Cancer Cell Cytotoxicities of 1-(4-Substitutedbenzoyl)-4-(4-chlorobenzhydryl)piperazine Derivatives

Abstract
:1. Introduction
2. Results and Discussion
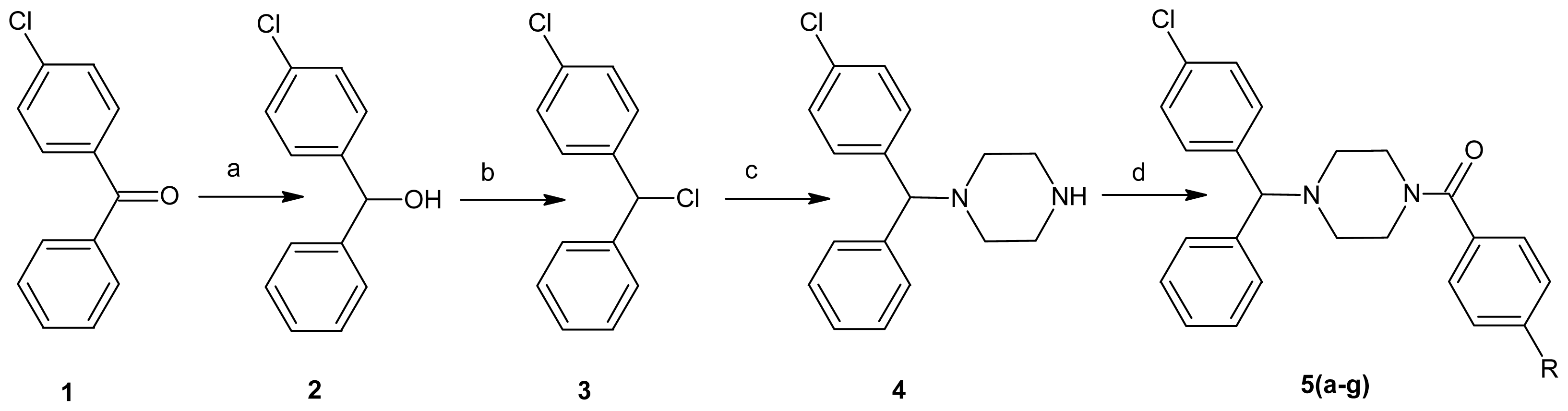
2.1. Chemistry
2.2. Cytotoxicity Analysis of the Compounds
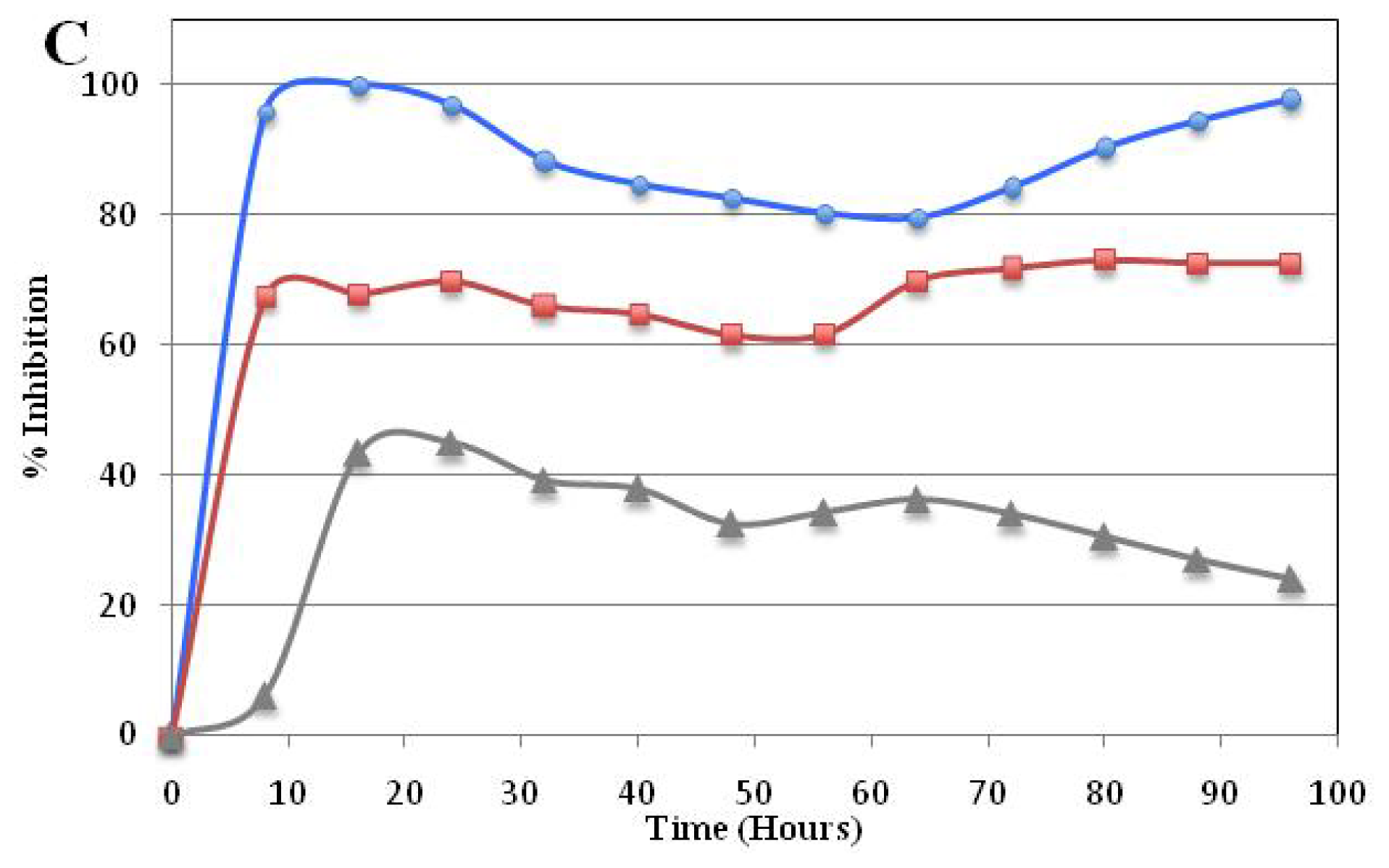
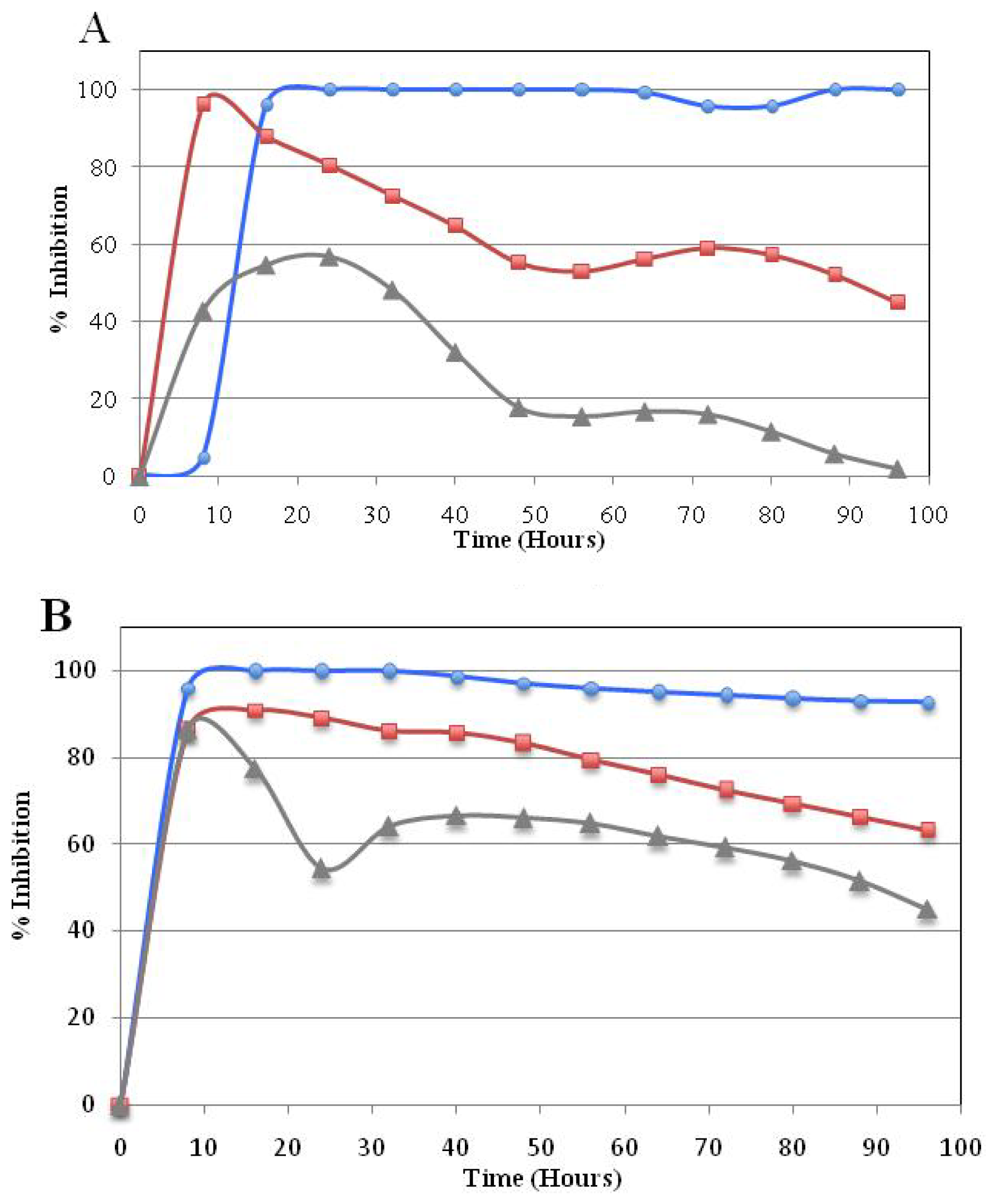
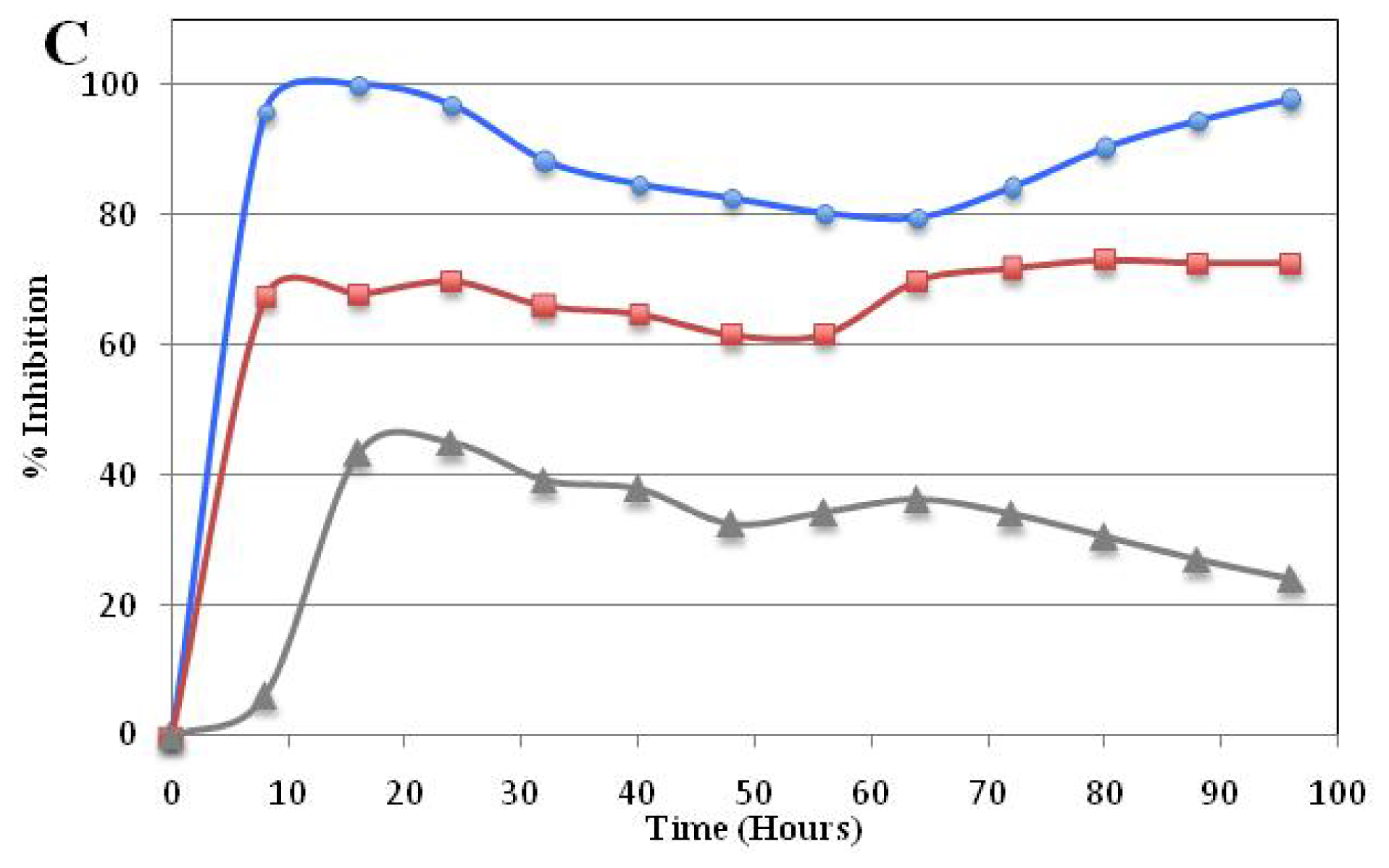
Real Time Cytotoxicity Analysis of Compound 5a
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of 1-(4-Chlorobenzhydryl)piperazine (4)
3.1.2. General Procedure for the Synthesis of 1-(4-Substitutedbenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salts 5(a–g)
3.1.2.1. 1-(4-Chlorobenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5a)
3.1.2.2. 1-(4-Fluorobenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5b)
3.1.2.3. 1-(4-Methoxybenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5c)
3.1.2.4. 1-(4-Bromobenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5d)
3.1.2.5. 1-(4-Nitrobenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5e)
3.1.2.6. 1-(4-Phenylbenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5f)
3.1.2.7. 1-(2,4-Difluorobenzoyl)-4-(4-chlorobenzhydryl)piperazine Hydrochloride Salt (5g)
3.2. Biology
3.2.1. Cell Culture
3.2.2. NCI-60 Sulphorhodamine B Assay
3.2.3. Time-dependent Cellular Response Profiles by Cell Electronic Sensing (xCELLigence)
4. Conclusion
Acknowledgments
- Conflict of InterestThe authors have declared no conflict of interest.
References
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar]
