, M.; Patting, M.; Erdmann, R.; Kapusta, P. TCSPC upgrade of a confocal FCS microscope. Rev. Sci. Instrum 2005, 76, 033106. [Google Scholar]
- Humpolícková, J.; Benda, A.; Sýkora, J.; Machán, R.; Kral, T.; Gasinska, B.; Enderlein, J.; Hof, M. Equilibrium dynamics of spermine-induced plasmid dna condensation revealed by fluorescence lifetime correlation spectroscopy. Biophys. J 2008, 94, L17–L19. [Google Scholar]
- Müller, B.K.; Zaychikov, E.; Bräuchle, C.; Lamb, D.C. Pulsed interleaved excitation. Biophys. J 2005, 89, 3508–3522. [Google Scholar]
- Paredes, J.M.; Orte, A.; Crovetto, L.; Alvarez-Pez, J.M.; Rios, R.; Ruedas-Rama, M.J.; Talavera, E.M. Similarity between the kinetic parameters of the buffer-mediated proton exchange reaction of a xanthenic derivative in its ground- and excited-state. Phys. Chem. Chem. Phys 2010, 12, 323–327. [Google Scholar]
- Ortega, A.; Amorós, D.; Torre, J.G.D.L. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic and residue-level models. Biophys. J 2011, 101, 892–898. [Google Scholar]
- Cohen, S.I.A.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Nucleated polymerization with secondary pathways III. Equilibrium behavior, filament length distribution and oligomer populations. J. Chem. Phys 2011, 135, 065107. [Google Scholar]
- Cheon, M.; Chang, I.; Mohanty, S.; Luheshi, L.M.; Dobson, C.M.; Vendruscolo, M.; Favrin, G. Structural reorganisation and potential toxicity of oligomeric species formed during the assembly of amyloid fibrils. PLoS Comput. Biol 2007, 3, e173. [Google Scholar]
- Sigurdson, C.J.; Nilsson, K.P.R.; Hornemann, S.; Manco, G.; Polymenidou, M.; Schwarz, P.; Leclerc, M.; Hammarström, P.; Wüthrich, K.; Aguzzi, A. Prion strain discrimination using luminescent conjugated polymers. Nat. Methods 2007, 12, 1023–1030. [Google Scholar]
- Nilsson, K.P.R.; Hammarström, P.; Ahlgren, F.; Herland, A.; Schnell, E.A.; Lindgren, M.; Westermark, G.T.; Inganäs, O. Conjugated polyelectrolytes—conformation-sensitive optical probes for staining and characterization of amyloid deposits. ChemBioChem 2006, 7, 1096–1104. [Google Scholar]
- Ham, T.J.V.; Esposito, A.; Kumita, J.R.; Hsu, S.-T.D.; Schierle, G.S.K.; Kaminski, C.F.; Dobson, C.M.; Nollen, E.A.A.; Bertoncini, C.W. Towards multiparametric fluorescent imaging of amyloid formation: Studies of a YFP model of α-synuclein aggregation. J. Mol. Biol 2010, 395, 627–642. [Google Scholar]
- Schierle, G.S.K.; Bertoncini, C.W.; Chan, F.T.S.; Goot, A.T.V.D.; Schwedler, S.; Skepper, J.; Schlachter, S.; Ham, T.V.; Esposito, A.; Kumita, J.R.; et al. A FRET sensor for non-invasive imaging of amyloid formation in vivo. Chem Phys Chem 2011, 12, 673–680. [Google Scholar]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils—current status. J. Chem. Biol 2010, 3, 1–18. [Google Scholar]
- Nilsson, K.P.R. Small organic probes as amyloid specific ligands–Past and recent molecular scaffolds. FEBS Lett 2009, 583, 2593–2599. [Google Scholar]
- Lindgren, M.; Hammarstrom, P. Amyloid oligomers: spectroscopic characterization of amyloidogenic protein states. FEBS J 2010, 277, 1380–1388. [Google Scholar]
- Lindgren, M.; Sörgjerd, K.; Hammarström, P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys. J 2005, 88, 4200–4212. [Google Scholar]
- Allsop, D.; Swanson, L.; Moore, S.; Davies, Y.; York, A.; El-Agnaf, O.M.A.; Soutar, I. Fluorescence anisotropy: A method for early detection of Alzheimer β-peptide (Aβ) aggregation. Biochem. Biophys. Res. Comm 2001, 285, 58–63. [Google Scholar]
- Sorgjerd, K.; Klingstedt, T.; Lindgren, M.; Kagedal, K.; Hammarström, P. Prefibrillar transthyretin oligomers and cold stored native tetrameric transthyretin are cytotoxic in cell culture. Biochem. Biophys. Res. Comm 2008, 377, 1072–1078. [Google Scholar]
- Hillger, F.; Nettels, D.; Dorsch, S.; Schuler, B. Detection and analysis of protein aggregation with confocal single molecule fluorescence spectroscopy. J. Fluoresc 2007, 17, 759–765. [Google Scholar]
- Haugland, R.P. Handbook of Fluorescent Probes and Research Products; Molecular Probes, Inc: OR, USA; p. 2002.