- Li, Q.; Xu, W. Novel anticancer targets and drug discovery in post genomic age. Curr. Med. Chem. Anticancer Agents 2005, 5, 53–55. [Google Scholar]
- Mencher, S.K.; Wang, L.G. Promiscuous drugs compared to selective drugs (promiscuity can be a virtue). BMC Clin. Pharmacol 2005, 5, 3–7. [Google Scholar]
- Jimeno, A.; Hidalgo, M. Multitargeted therapy: Can promiscuity be praised in an era of political correctness? Crit. Rev. Oncol. Hematol 2006, 59, 150–158. [Google Scholar]
- Berkheij, M.; van der Sluis, L.; Sewing, C.J.; den Boer, D.; Terpstra, J.W.; Hiemstra, H.; Bakker, W.I.I.; van den Hoogenband, A.; van Haarseveen, J.H. Synthesis of 2-substituted piperazines via direct α-lithiation. Tetrahedron Lett 2005, 46, 2369–2371. [Google Scholar]
- Guo, C.C.; Tong, R.B.; Li, K.L. Chloroalkyl piperazine and nitrogen mustard porphyrins: Synthesis and anticancer activity. Bioorg. Med. Chem 2004, 12, 2469–2475. [Google Scholar]
- Gillet, R.; Jeannesson, P.; Sefraoui, H.; Arnould-GueArin, M.; Kirkiacharian, L.S.; Jardillier, J.C.; Pieri, F. Piperazine derivatives of butyric acid as differentiating agents in human leukemic cells. Cancer Chemother. Pharmaco 1998, 41, 252–255. [Google Scholar]
- Gabriel, F.E.; Gu, J.; Slater, L.M.; Hara, K.; Jacobs, J.W. Tumor apoptosis induced by epoxide-containing piperazines, a new class of anti-cancer agents. Cancer Chemother. Pharmacol 2000, 45, 183–191. [Google Scholar]
- Hulme, C.; Ma, L.; Romano, J.; Morisette, M. Novel applications of ethyl glyoxalate with the Ugi MCR. Tetrahedron Lett 1999, 40, 5295–5299. [Google Scholar]
- Upadhayaya, R.S.; Sinha, N.; Jain, S.; Kishore, N.; Chandra, R.; Arora, S.K. Optically active antifungal azoles: Synthesis and antifungal activity of (2R,3S)-2-(2,4-difluorophenyl)-3-(5-[2-[4-aryl-piperazin-1-yl]-ethyl]-tetrazol-2-yl/1-yl)-1-[1,2,4]-triazol-1-yl-butan-2-ol. Bioorg. Med. Chem 2004, 12, 2225–2238. [Google Scholar]
- Choudhary, P.; Kumar, R.; Verma, K. Synthesis and antimicrobial activity of N-alkyl and N-aryl piperazine derivatives. Bioorg. Med. Chem 2006, 14, 1819–1826. [Google Scholar]
- Dorsey, B.D.; Levin, R.B.; McDaniel, S.L.; Vacca, J.P.; Guare, J.P.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; Schleif, W.A. The design of a potent and orally bioavailable HIV protease inhibitor. J. Med. Chem 1994, 37, 3443–3451. [Google Scholar]
- Askin, D.; Eng, K.K.; Rossen, K.; Purick, R.M.; Wells, K.M.; Volante, R.P.; Reider, P.J. Highly diasteroselective reaction of a chiral, non-racemic amide enolate with (S)-glycidyl tosylate. synthesis of the orally active HIV-1 protease inhibitor L-735,524. Tetrahedron Lett 1994, 35, 673–676. [Google Scholar]
- Rosen, K.; Weissman, S.A.; Sager, J.; Reamer, R.A.; Askin, D.; Volante, R.P.; Reider, P.J. Asymmetric Hydrogenation of tetrahydropyrazines: Synthesis of (S)-piperazine 2-tert-butylcarboxamide, an intermediate in the preparation of the HIV protease inhibitor indinavir. Tetrahedron Lett 1995, 36, 6419–6422. [Google Scholar]
- Pyridine Derivatives Having Antidepressant Activity U.S. Patent 3,865,828, 11 February 1975.