- LeVine, H., III. [18] Quantification of β-sheet amyloid fibril structures with thioflavin T. In Methods in Enzymology; Ronald, W., Ed.; Academic Press: New York, NY, USA, 1999; Volume 309, pp. 274–284. [Google Scholar]
- Edel, J.B.; Eid, J.S.; Meller, A. Accurate single molecule fret efficiency determination for surface immobilized DNA using maximum likelihood calculated lifetimes. J. Phys. Chem. B 2007, 111, 2986–2990. [Google Scholar]
- Haustein, E.; Schwille, P. Fluorescence correlation spectroscopy: Novel variations of an established technique. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 151–169. [Google Scholar]
- Burnett, G.R.; Rees, G.D.; Steytler, D.C.; Robinson, B.H. Fluorescence correlation spectroscopy of water-in-oil microemulsions: An application in specific characterisation of droplets containing biomolecules. Colloid Surf. A 2004, 250, 171–178. [Google Scholar]
- Serdyuk, I.N.; Zaccai, N.R.; Zaccai, J. Methods in Molecular Biophysics. Structure, Dynamics, Function; Cambridge University Press: Cambridge UK; p. 2007.
- Krouglova, T.; Vercammen, J.; Engelborghs, Y. Correct Diffusion coefficients of proteins in fluorescence correlation spectroscopy. Application to tubulin oligomers induced by Mg2+ and paclitaxel. Biophys. J 2004, 87, 2635–2646. [Google Scholar]
- Giurleo, J.T.; He, X.; Talaga, D.S. β-lactoglobulin assembles into amyloid through sequential aggregated intermediates. J. Mol. Biol 2008, 381, 1332–1348. [Google Scholar]
- He, X.; Giurleo, J.T.; Talaga, D.S. Role of small oligomers on the amyloidogenic aggregation free-energy landscape. J. Mol. Biol 2010, 395, 134–154. [Google Scholar]
- Carulla, N.; Caddy, G.L.; Hall, D.R.; Zurdo, J.; Gairi, M.; Feliz, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Molecular recycling within amyloid fibrils. Nature 2005, 436, 554–558. [Google Scholar]
- Chen, J.; Irudayaraj, J. Fluorescence lifetime cross correlation spectroscopy resolves EGFR and antagonist interaction in live cells. Anal. Chem 2010, 82, 6415–6421. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paredes, J.M.; Casares, S.; Ruedas-Rama, M.J.; Fernandez, E.; Castello, F.; Varela, L.; Orte, A. Early Amyloidogenic Oligomerization Studied through Fluorescence Lifetime Correlation Spectroscopy. Int. J. Mol. Sci. 2012, 13, 9400-9418. https://doi.org/10.3390/ijms13089400
Paredes JM, Casares S, Ruedas-Rama MJ, Fernandez E, Castello F, Varela L, Orte A. Early Amyloidogenic Oligomerization Studied through Fluorescence Lifetime Correlation Spectroscopy. International Journal of Molecular Sciences. 2012; 13(8):9400-9418. https://doi.org/10.3390/ijms13089400
Chicago/Turabian StyleParedes, Jose M., Salvador Casares, Maria J. Ruedas-Rama, Elena Fernandez, Fabio Castello, Lorena Varela, and Angel Orte. 2012. "Early Amyloidogenic Oligomerization Studied through Fluorescence Lifetime Correlation Spectroscopy" International Journal of Molecular Sciences 13, no. 8: 9400-9418. https://doi.org/10.3390/ijms13089400