- Yoshida, M.; Maehara, Y.; Sugimachi, K. MST-16, a novel bis-dioxopiperazine anticancer agent, ameliorates doxorubicin-induced acute toxicity while maintaining antitumor efficacy. Clin. Cancer Res 1999, 5, 4295–4300. [Google Scholar]
- Matulenko, M.A.; Hakeem, A.A.; Kolasa, T.; Nakane, M.; Terranova, M.A.; Uchic, M.E.; Miller, L.N.; Chang, R.; Donnelly-Roberts, D.L.; Namovic, M.T.; et al. Synthesis and functional activity of (2-aryl-1-piperazinyl)-N-(3-methylphenyl)acetamides: Selective dopamine D4 receptor agonists. Bioorg. Med. Chem 2004, 12, 3471–3483. [Google Scholar]
- Glase, S.A.; Akunne, H.C.; Georgic, L.M.; Haffner, T.G.; Mackenzie, R.G.; Manley, P.J.; Pugsley, T.A.; Wise, L.D. Substituted [(4-phenylpiperazinyl)-methyl]benzamides: Selective dopamine D4 agonists. J. Med. Chem 1997, 40, 1771–1772. [Google Scholar]
- Perrone, R.; Berardi, F.; Colabufo, N.A.; Leopoldo, M.; Tortorella, V. A structure-affinity relationship study on derivatives of N-[2-[4-(4-chlorophenyl)piperazin-1-yl]ethyl]-3-methoxybenzamide, a high-affinity and selective D4 receptor ligand. J. Med. Chem 2000, 43, 270–277. [Google Scholar]
- Chern, J.H.; Shia, K.S.; Hsu, T.A.; Tai, C.L.; Lee, C.C.; Lee, Y.C.; Chang, C.S.; Tseng, S.N.; Shih, S.R. Design, synthesis, and structure-activity relationship of pyrazolo[3,4-d]pyrimidines: A novel class of potent enterovirus inhibitors. Bioorg. Med. Chem. Lett 2004, 14, 2519–2525. [Google Scholar]
- Kimura, M.; Masudaa, T.; Yamadaa, K.; Mitania, M.; Kubota, N.; Kawakatsua, N.; Kishii, K.; Inazub, M.; Kiuchic, Y.; Oguchid, K.; et al. Syntheses of novel diphenyl piperazine derivatives and their activities as inhibitors of dopamine uptake in the central nervous system. Bioorg. Med. Chem 2003, 11, 1621–1630. [Google Scholar]
- Kimura, M.; Masudaa, T.; Yamadaa, K.; Mitania, M.; Kubota, N.; Kawakatsua, N.; Kishii, K.; Inazu, M.; Kiuchi, Y.; Oguchi, K.; et al. Novel diphenylalkyl piperazine derivatives with high affinities for the dopamine transporter. Bioorg. Med. Chem 2003, 11, 3953–3963. [Google Scholar]
- Kimara, M.; Masuda, T.; Yamada, K. Antioxidative activities of novel diphenylalkyl piperazine derivatives with high affinities for the dopamine transporter. Bioorg. Med. Chem. Lett 2004, 14, 4287–4290. [Google Scholar]
- Amin, E.A.; Welsh, W.J. Three-dimensional quantitative structure—Activity relationship (3D-QSAR) models for a novel class of piperazine-based stromelysin-1 (MMP-3) inhibitors: Applying a “divide and conquer” strategy. J. Med. Chem 2003, 44, 3849–3855. [Google Scholar]
- Braybrooke, J.P.; O’Byrne, K.J.; Propper, D.J.; Blann, A.; Saunders, M.; Dobbs, N.; Han, C.; Woodhull, J.; Mitchell, K.; Crew, J.; et al. A phase II study of razoxane, an antiangiogenic topoisomerase II inhibitor, in renal cell cancer with assessment of potential surrogate markers of angiogenesis. Clin. Cancer Res 2000, 6, 4697–4704. [Google Scholar]
- Wilson, W.D.; Barton, H.J.; Tanious, F.A.; Kong, S.B.; Strekowski, L. The interaction with DNA of unfused aromatic systems containing terminal piperazino substituents: Intercalation and groove-binding. Biophys. Chem 1990, 35, 227–243. [Google Scholar]
- Sampson, J.J.; Donkor, I.O.; Huang, T.L.; Adunyah, S.E. Novel piperazine induces apoptosis in U937 cells. Int. J. Biochem. Mol. Biol 2011, 2, 78–88. [Google Scholar]
- Narendra Sharath Chandra, J.N.; Sadashiva, C.T.; Kavitha, C.V.; Rangappa, K.S. Synthesis and in vitro antimicrobial studies of medicinally important novel N-alkyl and N-sulfonyl derivatives of 1-[bis(4-fluorophenyl)-methyl]piperazine. Bioorg. Med. Chem 2006, 14, 6621–6627. [Google Scholar]
- Ananda Kumar, C.S.; Nanjuda Swamy, S.; Thimmegawda, N.R.; Benaka Prasad, S.B.; Yip, G.W.; Rangappa, K.S. Synthesis and evaluation of 1-benzhydryl-sulfonyl-piperazine derivatives as inhibitors of MDA-MB-231 human breast cancer cell proliferation. Med. Chem. Res 2007, 16, 179–187. [Google Scholar]
- Ananda Kumar, C.S.; Benaka Prasad, S.B.; Viyana, K.; Chandrappa, S.; Thimmegawda, R.; Sunil Kumar, Y.C.; Sanjay, S.; Rangappa, K.S. Synthesis and in vitro antiproliferative activity of novel 1-benzhydrylpiperazine derivatives against human cancer cell lines. Eur. J. Med. Chem 2009, 44, 1223–1229. [Google Scholar]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer. Inst 1990, 4, 1107–1112. [Google Scholar]
- Kirstein, S.L.; Atienza, J.M.; Xi, B.; Zhu, J.; Yu, N.; Wang, X.; Xu, X.; Abassi, Y.A. Live cell quality control and utility of real-time cell electronic sensing for assay development. Assay Drug Dev. Technol 2006, 4, 545–553. [Google Scholar]
- Buontempo, F.; Ersahin, T.; Missiroli, S.; Senturk, S.; Etro, D.; Ozturk, M.; Capitani, S.; Cetin-Atalay, R.; Neri, M.L. Inhibition of Akt signaling in hepatoma cells induces apoptotic cell death independent of Akt activation status. Invest. New Drugs 2010. [Google Scholar] [CrossRef]
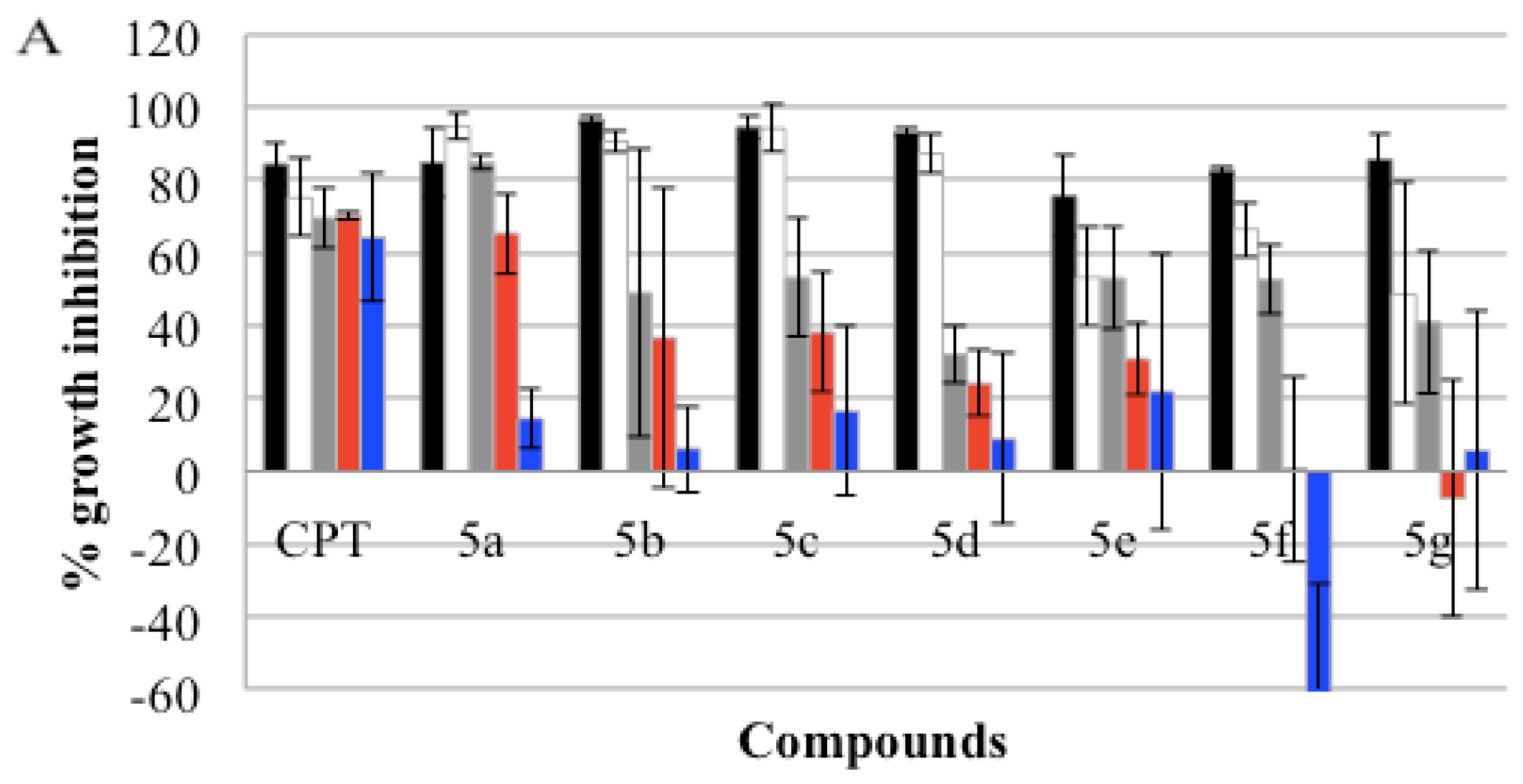
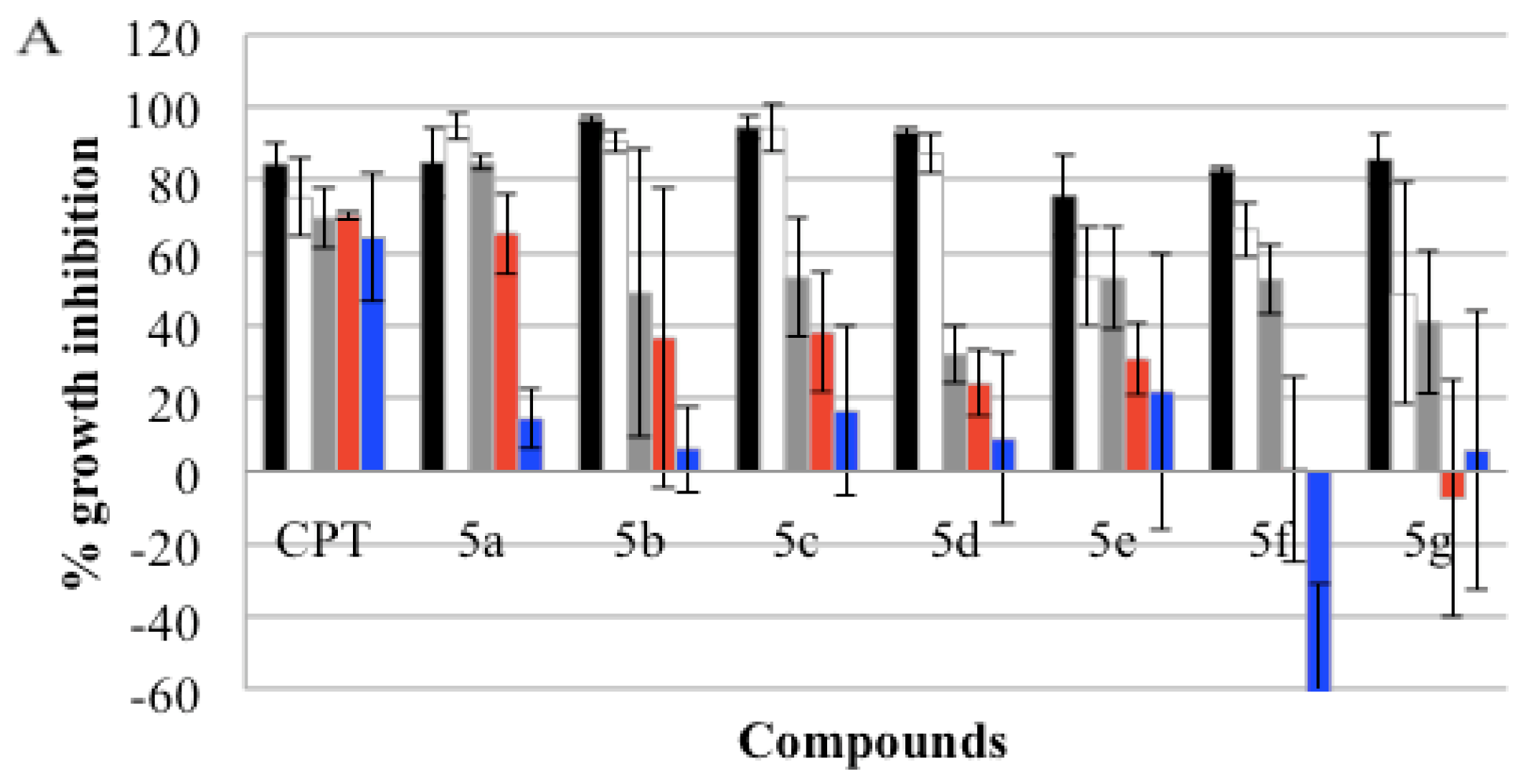
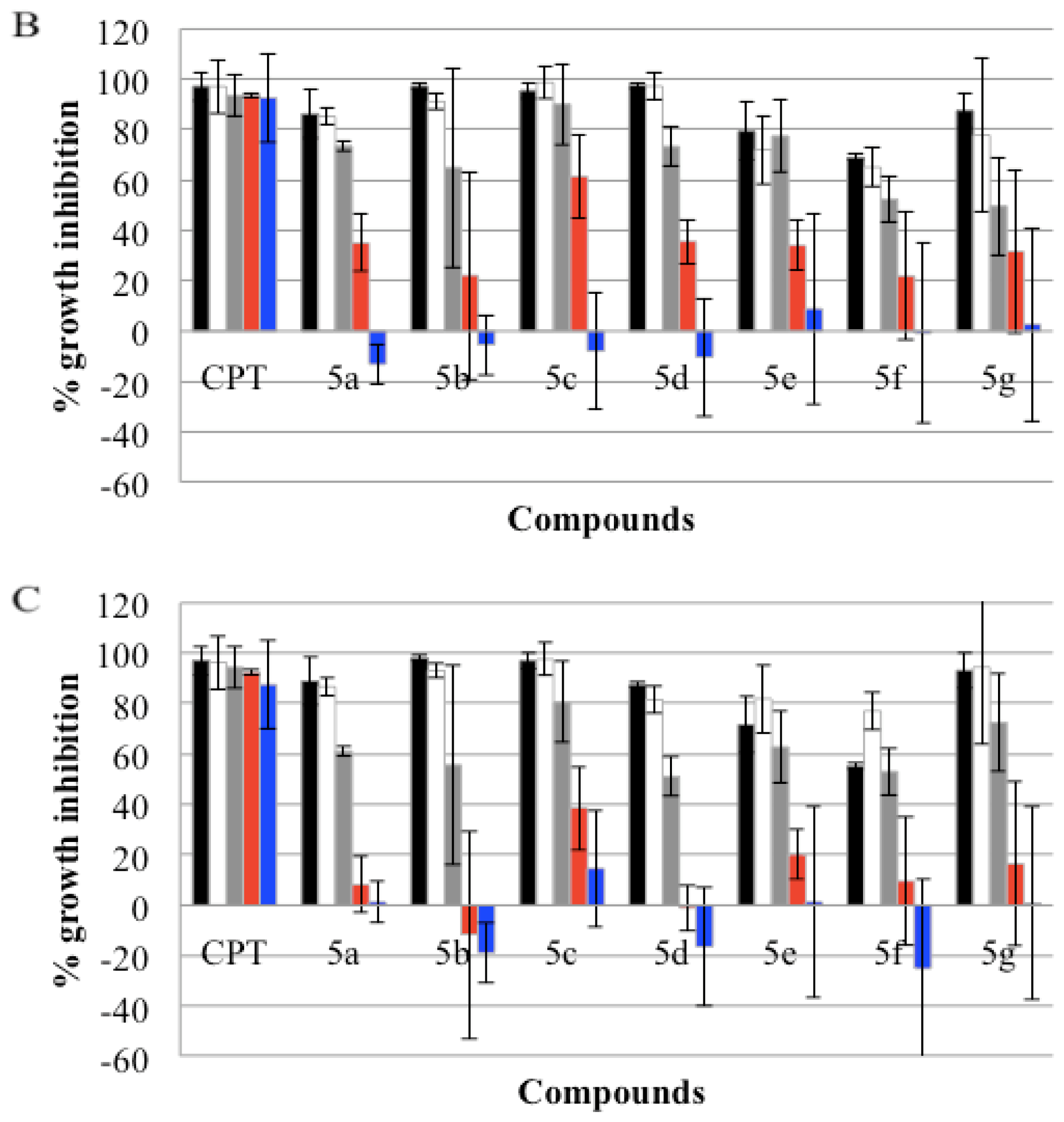
 ), GI50(
), GI50(
 ), and GI25 (
), and GI25 (
 ) concentrations, which were calculated from Table 2. Real-time cell growth surveillance by the cell impedance-based xCELLigence data acquisition system was performed in triplicate.
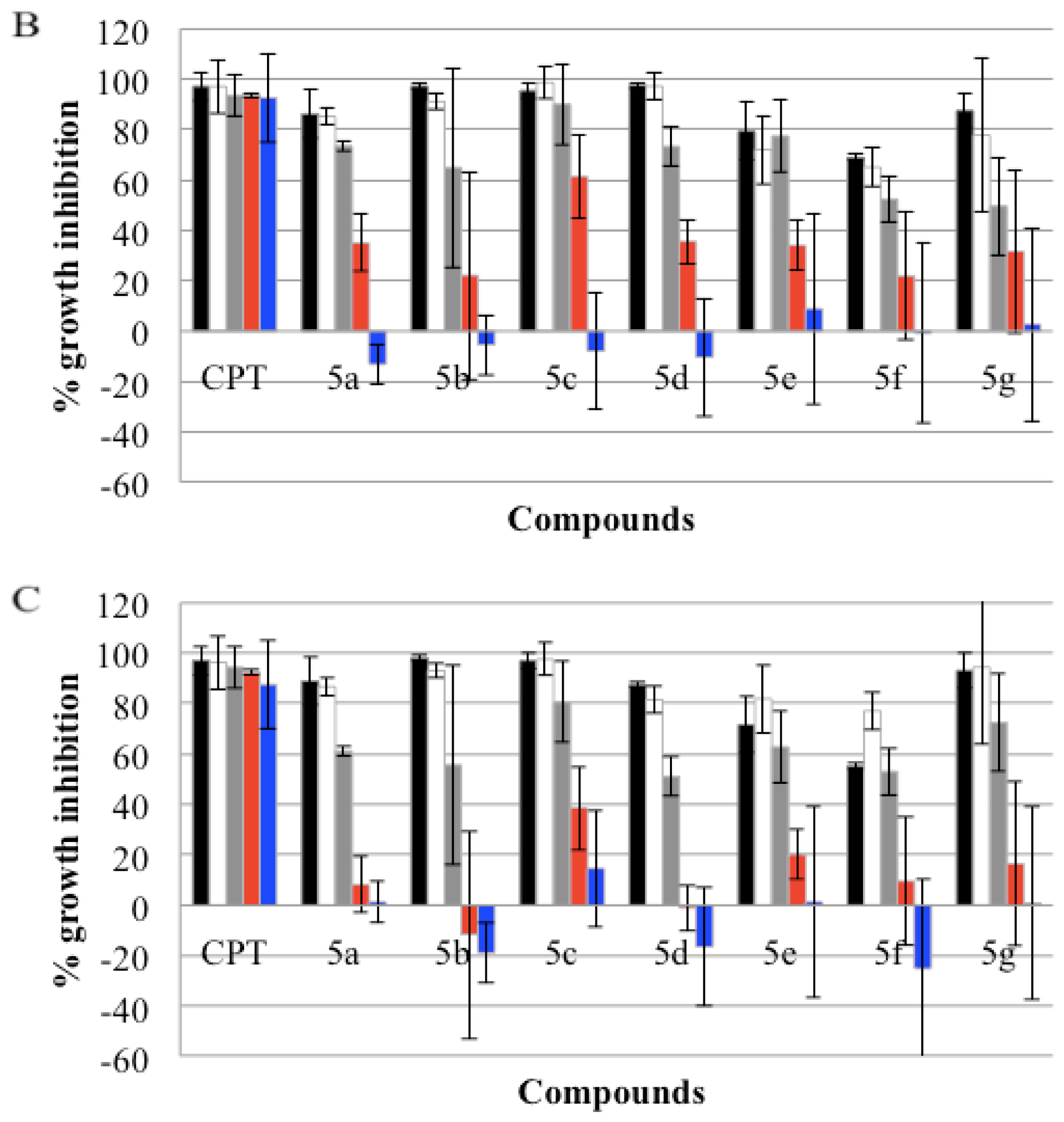
), GI50(
), and GI25 (
) concentrations, which were calculated from Table 2. Real-time cell growth surveillance by the cell impedance-based xCELLigence data acquisition system was performed in triplicate.
) concentrations, which were calculated from Table 2. Real-time cell growth surveillance by the cell impedance-based xCELLigence data acquisition system was performed in triplicate.
), GI50(
), and GI25 (
) concentrations, which were calculated from Table 2. Real-time cell growth surveillance by the cell impedance-based xCELLigence data acquisition system was performed in triplicate.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
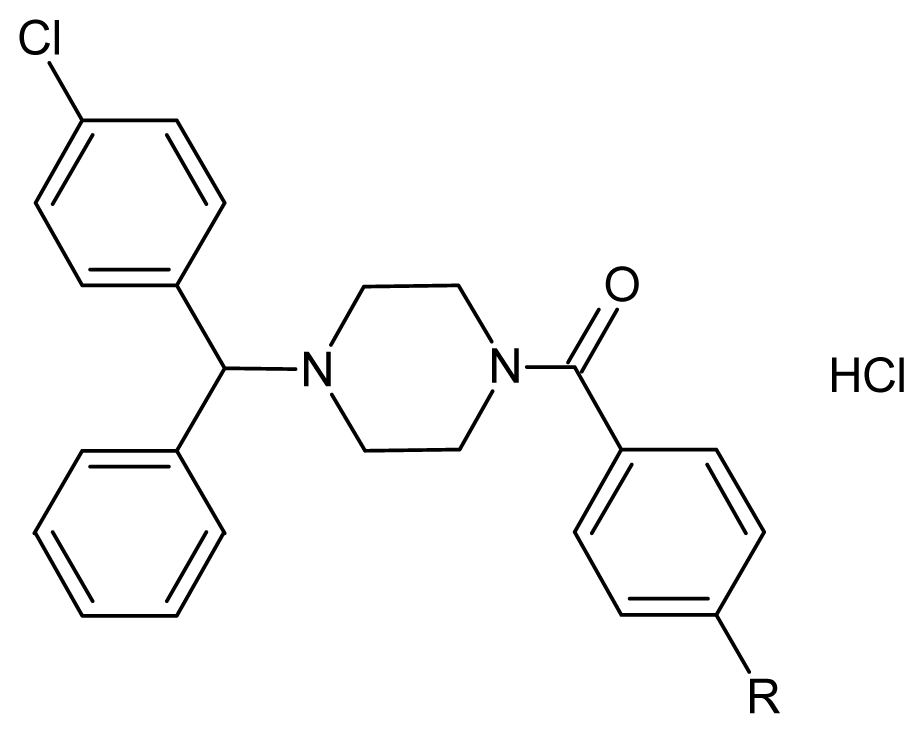{kind=link}
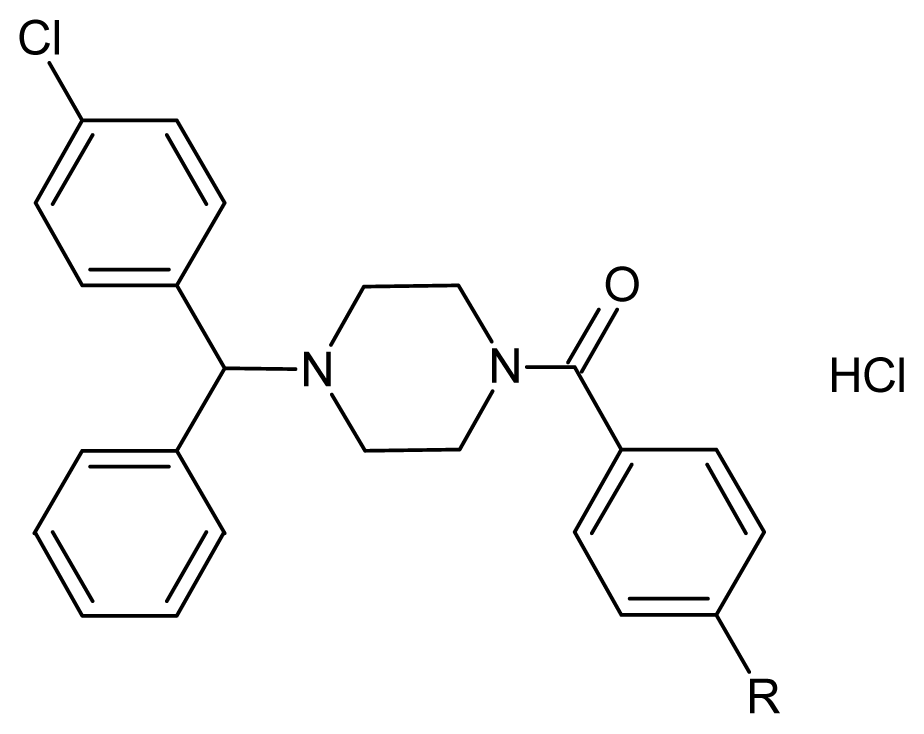 | ||||
|---|---|---|---|---|
| Compounds | R | Formula | Yield (%) | m.p. (°C) |
| 5a a | Cl | C24H23Cl3N2O | 75 | 172.8 |
| 5b a | F | C24H23Cl2FN2O | 81 | 144. 2 |
| 5c | OCH3 | C25H26Cl2N2O2 | 73 | 132.3 |
| 5d | Br | C24H23BrCl2N2O | 75 | 155.3 |
| 5e | NO2 | C24H23Cl2N3O3 | 76 | 153.4 |
| 5f | Ph | C30H28Cl2N2O | 70 | 152.1 |
| 5g a | 2,4-di F | C24H22Cl2F2N2O | 90 | 215.5 decomp |
| 5a | 5b | 5c | 5d | 5e | 5f | 5g | CPT | 5-F Uracil | |
|---|---|---|---|---|---|---|---|---|---|
| HUH7 | 4.64 | 8.43 | 7.37 | 10.29 | 11.80 | 15.46 | 16.35 | 0.15 | 30.66 |
| FOCUS | 4.15 | 11.19 | 6.06 | 10.81 | 10.04 | 11.41 | 7.77 | 1> | 7.69 |
| MAHLAVU | 7.59 | 9.86 | 7.35 | 10.97 | 14.82 | 29.46 | 7.00 | 1> | 9.97 |
| HEPG2 | 9.37 | 7.32 | 7.22 | 13.00 | 13.72 | 18.93 | 14.61 | 0.00 | 5.07 |
| HEP3B | 2.49 | 6.82 | 1.67 | 5.59 | 2.59 | 8.58 | 7.03 | 3.61 | 15.22 |
| MCF7 | 9.12 | 9.05 | 6.09 | 8.07 | 8.47 | 13.83 | 10.07 | 1> | 3.51 |
| BT20 | 18.82 | 16.92 | 11.62 | 42.71 | 33.94 | 120.52 | 18.91 | 0.07 | 47.30 |
| T47D | 1.91 | 6.36 | 0.44 | 6.45 | 0.31 | 8.78 | 0.85 | 1> | 8.91 |
| CAMA-1 | 1.48 | 9.78 | 1.22 | 8.07 | 4.99 | 13.62 | 2.73 | 0.07 | 1.28 |
| HCT116 | 10.23 | 11.49 | 6.18 | 12.55 | 10.89 | 16.33 | 8.68 | 1> | 18.67 |
| KATO-3 | 11.11 | 10.64 | 10.07 | 17.99 | 17.77 | 149.55 | 13.84 | 1> | ND |
| MFE-296 | 24.97 | 16.51 | 9.73 | 34.71 | 24.59 | 321.84 | 18.72 | 1> | 30.68 |
| MCF-12A | 5.12 | 8.06 | 6.6 | 23.26 | 13.91 | 299.66 | 12.3 | 1> | ND |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yarim, M.; Koksal, M.; Durmaz, I.; Atalay, R. Cancer Cell Cytotoxicities of 1-(4-Substitutedbenzoyl)-4-(4-chlorobenzhydryl)piperazine Derivatives. Int. J. Mol. Sci. 2012, 13, 8071-8085. https://doi.org/10.3390/ijms13078071
Yarim M, Koksal M, Durmaz I, Atalay R. Cancer Cell Cytotoxicities of 1-(4-Substitutedbenzoyl)-4-(4-chlorobenzhydryl)piperazine Derivatives. International Journal of Molecular Sciences. 2012; 13(7):8071-8085. https://doi.org/10.3390/ijms13078071
Chicago/Turabian StyleYarim, Mine, Meric Koksal, Irem Durmaz, and Rengul Atalay. 2012. "Cancer Cell Cytotoxicities of 1-(4-Substitutedbenzoyl)-4-(4-chlorobenzhydryl)piperazine Derivatives" International Journal of Molecular Sciences 13, no. 7: 8071-8085. https://doi.org/10.3390/ijms13078071